

Координационная химия, 2019, T. 45, № 6, стр. 366-371
Синтез, строение и гаптотропные взаимопревращения циклогептатриенил-ацетонитрил-карбонильных комплексов вольфрама
И. В. Скабицкий 1, *, П. В. Русина 1, А. А. Пасынский 1, Ю. В. Торубаев 1, С. Г. Сахаров 1
1 Институт общей и неорганический химии им. Н.С. Курнакова РАН
Москва, Россия
* E-mail: skabitskiy@gmail.com
Поступила в редакцию 29.10.2018
После доработки 23.11.2018
Принята к публикации 03.12.2018
Аннотация
Получены и рентгеноструктурно исследованы циклогептатриенильные комплексы вольфрама (η7-C7H7)W(CO)2I (I), [(η3-C7H7)W(CO)2(CH3CN)3]PF6 (II) и [(η7-C7H7)W(CO)2(CH3CN)]PF6 (III) (CIF files CCDC № 1875096 (I), 1875097 (II), 1875098 (III)), охарактеризованные ранее только спектральными методами. Установлено, что трис-ацетонитрильный комплекс II способен терять два ацетонитрильных лиганда как в координирующих, так и в некоординирующих растворителях с η3 → η7 изменением гаптности циклогептатриенильного кольца в III. Обратный процесс присоединения лигандов с η7 → η3 изменением гаптности циклогептатриенильного кольца происходит при растворении моноацетонитрильного комплекса III в ацетонитриле. Электронокомпенсирующее η3 ↔ η7 изменение гаптности тропилиевого лиганда оказывается обратимым.
Проблема изменения гаптности π-координированных циклических лигандов – часть проблемы изменения дентатности лигандов в координационной химии в зависимости от заполненности вакантных орбиталей и от стерики межлигандных взаимодействий. В частности, для циклогептатриенильного лиганда (C7H7) наблюдались комплексы с η7-, η5-, η3- и η1-типами координации [1]. Особое внимание было уделено переходам η7 → η3 [2–4] для комплексов вольфрама [(η7-C7H7)]+W(MeCN)(CO)2 (схема 1 ).
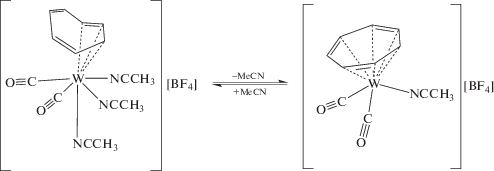
Схема 1 .
В частности, было показано [3], что процесс превращения [(η7-C7H7)W(MeCN)(CO)2]+ требует L = CO, т.е. сильного π-акцепторного лиганда, причем присоединение двух молекул L' = MeCN приводит к изменению окраски от темно-зеленой до оранжево-желтой, резкому понижению частот валентных колебаний СО (от 2022 и 1969 до 1956 и 1880 см–1) и характеристическому изменению спектра ПМР со смещением сигнала протонов циклогептатриенильного кольца с 5.68 до 5.08 м.д. и сигнала координированного ацетонитрила с 2.50 до 2.30 и 2.26 м.д. [3].
В настоящей работе мы получили монокристаллы соединений (η7-C7H7)W(CO)2I (I), [(η3-C7H7)W(CO)2(CH3CN)3]PF6 (II) и [(η7-C7H7)W(CO)2 -(CH3CN)]PF6 (III) и изучили переходы между этими комплексами в растворах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу и выделению комплексов I–III проводили в атмосфере чистого аргона в абсолютированных растворителях. Комплекс I синтезировали по методике [5].
Реакцию с использованием соединений серебра проводили при комнатной температуре в отсутствии света во избежание попадания продуктов разложения иодида серебра в реакционную смесь. Для элементного анализа использовали CHNS-анализатор EA3000 фирмы EuroVector. ИК-спектры регистрировали на Фурье-спектрометре BrukerAlpha с использованием приставки НПВО Platinum ATR. Спектры ЯМР 1H и 13С записывали на спектрометре Bruker AV 400 c рабочими частотами 400.13, 75.4 МГц соответственно и внутренней стабилизацией по дейтерию при 303 K. Хим. сдвиги ядер 1H и 13С приведены относительно ТМС.
Синтез [(C7H7)W(CO)2(CH3CN)3]PF6 (II). К зеленому раствору (C7H7)W(CO)2I (I) (0.229 г., 0.50 ммоль) в ацетонитриле (13 мл) добавляли раствор AgPF6 (0.424 г, 1.67 ммоль) в 9 мл ацетонитрила. Реакционную смесь перемешивали в течение 20 мин. Обрaзовавшийся оранжевый раствор отфильтровывали от осадка AgI, упаривали без нагревания при пониженном давлении до половины объема и добавляли по каплям 3 мл диэтилового эфира. Выпавший при выдерживании при –25°С в течение 4 сут оранжевый осадок отделяли декантацией, промывали 5 мл эфира и высушивали в вакууме. Выход 0.26 г (86%).
| Найдено, %: | С 32.41; | H 2.18; | N 6.94. |
| Для C15H16F6N3O2PW· 0.5(С2Н5)2О (M = 636) | |||
| вычислено, %: | С 32.10; | Н 3.33; | N 6.61. |
ИК-спектр (ν, cм–1): 3027 сл. уш, 2950 сл, 2931 оч. сл, 2319 сл, 2290 сл, 1944 ср, 1872 с, 1434 оч. сл, 1409 сл. уш, 1367 оч. сл, 1030 оч. сл, 881 оч. сл, 831 оч. с., уш, 725 ср, 625 сл, 556 с, 519 сл, 477 сл, 469 сл, 435 сл, 416 оч. сл. ЯМР 1H (CD3CN; δ, м.д.): 1.98 (с. 9H, MeCNнекоорд, 5.18 (с., 7H). ЯМР 1H (d6-ацетон; δ, м.д.): 2.69 (с. 3H, CH3CN) 6.00 (с., 7H C7H7). ЯМР 13C (d6-ацетон, δ, м.д.): 94.9 (C7H7), 210.0 (CO).
Синтез [(C7H7)W(CO)2(CH3CN)]PF6 (III). А) К навеске 0.26 г (0.43 ммоль) оранжевого порошка [(C7H7)W(CO)2(CH3CN)3]PF6 добавляли 10 мл хлористого метилена, выдерживали смесь 1 ч без перемешивания до изменения окраски на зеленую. Растворитель удаляли в вакууме, зеленый остаток растворяли в 15 мл хлористого метилена, отфильтровывали, добавляли по каплям 5 мл толуола и концентрировали при пониженном давлении до начала помутнения раствора. Выпавший при выдерживании при –25°С в течение недели темно-зеленый осадок отделяли декантацией, промывали 5 мл толуола и высушивали в вакууме. Выход 91 мг (41%).
| Найдено, %: | С 26.01; | H 2.24; | N 2.53. |
| Для C11H10F6NO2PW (M = 517) | |||
| вычислено, %: | С 25.55; | Н 1.95; | N 2.71. |
ИК-спектр (ν, cм–1): 3077 оч. сл., уш, 3010 оч. сл, 2943 оч. сл, 2442 оч. сл., уш, 2322 оч. сл, 2002 ср, 1954 с, 1475 сл, 1433 сл, 1415 оч. сл, 1361 оч. сл, 1249 оч. сл, 1056 оч. сл, 1033 оч. сл, 955 оч. сл. 897 оч. сл, 877 оч. сл, 864 сл, 822 оч. с., уш, 808 оч. с, 554 с, 517 сл, 503 сл, 491 сл, 464 сл, 423 с.
Б) К навеске 0.104 г (0.2 ммоль) комплекса II добавляли 6 мл толуола и кипятили 30 мин с обратным холодильником до изменения окраски осадка на зеленую. Затем раствор выдерживали при –25°С в течение 10 мин, раствор декантировали, черно-зеленые кристаллы промывали 5 мл гептана и высушивали в вакууме. Выход 0.027 г. (30%).
ИК-спектр полностью совпадает со спектром образца, полученного в синтезе А.
Превращение III в II. К 0.091 г (0.2 ммоль) зеленого порошка III добавляли 6 мл ацетонитрила и выдерживали 1 ч без перемешивания. При этом наблюдалось полное растворение вещества и изменение цвета раствора от зеленого до оранжевого. Реакционную смесь упаривали при пониженном давлении до половины объема. Добавляли по каплям диэтиловый эфир (~3 мл) до начала помутнения раствора. Выдерживали при –25°С неделю. Декантировали раствор, промывали оранжевый осадок эфиром (5 мл) и высушивали в вакууме. Выход 0.048 г (46%).
ИК и ЯМР данные полностью совпадают с данными для соединения II.
РСА выполнен на дифрактометре Bruker APEX II CCD. Поглощение учтено методом множественного измерения эквивалентных отражений по программе SADABS [6]. Структуры I–III определены прямым методом и уточнены методом наименьших квадратов относительно F 2 в анизотропном приближении неводородных атомов с использованием пакетов программ SHELX-2014 [7] и OLEX2 [8]. Кристаллографические данные и параметры уточнения структур I–III приведены в табл. 1. Основные длины связей и валентные углы в I–III приведены в подписях к рис. 1–3.
Таблица 1.
Кристаллографические данные и параметры уточнения структуры комплексов I–III
| Параметр | Значение | ||
|---|---|---|---|
| I | II | III | |
| Брутто-формула | C18H14I2O4W2 | C15H16F6N3O2PW | C11H10F6NO2PW |
| M | 915.79 | 599.13 | 517.02 |
| Излучение (λ, Å) | MoKα (λ = 0.71073) | ||
| Температура съемки, K | 150 | 120 | 150 |
| Сингония | Моноклинная | Моноклинная | Моноклинная |
| Пр. гр. | P21/m | P21/c | P21/c |
| а, Å | 7.2133(8) | 14.7893(7) | 11.4294(4) |
| b, Å | 10.514(1) | 8.3359(4) | 10.8123(4) |
| c, Å | 13.780(1) | 16.0951(7) | 11.9302(4) |
| β, град | 104.324(2) | 90.6202(7) | 100.3769(5) |
| V, Å3 | 1012.6(2) | 1984.1(2) | 1450.20(9) |
| Z | 2 | 4 | 4 |
| ρ(выч.), г/cм–3 | 3.004 | 2.006 | 2.368 |
| μ, мм–1 | 14.420 | 5.972 | 8.147 |
| F(000) | 816.0 | 1144.0 | 968.0 |
| Интервал сканирования θ, град | 4.932–59.158 | 5.062–61.016 | 3.622–63.984 |
| Тип сканирования | ω | ||
| Независимых отражений (N1) | 2958 | 6051 | 4935 |
| Rint | 0.0265 | 0.0279 | 0.0206 |
| Отражений с I > 2σ(I) (N2) | 2664 | 5358 | 4620 |
| Число уточняемых параметров | 127 | 256 | 200 |
| GOОF (F 2) | 1.051 | 1.040 | 1.065 |
| R1 для N2 | 0.0206 | 0.0164 | 0.0163 |
| wR2 для N1 | 0.0430 | 0.0349 | 0.0405 |
| Δρmax/Δρmin, e Å–3 | 0.90/–1.02 | 0.68/–0.49 | 0.71/–1.11 |
Рис. 1.
Молекулярная структура комплекса I. Важнейшие расстояния: W(1)–I(1) 2.8191(4), W(1)–(5) 2.003(4), C(1)C(2)i 1.403(5), C(1)–C(2) 1.403(5), C(2)–C(3) 1.432(5), C(3)–C(4) 1.381(5), C(4)–C(4)i 1.439(7), O(1)–C(5) 1.144(4) Å.
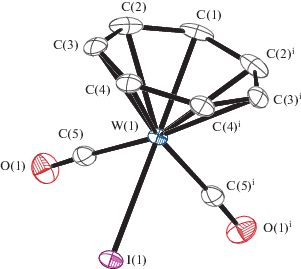
Рис. 2.
Молекулярная структура катиона комплекса II. Важнейшие расстояния и углы: W(1)–N(1) 2.149(2), W(1)–N(2) 2.193(2), W(1)–N(3) 2.202(2), W(1)–C(1) 2.367(2), W(1)–C(2) 2.193(2), W(1)–C(3) 2.367(2), W(1)–C(4) 3.380(2), W(1)–C(5) 4.032(2), W(1)–C(6) 4.033(2), W(1)–C(7) 3.387(2), W(1)–C(8) 1.968(2), W(1)–C(9) 1.972(2), O(1)–C(8) 1.151(2), O(2)–C(9) 1.154(2), C(1)–C(7) 1.466(3), C(2)–C(3) 1.423(3), C(3)–C(4) 1.462(3), C(4)–C(5) 1.346(3), C(5)–C(6) 1.436(3), C(6)–C(7) 1.339(3), C(1)–C(2) 1.425(3), N(1)–C(10) 1.136(2), N(2)–C(12) 1.136(2), N(3)–C(14) 1.134(3) Å и C(8)W(1)C(9) 84.21(8)°, N(1)W(1)N(2) 80.17(6)°, N(1)W(1)N(3) 78.26(6)°, N(2)W(1)N(3) 81.27(6)°.
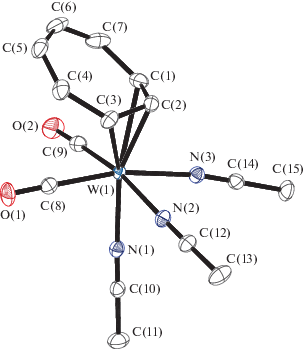
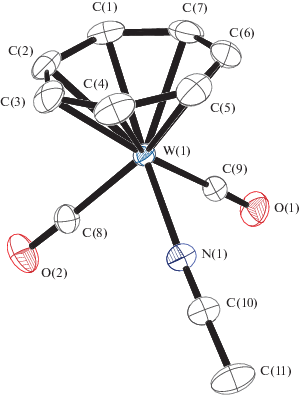
Рис. 3.
Молекулярная структура катиона комплекса III. Важнейшие расстояния и углы: W(1)–N(1) 2.121(2), W(1)–C(1) 2.320(2), W(1)–C(2) 2.277(2), W(1)–C(3) 2.337(2), W(1)–C(4) 2.332(2), W(1)–C(5) 2.327(2), W(1)–C(6) 2.336(2), W(1)–C(7) 2.278(2), W(1)–C(8) 2.012(2), W(1)–C(9) 2.011(2), O(2)–C(8) 1.137(3), O(1)–C(9) 1.138(2), C(1)–C(7) 1.403(4), C(2)–C(3) 1.424(3), C(3)–C(4) 1.391(4), C(4)–C(5) 1.429(4), C(5)–C(6) 1.395(4), C(6)–C(7) 1.430(4), C(1)–C(2) 1.409(3), N(1)–C(10) 1.137(3) Å и C(9)W(1)C(8) 81.60(8)°, C(8)W(1)N(1) 89.38(7)°, C(9)W(1)N(1) 86.22(7)°.
Координаты атомов и другие параметры структур депонированы в Кембриджском банке структурных данных (ССВС № 1875096 (I), 1875097 (II), 1875098 (III); http://www.ccdc.cam.ac.uk/data_request/cif).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
При реакции зеленого 18-электронного комплекса I [5] с избытком гексафторфосфата серебра в ацетонитриле происходит отщепление иодистого серебра с образованием катионного 18-электронного комплекса II, выделенного в виде оранжевых кристаллов из смеси ацетонитрил–эфир (1 : 2) (схема 2 ).
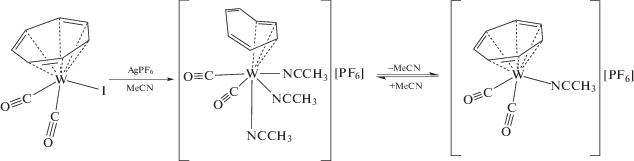 Схема 2.
Схема 2.При этом происходит смещение двух интенсивных полос СО (1995, 1939 см–1) в низкочастотную область (1944, 1872 см–1).
По данным РСА, катион в комплексе II содержит три ацетонитрильных лиганда. В нем существенно меняется геометрия тропилиевого лиганда и длины связей W–C(C7H7). В отличие от структуры комплекса I, содержащей плоский фрагмент С7Н7 (рис. 1), в II четыре атома углерода выходят из плоскости и оказываются на несвязывающих расстояниях от атома W (W–C 4.032(2) и 4.033(2) Å), т.е. в процессе реакции происходит изменение гаптности η7 → η3. Меняется также геометрия комплексов: II имеет псевдо-октаэдрическую структуру, III – тип “трехногой табуретки”.
При растворении II в хлористом метилене, ацетоне, ТГФ или при кипячении в толуоле цвет раствора изменяется от оранжевого до зеленого, в котором при повышенной концентрации заметен вишневый оттенок (дихроизм). При этом катион в комплексе II теряет две молекулы ацетонитрила с образованием 18-электронного III (схема 2 ), содержащего, помимо двух СО, один ацетонитрильный лиганд и η7-координированный тропилиевый лиганд. Возможна и обратная реакция, в которой моноацетонитрильный комплекс при растворении в ацетонитриле снова образует триацетонитрильный II. При этом, по данным ИК-спектроскопии, возрастает частота валентных колебаний СО.
В спектре ЯМР 1Н раствора триацетонитрильного комплекса II (в CD3CN) наблюдаются только сигналы некоординированного ацетонитрила (1.98 м.д), а в области ароматических протонов наблюдается уширенный синглет 5.18 м.д., по-видимому, в связи с быстрым скольжением η3-координированного тропилия относительно атома вольфрама. По этой же причине не удается зарегистрировать соответствующий сигнал в спектре 13C. При растворении комплекса II в d6-ацетоне происходит изменение окраски на зеленую, по-видимому, в связи с отщеплением части ацетонитрильных лигандов с образованием катиона III. Это приводит к заметному смещению 1H сигнала тропилиевого фрагмента (6.00 м.д.), а в спектре ЯМР 13С наблюдаются сигналы, соответствующие тропилиевой (94.9 м.д.) и CO группам (210 м.д.).
На основании данных РСА, для I–III можно сделать вывод, что циклогептатриенильный лиганд претерпевает электронокомпенсирующее изменение гаптности η7 ↔ η3 ↔ η7. Расстояния между атомами углерода в циклогептатриениле при этом также меняются. Например, длина связи C(1)–C(7) и C(3)–C(4) в II и III различается на ~0.6 Å, что связано с изменением типа координации тропилиевого лиганда. В то же время II и III имеют в группировках W(СО)2 схожее строение и длины связей, и также незначительно различаются длины связей W с атомами углерода тропилиевого лиганда в молекулах I–III.
Следует заметить, что для перметилированных трикарбонильных комплексов η5-Ме7С7W(CO)3-(MeCN)+ (IV) и η5-Ме7С7W(CO)3I (V) наблюдался электронокомпенсирующий переход гаптности η7 ↔ η5 [9] (схема 3 ).
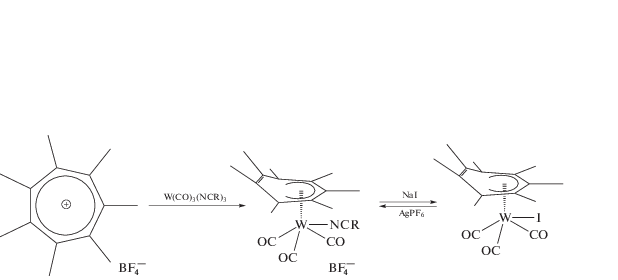
Схема 3 .
По-видимому, это означает, что потеря ароматичности семичленного цикла не является существенным фактором. С этой точки зрения интересно сопоставить тропилиевые комплексы с гетерометаллическими тетраэдрическими кластерами Ср3Cr3S4ML, содержащими фрагмент Ср3Cr3(μ3-S)- (μ-S)3, который связывается с металлсодержащими фрагментами ML = CpCr, CpV•, CpNb• [10] и Со(СО) [11], как семиэлектронодонорный лиганд, подавая четыре электрона от трех μ-S-мостиков и три электрона за счет связей Cr–М. Например, в электрононасыщенном кластере Ср3Cr3S4Со(СО) все связи Со–Cr и Со–S одинарные (в среднем, 2.658 и 2.163 Å) [11], тогда как введение к атому кобальта двух дополнительных СО в сольватированном кластере Ср3Cr3(μ3-О)(μ3-S)3Со(СО)3 · СН3СООН · 0.5C6H6 резко разрыхляет как связи Со–Cr (до 3.216(6), 3.238(6) и 2.892(6) Å), так и Со–S (до 2.48 Å) [12]. Следует подчеркнуть, что такой эффект наблюдается только при попадании лишних электронов от дополнительных молекул СО на разрыхляющие орбитали связей кобальта с хром-сульфидным остовом, как это происходит и в комплексах II, IV и V относительно связей вольфрам–тропилий.
Таким образом, получены и рентгеноструктурно исследованы циклогептатриенильные комплексы вольфрама [(η7-C7H7)W(CO)2I, [(η3-C7H7)W(CO)2-(CH3CN)3]PF6 и [(η7-C7H7)W(CO)2(CH3CN)]PF6, охарактеризованные ранее только спектральными методами. Установлено, что трис-ацетонитрильный комплекс [(η3-C7H7)W(CO)2(CH3CN)3]PF6 способен терять двa ацетонитрильных лиганда как в координирующих, так и в некоординирующих растворителях с η3 → η7 изменением гаптности циклогептатриенильного кольца. Обратный процесс присоединения лигандов с η7 → η3 изменением гаптности циклогептатриенильного кольца происходит при растворении моноацетонитрильного комплекса [(η7-C7H7)W(CO)2(CH3CN)]PF6 в ацетонитриле. Таким образом, тропилиевый лиганд претерпевает обратимое электронокомпенсирующее η3 ↔ η7 изменение гаптности, сопоставленное с аналогичными изменениями связей фрагментов Со(СО) и Со(СО)3 с хром-сульфидным остовом в гетерометаллических тетраэдрических кластерах.
Исследование выполнено при финансовой поддержке Президиума РАН (проект № I.35) и Российского фонда фундаментальных исследований (грант № 16-03-00798). Синтез соединений выполнен в рамках государственного задания ИОНХ РАН в области фундаментальных научных исследований. Исследования проводили с использованием оборудования ЦКП ФМИ ИОНХ РАН.
Список литературы
Campen A.K., Narayanaswamy R., Rest A.J. // Dalton Trans. 1990. P. 823.
Brown R.A., Endud S., Friend J. et al. // J. Organomet. Chem. 1988. V. 339. P. 283.
Breeze R., Plant M.S., Ricalton A. et al. // J. Organomet. Chem. 1988. V. 356. P. 343.
Hinchliffe J.R., Ricalton A., Whiteley M.W. // Polyhedron. 1991. V. 10. P. 267.
King R.B., Fronzaglia A. // Inorg. Chem. 1966. V. 5. P. 1837.
SADABS. Version 2008/1. Madison (WI, USA): Bruker AXS Inc., 2008.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Ap-pl. Cryst. 2009. V. 42. P. 339.
Tamm M., Dreßel B., Fröhlich R., Berganderb K. // Chem. Commun. 2000. P. 1731.
Pasynskii A.A., Eremenko I.L., Orazsakhatov B. et al. // J. Organomet. Chem. 1981. V. 216. P. 211.
Pasynskii A.A., Eremenko I.L., Orazsakhatov B. et al. // J. Organomet. Chem. 1981. V. 214. P. 367.
Eremenko I.L., Pasynskii A.A., Gasanov G.Sh. et al. // J. Organomet. Chem. 1984. V. 275. P. 71.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия