

Координационная химия, 2019, T. 45, № 6, стр. 341-349
Спиновое состояние 2,6-ди(5-амино-1H-пиразол-3-ил)пиридиновых комплексов железа(II) и кобальта(II) в растворе и кристалле
А. А. Павлов 1, И. А. Никовский 1, А. В. Полежаев 1, 2, Д. Ю. Алешин 1, 3, Е. К. Мельникова 1, 4, Я. А. Панкратова 4, Ю. В. Нелюбина 1, 5, *
1 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
2 Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
3 Российский химико-технологический университет им. Д.И. Менделеева
Москва, Россия
4 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
5 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
* E-mail: unelya@ineos.ac.ru
Поступила в редакцию 13.11.2018
После доработки 17.01.2019
Принята к публикации 24.01.2019
Аннотация
Прямой темплатной реакцией получены комплексы кобальта(II) и железа(II) с 2,6-ди(5-амино-1H-пиразол-3-ил)пиридином (L) – CoL2(ClO4)2 (I) и FeL2(ClO4)2 (II), выделенные в индивидуальном виде и охарактеризованные методами элементного анализа, спектроскопии ЯМР и термогравиметрии. Структура комплексов I, II (в том числе комплекса железа(II), полученного в виде двух новых сольватных форм (FeL2(ClO4)2 ∙ 2(C2H5)2O ∙ H3CN (IIa) и FeL2(ClO4)2 ∙ (C2H5)2O ∙ CH3CN ∙ 0.75H2O (IIb)) подтверждена методом РСА (CIF files CCDC № 1876396, 1876397, 1876398). Полученные данные в кристалле (методом РСА) и в растворе (при помощи предложенного подхода к анализу парамагнитных сдвигов в спектрах ЯМР 1H) позволили установить, что ион металла в комплексах находится в высокоспиновом состоянии (S = 3/2 для Co(II) и S = 2 для Fe(II)) и не претерпевает температурно-индуцируемого спинового перехода в диапазоне 120–300 К.
Молекулярные системы, обладающие бистабильностью, т.е. способностью существовать в двух различных электронных состояниях при определенных условиях [1], лежат в основе передовых концепций сверхплотного хранения информации и квантовых компьютеров [2], переключателей, сенсоров и других молекулярных устройств и материалов [3–5]. Типичные примеры таких бистабильных систем – комплексы переходных металлов с d4–d7-электронной конфигурацией (главным образом железа(II), железа(III) и кобальта(II) [6]), способные переходить из одного спинового состояния в другое при приложении подходящего внешнего возмущения (температуры, давления, света, магнитного и электрического полей и т.п.) [6, 7]. Так как возможность протекания спинового перехода определяется локальным окружением иона металла, его параметры в значительной степени зависят от взаимодействий между молекулами комплекса, благодаря которым в кристаллическом образце возможен резкий спиновый переход с гистерезисом [6]. При этом даже небольшие изменения на периферии молекулы могут приводить к изменению заселенностей спиновых состояний [8, 9]. В некоторых случаях сдвиг спинового равновесия является следствием разного фазового состояния исследуемого соединения (монокристалл, поликристаллический порошок, раствор) [10], различий в кристаллической упаковке (разные полиморфные модификации [11]) и даже влияния растворителя [11–13].
Наиболее изучены соединения, претерпевающие температурно-индуцированный спиновый переход – 2,6-ди(пиразол-1-ил)пиридиновые комплексы железа(II) [14, 15] благодаря их синтетической доступности и возможности контролировать спиновое состояние иона металла выбором подходящего заместителя [16]. Обнаружение аналогичных корреляций “структура–свойство” для комплексов с изомерными лигандами – 2,6-ди(1H-пиразол-3-ил)пиридинами (не уступающими им по возможностям функционализации [17], однако более подверженными влиянию окружения на спиновое состояние иона металла [5]) требует не только получения большего числа их производных, чем известно к настоящему моменту [17], но и изучения их свойств в разных фазовых состояниях и в виде разных полиморфных/сольватоморфных модификаций.
Ранее для комплекса железа(II) с 2,6-ди(5-амино-1H-пиразол-3-ил)пиридином (L) [18], полученного в виде дигидрата FeL2(ClO4)2 ∙ 2H2O, наблюдался температурно-индуцированный спиновый переход в поликристаллическом образце, при котором основная масса образца оставалась в высокоспиновом состоянии.
В настоящей работе мы получили ранее неизвестный комплекс CoL2(ClO4)2 (I), а также комплекс FeL2(ClO4)2 (II) в виде двух новых сольватных форм, одновременно выпадающих в процессе кристаллизации (схема 1 ) и проанализировали их спиновое состояние в растворе и в кристалле.
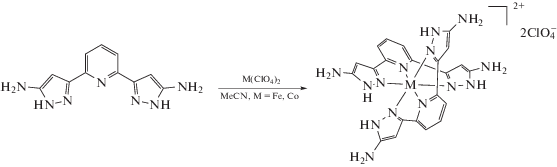
Схема 1 .
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции, связанные с синтезом комплексов, выполняли на воздухе с использованием коммерчески доступных органических растворителей и реагентов. Co(ClO4)2 ∙ 6H2O [19] и лиганд L [8, 20, 21] получали по ранее описанным методикам. Анализ на содержание углерода, азота и водорода проводили на микроанализаторе CarloErba, модель 1106. Термогравиметрический анализ комплексов I и II проводили на приборе NetzchTG 209 F1 Libra в диапазоне 20–500°С со скоростью нагрева 7 К/мин в атмосфере аргона.
Синтез CoL2(ClO4)2 (I). 22.7 мг (0.0622 ммоль) Co(ClO4)2 ∙ 6H2O и 30 мг (0.124 ммоль) L растворяли/суспендировали в 10 мл сухого ацетонитрила с образованием темно-коричневого раствора. Реакционную смесь перемешивали 1 ч при комнатной температуре и наблюдали образование осадка. Полученный осадок отделяли и очищали перекристаллизацией из системы ацетонитрил–диэтиловый эфир в соотношении 1 : 1. Выход продукта 29.5 мг (64%).
ЯМР 1H (CD3CN; δ, м.д.): 1.38 (уш. с., 2H, 4-Py), 21.29 (уш. с., 8H, NH2), 28.52 (уш. с., 4H, 3-Py), 54.58 (уш. с., 4H, 4-Pz), 82.34 (уш. с., 4H, NH).
Синтез FeL2(ClO4)2 (II). 15.8 мг (0.0622 ммоль) Fe(ClO4)2 и 30 мг (0.124 ммоль) L растворяли/суспендировали в 10 мл сухого ацетонитрила с образованием темно-коричневого раствора. Реакционную смесь перемешивала 1 ч, выпадал осадок целевого продукта. Полученный осадок отделяли и очищали перекристаллизацией из системы ацетонитрил–диэтиловый эфир в соотношении 1 : 1. Выход продукта 25.2 мг (55%).
ЯМР 1H (CD3CN; δ, м.д.): 9.7 (уш. с., 8H, NH2), 27.64 (уш. с., 4H, NH), 30.07 (уш. с., 2H, 4-Py), 52.11 (уш. с., 4H, 3-Py), 60.71 (уш. с., 4H, 4-Pz).
РСА монокристаллов комплексов I и II (в виде двух сольватоморфов IIa и IIb, полученных медленным испарением на воздухе из смеси растворителей ацетонитрил–диэтиловый эфир (соотношение 1 : 1), проведен на дифрактометре Bruker APEX2 DUO CCD (MoKα-излучение, графитовый монохроматор, ω-сканирование) при 120 К. Структуры расшифрованы прямым методом и уточнены МНК в анизотропном полноматричном приближении по $F_{{hkl}}^{{\text{2}}}.$ Атомы водорода NH- и NH2-групп, а также сольватных молекул воды (предположительно “захваченных” из воздуха в процессе кристаллизации), локализованы из разностных синтезов Фурье электронной плотности и уточнены в изотропном приближении по модели наездника. Основные кристаллографические данные и параметры уточнения представлены в табл. 1. Все расчеты проведены по комплексу программ SHELXTL PLUS [22].
Таблица 1.
Основные кристаллографические данные и параметры уточнения структур I, IIa и IIb
| Параметр | Значение | ||
|---|---|---|---|
| I | IIa | IIb | |
| Брутто формула | C28H36.12N15O9.56Cl2Co | C32H45N15O10Cl2Fe | C28H36.5N15O9.75Cl2Fe |
| М | 865.54 | 926.58 | 865.97 |
| T, K | 120 | 120 | 120 |
| Сингония | Моноклинная | Ромбическая | Моноклинная |
| Пр. гр. | P21/n | Pbcn | P21/n |
| Z | 4 | 4 | 4 |
| a, Å | 15.1212(9) | 14.226(1) | 14.941(2) |
| b, Å | 14.4842(9) | 23.453(2) | 14.729(2) |
| c, Å | 18.134(1) | 12.787(1) | 18.031(2) |
| β, град | 111.9950(10) | 90 | 111.193(2) |
| V, Å3 | 3682.6(4) | 4266.3(6) | 3699.8(6) |
| ρ(выч.), г см–3 | 1.561 | 1.443 | 1.555 |
| μ, см–1 | 6.86 | 5.50 | 6.27 |
| F(000) | 1786 | 1928 | 1790 |
| 2θmax, град | 52 | 58 | 52 |
| Число измеренных отражений | 71 353 | 66 925 | 35 867 |
| Число независимых отражений | 7245 | 5677 | 7269 |
| Число отражений с I > 2σ(I) | 6063 | 4441 | 5408 |
| Количество уточняемых параметров | 543 | 276 | 527 |
| R1 | 0.0579 | 0.0380 | 0.0636 |
| wR2 | 0.1530 | 0.1048 | 0.1775 |
| GOОF | 1.076 | 1.049 | 1.019 |
| Остаточная электронная плотность (Δρmin/Δρmax), e Å–3 | –0.722/1.631 | –0.366/0.516 | –0.939/1.894 |
Структурные данные для I, IIa и IIb депонированы в Кембриджском банке структурных данных (CCDC № 1876396, 1876397, 1876398 соответственно; http://www.ccdc.cam.ac.uk/).
Спектры ЯМР 1Н комплексов I и II регистрировали в CD3CN на спектрометре Bruker Avance 600 (600.22 МГц). Значения химических сдвигов (δ, м.д.) в спектрах определяли относительно остаточного сигнала растворителя (1H 1.94 м.д.). Спектры регистрировали с использованием следующих параметров: диапазон спектра 200 м.д., время регистрации 0.5 с, длительность релаксационной задержки 0.5 с, длительность импульса 6.5 мкс, количество накоплений 128. Для повышения соотношения сигнал/шум полученные спады свободной индукции обрабатывали при помощи экспоненциального взвешивания с коэффициентом до 3 Гц.
Квантово-химические расчеты для комплексов I и II проводили с помощью программного пакета ORCA (версия 4) [23] в рамках теории функционала плотности (DFT). Геометрию катионов [CoL2]2+ и [FeL2]2+ оптимизировали без наложения симметрийных ограничений с использованием негибридного функционала PBE0 [24] и базисного набора def2-TZVP [25]. В качестве начального приближения использовали структуры, полученные из рентгенодифракционных данных. Эффекты сольватации учитывали в рамках модели CPCM программного пакета ORCA (версия 4) [23]. В качестве растворителя выбирали ацетонитрил, в растворе которого регистрировали спектры ЯМР. На основе полученной геометрии для комплексов I и II рассчитывали g-тензор и тензоры сверхтонкого взаимодействия для протонов с использованием гибридного функционала PBE0 [24] и базисного набора def2-TZVP [25] с добавлением дополнительных примитивов Гаусса с высокой степенью экспоненты для более точного описания электронной плотности в районе ядра.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Комплексы I и II получены с высоким выходом прямой реакцией в ацетонитриле при комнатной температуре. Из-за низкой растворимости в ацетонитриле и в большинстве других органических растворителей оба комплекса выпадают в осадок в процессе синтеза. Комплексы I и II выделены в индивидуальном виде и охарактеризованы методами элементного анализа и спектроскопии ЯМР. Их строение подтверждено методом РСА при 120 К.
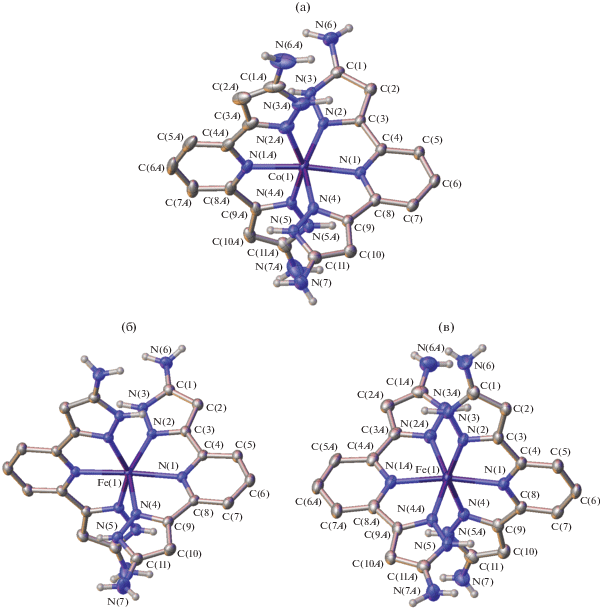
Комплекс I (рис. 1а) кристаллизуется в виде сольвата с ацетонитрилом, диэтиловым эфиром и водой, молекулы которой предположительно захвачены из воздуха в процессе кристаллизации комплекса из смеси ацетонитрил–диэтиловый эфир. В кристалле молекулы растворителей удерживаются за счет водородных связей с NH- и NH2-группами катиона [CoL2]2+ (N···O 2.820(4)–3.205(7) Å, NHO 136°–167°; N···N 2.871(7) Å, NHN 152°). Катионы образуют водородные связи (ВС) с перхлорат-анионами (N···O 3.018(8)–3.449(10) Å, NHO 126°–173°), связанными, в свою очередь, с сольватной молекулой воды (O···O 2.755(11)–3.169(6) Å, OHO 127°–166°). Кристаллизация комплекса II в таких же условиях привела к получению механической смеси двух типов кристаллов – сольватоморфов IIa и IIb (pис. 1б и 1в), отличающихся природой и соотношением сольватных молекул друг от друга (FeL2(ClO4)2 ∙ 2(C2H5)2O ∙ H3CN (IIa) и FeL2(ClO4)2 ∙ (C2H5)2O ∙ CH3CN ∙ 0.75H2O (IIb)) и от ранее охарактеризованного кристаллосольвата II с диэтиловым эфиром и нитрометаном FeL2(ClO4)2 ∙ 2(C2H5)2O ∙ CH3NO2 [18]. Форма IIa содержит одну молекулу ацетонитрила и две молекулы диэтилового эфира на одну формульную единицу комплекса. Форма IIb представляет собой сольват с ацетонитрилом, диэтиловым эфиром и водой в соотношении 1 : 1 : 0.75. В сольватоморфе IIa молекулы диэтилового эфира удерживаются в кристалле ВС с двумя NH-группами катиона [FeL2]2+ (N···O 2.729(2) Å, NHO 168(1)°), тогда как остальные NH- и NH2-группы связаны с перхлорат-анионами (N···O 2.898(2)–3.190(2) Å, NHO 146°–171°). В сольватоморфе IIb молекулы растворителей всех трех типов: диэтилового эфира, ацетонитрила и воды – вовлечены в ВС с одной NH2- и двумя NH-группами катиона [FeL2]2+ (N···O 2.845(4)–2.879(8) Å, NHO 154°–167°; N···N 2.966(9) Å, NHN 147°), а перхлорат-анионы – с его оставшимися NH- и NH2-группами (N···O 2.905(6)–3.407(9) Å, NHO 130°–171°). Молекула воды также образует ВС с молекулой ацетонитрила и одним из перхлорат-анионов, выступая в качестве донора протона (O···N 2.837(14) Å, OHN 143°; O···O 3.244(10) Å, OHO 152°).
Рис. 1.
Общий вид катионов [ML2]2+ (M = Co, Fe) в кристаллах I (а), IIa (б) и IIb (в) по рентгенодифракционным данным в представлении атомов эллипсоидами тепловых колебаний (p = 50%). Перхлорат-анионы, молекулы растворителей и атомы водорода (за исключением принадлежащих NH и NH2 группам) не показаны. В кристалле IIa (б) катион [FeL2]2+ занимает частное положение на оси 2, проходящей через ион железа(II).
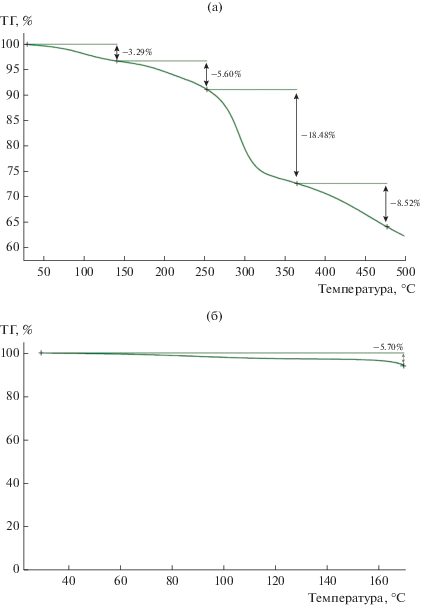
Полученные из рентгенодифракционного эксперимента данные о наличии в кристаллах I, IIa и IIb молекул растворителей и их природе согласуются с результатами ТГА мелкокристаллических образцов соответствующих комплексов (рис. 2). Так, для комплекса кобальта I при нагревании наблюдается потеря массы в ~8% в диапазоне от 20 до 200°С (pис. 2a), что соответствует потере молекул воды и ацетонитрила. Молекула диэтилового эфира предположительно удалена из кристаллов при высушивании образца в вакууме. Интенсивное разложение комплекса начинается при 220°С и, по всей видимости, сопровождается разложением лиганда L. В диапазоне 220–350°С комплекс I теряет порядка 30% массы. Кристаллический образец комплекса железа II, представляющий собой смесь двух сольватоморфов IIa и IIb, одновременно полученных в процессе кристаллизации из системы ацетонитрил–диэтиловый эфир на воздухе, теряет ~5% массы в диапазоне 20–150°С (pис. 2б), что связано с потерей молекул растворителя. При достижении 168°С комплекс II разлагается со взрывом и разрушением тигля.
По данным РСА, проведенного для монокристаллов I, IIa и IIb при 120 К, атомы азота 2,6-ди(пиразол-3-ил)пиридиновых лигандов координированы к иону металла (КЧ 6): расстояния 2.079(3)–2.167(3) и 2.136(3)–2.199(3) Å (табл. 2) типичны для ионов кобальта(II) и железа(II) в высокоспиновом состоянии (2.0–2.3 Å [6]). Во всех случаях также наблюдается искажение координационного полиэдра (КП) MN6 (M = Co, Fe) [26], который у низкоспинового иона железа(II) представляет собой октаэдр. В частности, значения угла N(1)MN(1)' и двугранного угла θ между среднеквадратичными плоскостями двух лигандов, равные 90° и 180° для идеального октаэдра, в кристаллах I, IIa и IIb составляют 83.3(4)°–85.9(2)° и 169.60(13)°–176.56(8)° соответственно. Стоит отметить, что для ранее описанного кристаллосольвата II с диэтиловым эфиром и нитрометаном FeL2(ClO4)2 ∙ 2(C2H5)2O ∙ CH3NO2 [18] аналогичные значения составляют 87.91(1)° и 177.33(6)°.
Таблица 2.
Основные геометрические параметры комплексов в кристаллах I, IIa и IIb*
| Комплекс | Связь | d, Å | Угол | ω, град |
|---|---|---|---|---|
| I | Co–N(Py) | 2.079(3) | θ | 84.5(3) |
| Co–N(Pz) | 2.121(3)–2.167(3) | N(1)CoN(1) | 173.16(12) | |
| IIа | Fe–N(Py) | 2.1532(14) | θ | 85.9(2) |
| Fe–N(Pz) | 2.1831(15)–2.1955(14) | N(1)FeN(1) | 176.56(8) | |
| IIb | Fe–N(Py) | 2.136(3) | θ | 83.3(4) |
| Fe–N(Pz) | 2.154(3)–2.199(3) | N(1)FeN(1) | 169.60(13) |
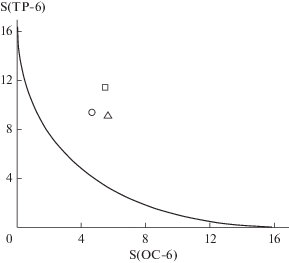
Наблюдаемое в кристаллах I, IIa и IIb искажение КП MN6 можно графически представить при помощи так называемых “мер симметрии” (или continuous symmetry measures в англоязычной литературе [27]), описывающих его отклонения от идеального октаэдра S(OC-6) и идеальной тригональной призмы S(TP-6) (рис. 3). Чем эти значения меньше, тем лучше форма КП (например, получаемая в рентгенодифракционном эксперименте) описывается соответствующим многогранником. В случае исследуемых комплексов I и II величины октаэдрической S(OC-6) и тригонально-призматической S(TP-6) “мер симметрии” (рис. 3), оцененные на основе РСА для I, IIa и IIb, составляют 9.063–9.377 и 4.665–5.661 соответственно, что указывает на заметное искажение КП MN6 в сторону тригональной призмы. Они также попадают в диапазон значений “мер симметрии” S(OC-6) и S(TP-6), характерных для высокоспиновых комплексов железа(II) с изомерными 2,6-ди(пиразол-1-ил)пиридиновыми лигандами [17].
Рис. 3.
Графическое представление “мер симметрии” S(TP-6) и S(OC-6) – отклонений формы полиэдра MN6 в кристаллах I (◻), IIa (⚪) и IIb (△) от идеальной тригональной призмы TP-6 и идеального октаэдра OC-6 соответственно. Черная линия представляет собой путь наименьшего искажения геометрии полиэдра при переходе между указанными многогранниками.
Таким образом, полученные рентгенодифракционные данные однозначно указывают на то, что ион кобальта(II) и железа(II) в комплексах I и II находится в высокоспиновом состоянии при 120 K, и спиновый переход в кристалле не наблюдается ни в том, ни в другом случае.
Высокоспиновое состояние этих комплексов в растворе при комнатной температуре также подтверждено данными спектроскопии ЯМР. В частности, сигналы в спектре ЯМР 1Н для комплекса II (рис. 4) лежат за пределами диамагнитной области (0–15 м.д.), что однозначно указывает на высокоспиновое состояние иона железа(II) (SВС = 2), поскольку его низкоспиновое состояние диамагнитно. Оба спиновых состояния иона кобальта(II) парамагнитны (SНС = 1/2, SВС = 3/2), однако в первом случае значения химических сдвигов, как правило, лежат вблизи диамагнитной области, а во втором достигают нескольких десятков м.д. Таким образом, полученный для I спектр ЯМР 1Н (pис. 4) соответствует высокоспиновому комплексу кобальта(II).
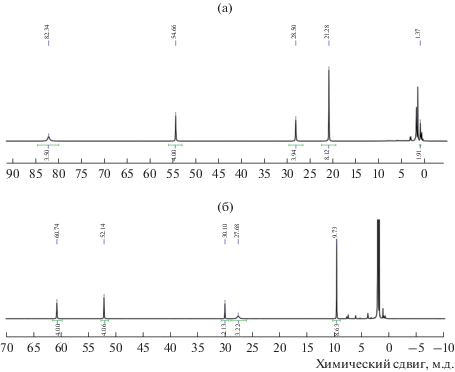
Для более надежной интерпретации данных спектроскопии ЯМР 1Н о спиновом состоянии двух комплексов в растворе использован ранее предложенный нами оригинальный подход к анализу парамагнитных сдвигов [28, 29]. Он основан на том, что в зависимости от спинового состояния иона металла ядра комплекса имеют резко отличающиеся значения химических сдвигов, которые для парамагнитных соединений складываются из трех составляющих – диамагнитной, контактной и псевдоконтактной.
(1)
$\delta = {{\delta }_{{{\text{д и а }}}}} + {{\delta }_{{{\text{к о н }}}}} + {{\delta }_{{{\text{п к }}}}},$Вклад δдиа обусловлен экранированием ядра орбитальным движением спаренных электронов, и за него можно принять соответствующее значение химического сдвига в спектре ЯМР для диамагнитного аналога, например изоструктурного комплекса диамагнитного металла или даже исходного лиганда. Вклад δкон определяется перераспределением спиновой плотности на ядро и пропорционален ей:
(2)
${{\delta }_{{{\text{к о н }}}}} = \frac{{4\pi \mu _{B}^{2}}}{{9kT}}{{g}^{{{\text{iso}}}}}\rho ,$(3)
$\begin{gathered} {{\delta }_{{{\text{п к }}}}} = \frac{1}{{12\pi {{r}^{3}}}} \times \\ \times \,\,\left[ {\Delta {{\chi }_{{{\text{а х }}}}}\left( {3{{{\cos }}^{2}}\theta - 1} \right) + \frac{3}{2}\Delta {{\chi }_{{rh}}}{{{\sin }}^{2}}\theta \cos 2\varphi } \right], \\ \end{gathered} $Поскольку комплексы I и II имеют аксиальную симметрию, ромбическая составляющая Δχrh в уравнении (3) обращается в нуль, и выражение для химического сдвига принимает следующий вид:
(4)
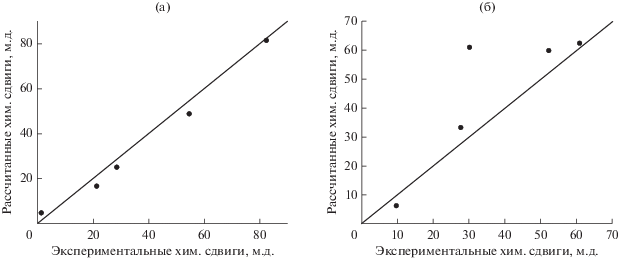
$\begin{gathered} \delta = \frac{1}{{12\pi {{r}^{3}}}} \times \\ \times \,\,\left[ {\Delta {{\chi }_{{{\text{а х }}}}}\left( {3{{{\cos }}^{2}}\theta - 1} \right)} \right] + \frac{{4\pi \mu _{B}^{2}}}{{9kT}}{{g}^{{{\text{iso}}}}}\rho + {{\delta }_{{{\text{д и а }}}}}. \\ \end{gathered} $Рассчитанные таким образом химические сдвиги для комплексов I и II с учетом высокоспинового состояния иона кобальта(II) (SВС = 3/2) и железа(II) (SВС = 2), использованного для оценки вкладов δкон в соответствии с уравнением (2), и вклада δдиа, взятого из спектра ЯМР 1Н для 2,6-ди(пиразол-3-ил)пиридинового лиганда (схема 1 ), хорошо согласовались с экспериментальными (рис. 5). Это подтверждает предположение о высокоспиновом состоянии комплексов I и II, сделанное на основе анализа химических сдвигов в спектрах ЯМР 1Н, и указывает на “молекулярную” природу данного эффекта.
Рис. 5.
Сравнение экспериментальных и рассчитанных химических сдвигов в спектрах ЯМР 1Н для комплексов I (a) и II (б) в d3-ацетонитриле при комнатной температуре.
Таким образом, мы синтезировали и охарактеризовали комплексы кобальта(II) и железа(II) с 2,6-ди(5-амино-1H-пиразол-3-ил)пиридином. Полученные для них (в том числе для комплекса II в виде двух новых сольватоморфов FeL2(ClO4)2 ∙ ∙ 2(C2H5)2O ∙ CH3CN и FeL2(ClO4)2 ∙ (C2H5)2O ∙ ∙ CH3CN ∙ 0.75(H2O)) рентгенодифракционные данные, в первую очередь длины связей M–N и тригонально-призматическое искажение КП MN6, однозначно указывают на то, что ионы металлов в кристалле при 120 К находятся в высокоспиновом состоянии (S = 3/2 для Со(II) и S = 2 для Fe(II)). Отсутствие температурно-индуцируемого спинового перехода в обоих комплексах при 120–300 К подтверждено данными ЯМР 1Н спектроскопии при комнатной температуре, в том числе при помощи оригинального подхода к анализу парамагнитных сдвигов на основе квантово-химических расчетов.
Список литературы
Garcia Y. // Spin Crossover in Transition Metal Compounds I. Springer Science & Business Media, 2004. P. 229.
Ferrando-Soria J., Vallejo J., Castellano M. et al. // Coord. Chem. Rev. 2017. V. 339. P. 17.
Hayami S., Holmes S.M., Halcrow M.A. // J. Mater. Chem. C. 2015. V. 3. № 30. P. 7775.
Molnár G., Rat S., Salmon L. et al. // Adv. Mater. 2017. V. 30. № 5. P. 1703862.
Kumar K.S., Ruben M. // Coord. Chem. Rev. 2017. V. 346. P. 176.
Spin-Crossover Materials: Properties and Applications / Ed. Halcrow M.A. John Wiley & Sons, Ltd., 2013.
Gütlich P., Gaspar A.B., Garcia Y. // Beilstein J. Org. Chem. 2013. V. 9. P. 342.
Judge J.S., Baker W. // Inorg. Chim. Acta. 1967. V. 1. P. 68.
Harris C.M., Lockyer T.N., Martin R.L. et al. // Aust. J. Chem. 1969. V. 22. № 10. P. 2105.
Novikov V.V., Ananyev I.V., Pavlov A.A. et al. // J. Phys. Chem. Lett. 2014. V. 5. № 3. P. 496.
Bartual-Murgui C., Codina C., Roubeau O., Guillem A. // Chem. Eur. J. 2016. V. 22. № 36. P. 12767.
Hathcock D.J., Stone K., Madden J., Slattery S.J. // Inorg. Chim. Acta. 1998. V. 282. № 2. P. 131.
Constable E.C., Housecroft C.E., Kulke T. et al. // Dalton Trans. 2001. № 19. P. 2864.
Halcrow M.A. // Coord. Chem. Rev. 2005. V. 249. № 25. P. 2880.
Halcrow M.A. // Coord. Chem. Rev. 2009. V. 253. № 21. P. 2493.
Kershaw Cook L.J., Kulmaczewski R., Mohammed R. et al. // Angew. Chem. Int. Ed. 2016. V. 55. № 13. P. 4327.
Kershaw Cook L.J., Mohammed R., Sherborne G. et al. // Coord. Chem. Rev. 2015. V. 289–290. P. 2.
Roberts T.D., Little M.A., Kershaw L.J., Halcrow M.A. // Dalton Trans. 2014. V. 43. P. 7577.
Hynes M.J., O’Shea M.T. // Dalton Trans. 1983. № 2. P. 331.
Cook B.J., Polezhaev A.V., Chen C.-H. et al. // Eur. J. Inorg. Chem. 2017. V. 2017. № 34. P. 3999.
Polezhaev A.V., Chen C.-H., Kinne A.S. et al. // Inorg. Chem. 2017. V. 56. № 16. P. 9505.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. № 1. P. 112.
Neese F. // Wiley Interdiscipl. Rev.: Comput. Mol. Sci. 2012. V. 2. № 1. P. 73.
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. P. 6158.
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. № 18. P. 3297.
Alvarez S. // J. Am. Chem. Soc. 2003. V. 125. № 22. P. 6795.
Alvarez S. // Chem. Rev. 2015. V. 115. P. 13447.
Pavlov A.A., Denisov G.L., Kiskin M.A. et al. // Inorg. Chem. 2017. V. 56. № 24. P. 14759.
Павлов А.А., Белов А.С., Савкина С.А. и др. // Коорд. химия. 2018. Т. 44. № 4. С. 236 (Pavlov A.A., Belov A.S., Savkin S.A. et al. // Russ. J. Coord. Chem. 2018. V. 44. № 8. P. 489. https://doi.org/10.1134/S1070328418080067).
Novikov V.V., Pavlov A.A., Nelyubina Yu.V. at al. // J. Am. Chem. Soc. 2015. V. 137. № 31. P. 9792.
Bertini I., Luchinat C., Parigi G., Ravera E. NMR of Paramagnetic Molecules: Applications to Metallobiomolecules and Models. Elsevier, 2016. V. 2. P. 508.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия