

Координационная химия, 2019, T. 45, № 8, стр. 461-467
Взаимодействие Re3Br9 c P(CH2OH)3: многообразие способов координации гидроксиметилфосфиновых лигандов к кластерам
А. В. Анюшин 1, П. А. Абрамов 1, 2, М. Н. Соколов 1, 2, 3, *
1 Институт неорганической химии им. А.В. Николаева СО РАН
Новосибирск, Россия
2 Новосибирский государственный университет
Новосибирск, Россия
3 Казанский (Приволжский) федеральный университет
Казань, Россия
* E-mail: caesar@niic.nsc.ru
Поступила в редакцию 28.12.2018
После доработки 19.02.2019
Принята к публикации 14.03.2019
Аннотация
Взаимодействие Re3Br9 с P(CH2OH)3 (THP) в метаноле в инертной атмосфере приводит к образованию нового кластерного комплекса состава [Re3(µ2-Br)3Br3(µ3-P,O,O-P(CH2OH)(CH2O)2)2H] ⋅ 0.8H2O · 1.6MeOH (I). При доступе воздуха образуется другой продукт, [{Re3(µ2-Br)3Br2(OCH2)2PCH2OP(CH2OH)2}2{(P(CH2OH)(CH2O)2)}2] (II), в котором два треугольных кластера связаны мостиковыми лигандами – депротонированными молекулами ТНР; помимо этого, каждый кластер тетрадентатно координирован фосфино-фосфинитным лигандом, возникающим в результате окислительной конденсации двух молекул ТНР (CIF files CCDC 1829565 (I) и 1829566 (II)).
Получение устойчивых водорастворимых (гидрофильных) комплексов переходных металлов с целью их применения в катализе, биологии и медицине является одной из насущных задач современной препаративной координационной химии [1–6]. С этой целью обычно используют фосфиновые лиганды, содержащие гидроксильные или сульфонатные группы в органических заместителях [1, 7]. В частности, трис(гидроксиметил)фосфин образует устойчивые водорастворимые комплексы с Pd, Pt, Rh, Re, Ru, Ir [8–12]. Такие комплексы могут найти применение как терапевтические и диагностические агенты, а также катализаторы для “зеленой химии” [13–17]. Возможна координация ТНР и его аналогов к халькогенидным кластерам молибдена, платины, никеля, кобальта, железа, родия [18–26]. Особый интерес представляет синтез комплексов ТНР с рением, учитывая исключительно богатую координационную и кластерную химию этого металла в широком интервале степеней окисления. Катализаторы на основе Re и его соединений (включая кластеры) активны в различных реакциях, включая гидрогенолиз глицерина, восстановление дикарбоновых кислот в диолы, трансформации, затрагивающие фрагменты CHnCO2H, макроциклизация тиоэфиров, гидрогенолиз алканов и др. [27–35].
Интересными объектами в химии рения являются треугольные кластеры {Re3} с двойной связью рений–рений, к каковым относятся тригалогениды Re3X9 (X = Cl, Br, I) и их производные. Они сыграли важную историческую роль в становлении представления о кратных связях металл–металл и способны вступать в самые разные химические трансформации. Достаточно хорошо изучены комплексы с фосфинами и арсинами, которые легко образуются из Re3X9 и соответствующих лигандов и имеют состав [Re3X9(PR3)3] (X = = Cl, Br; PR3 = PMe3, PMe2Ph, PMePh2, PEt2Ph, PEtPh2, PPh3, AsPh3), [Re3Cl9L1.5] и [Re3Cl9(η1‑L)3] (L = бидентатные дифосфины) [36–41]. Логическим шагом было расширить круг этих соединений путем координации Р(CH2OH)3.
В настоящей работе мы сообщаем о двух продуктах взаимодействия THP с Re3Br9 – трехъядернoм кластере [Re3(µ2-Br)3Br3(µ3-P,O,O-P(CH2OH)(CH2O)2)2H] · 0.8H2O · 1.6СН3OH (I) и “димере”, составленном из двух треугольных кластеров [{Re3(µ2-Br)3Br2(µ4-P,P,O,O-(OCH2)2PCH2OP- (CH2OH)2}2{µ2-P,O-(P(CH2OH)(CH2O)2)}2] (II), который содержит редкий тип фосфино-фосфинитного лиганда (HOCH2)2PCH2OP(CH2OH)2.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Соединение Re3Br9 синтезировали по опубликованной методике [42]. Остальные реактивы получали из коммерческих источников и использовали без дополнительной очистки. Метанол очищали перегонкой над магнием. Элементный анализ проводили с помощью микроскопа Toshiba TM2000 и CHNS-анализатора vario MICRO cube. Спектры ЯМР 31P{1H} записывали на спектрометре BrukerAvance 500 при комнатной температуре относительно 85%-ной H3PO4 в качестве внешнего стандарта. Электроспрей масс-спектры получали на приборе QuattroLC (Micromass, Manchester, UK). В качестве газа-осушителя и рабочего газа использовали азот со скоростью потока 400 и 80 мл/ч соответственно. Растворы образцов впрыскивали через шприцевой насос со скоростью 6 мкл/мин. Капиллярное напряжение 3.5 кВ, напряжение в камере варьировали от –10 до –60 В. Состав каждого пика определяли сравнением рассчитанной и экспериментальной изотопной картины, используя программу MassLynx 3.5.
Синтез I. Навеску Re3Br9 (0.67 г, 0.52 ммоль) помещали в колбу Шленка, растворяли в 10 мл метанола, дегазировали и заполняли аргоном. К полученному раствору добавляли твердый THP (0.40 г, 3.23 ммоль) и перемешивали на магнитной мешалке в атмосфере аргона в течение 2 нед. Окраска раствора постепенно менялась на темно-зеленую. Раствор концентрировали под вакуумом и ставили на кристаллизацию. Через 2 нед. выпавшие коричневые кристаллы отделяли декантацией от маточного раствора и промывали холодным диэтиловым эфиром. Выход 0.17 г (25%).
| Найдено, %: | C 6.21; | H 1.57; | Br 36.08; | Re 42.06. |
| Для C7.6H23O8.4P2Br6Re3 | ||||
| вычислено, %: | C 6.78; | H 1.72; | Br 35.54; | Re 41.41. |
Электроспрей масс-спектр (m/z): 1281.1 (M+). Спектр ЯМР 31P{1H} (CD3OD; δ, м.д.): 36.8.
Синтез II. Навеску Re3Br9 (0.10 г, 78.3 μмоль) помещали в колбу Шленка, растворяли в 5 мл метанола, дегазировали и заполняли аргоном. К полученному раствору добавляли твердый THP (0.06 г, 4.70 ммоль) и перемешивали на магнитной мешалке в атмосфере аргона в течение 2 нед. Окраска раствора постепенно менялась на темно-зеленую. Раствор концентрировали под вакуумом и на воздухе переносили в ампулу (внутренний диаметр 4 мм) для ЯМР. Ампулу закрывали крышкой и оставляли для кристаллизации. Через 4 нед. Выпавшие зеленые кристаллы отделяли от маточного раствора и промывали диэтиловым эфиром. Выход 0.01 г (11%).
| Найдено, %: | C 7.49; | H 1.53; | Br 30.42; | Re 42.74. |
| Для C16H40O16P6Br10Re6 | ||||
| вычислено, %: | C 7.41; | H 1.56; | Br 30.84; | Re 43.12. |
РСА. Дифракционные массивы получены на приборах New Xcalibur (Agilent Technologies) (I, 130 K) и Bruker X8 Apex (II, 100 K) (MoKα-излучениe, λ = 0.71073 Å, φ- и ω-сканированиe узких фреймов 0.5°). Поправка на поглощение ввeдена эмпирически по программам SADABS [43] и SCALE3 ABSPACK (CrysAlisPro, Agilent Technologies, Version 1.171.37.35 (выпущена 13-08-2014 CrysAlis 171.NET)). Структуры решены прямым методом и уточнены методом полноматричного МНК по |F|2 в анизотропном приближении (SHELX 2014/7, ShelXle) [44, 45]. Атомы водорода фиксированы в рассчитанных позициях для CH2 и OH-групп в I. Для II атомы водорода гидроксильных групп добавлены к вычисленному из данных РСА составу соединения. Основные кристаллографические данные и параметры уточнения для I и II приведены в табл. 1, основные длины связей для I и II – в табл. 2.
Таблица 1.
Основные кристаллографические данные и парaметры уточнения для соединений I и II*
| Параметр | Значение | |
|---|---|---|
| I | II | |
| M | 1348.86 | 2590.60 |
| Сингония; пр. гр. | Тетрагональная; I41/acd | Моноклинная; C2/c |
| a, Å | 17.7135(5) | 10.1233(4) |
| b, Å | 17.7135(5) | 16.0065(7) |
| c, Å | 33.5816(12) | 28.0801(14) |
| β, град | 90 | 98.072(2) |
| V, Å3 | 10 536.8(7) | 4505.0(3) |
| Z | 16 | 4 |
| µ, мм–1 | 23.00 | 25.20 |
| Размер кристалла, мм | 0.22 × 0.06 × 0.06 | 0.15 × 0.10 × 0.06 |
| Коррекция поглощения | Мультискан CrysAlisPRO 1.171.38.41 (Rigaku Oxford Diffraction, 2015). Эмпирически, используя сферические гармоники, согласно алгоритму SCALE3 ABSPACK | Мультискан SADABS (Bruker-AXS, 2004) |
| Tmin, Tmax | 0.232, 1.000 | 0.544, 0.723 |
| Число измеренных/независимых/ наблюдаемых отражений с I > 2σ(I) |
14 231/3346/1750 | 18 023/5159/4163 |
| Rint | 0.086 | 0.050 |
| Пределы по θ, град | 3.8–29.7 | 2.4–27.5 |
| (sin θ/λ)max, Å–1 | 0.697 | 0.649 |
| Пределы h, k, l | –17 ≤ h ≤ 22, –24 ≤ k ≤ 16, –44 ≤ l ≤ 46 |
–10 ≤ h ≤ 13, –20 ≤ k ≤ 20, –36 ≤ l ≤ 36 |
| R(F 2 > 2σ(F 2)), wR(F 2); S | 0.060, 0.139; 1.01 | 0.039, 0.088; 1.02 |
| Число отражений/уточняемых параметров/ограничений | 3346/122/6 | 5159/244/0 |
| Весовая схема | w = 1/[σ2$(F_{{\text{o}}}^{2})$ + (0.053P)2 + + 211.8261P], P = ($F_{{\text{o}}}^{2}$ + $2F_{{\text{c}}}^{2}$)/3 | w = 1/[σ2$(F_{{\text{o}}}^{2})$ + (0.0288P)2 + + 154.836P], P = ($F_{{\text{o}}}^{2}$ + $2F_{{\text{c}}}^{2}$)/3 |
| Δρmax/Δρmin, e Å–3 | 5.22/–3.37 | 2.32/–1.35 |
Таблица 2.
Основные межатомные расстояния (Å) для I и II*
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| I | |||
| Br(1)–Re(1) | 2.5802(16) | Br(4)–Re(2) | 2.665(2) |
| Br(2)–Re(1) | 2.5265(19) | O(1)–Re(2) | 1.984(10) |
| Br(2)–Re(1)i | 2.5265(19) | O(2)–Re(1) | 2.128(11) |
| Br(3)–Re(1)i | 2.5302(16) | P(1)–Re(1)i | 2.365(4) |
| Br(3)–Re(2) | 2.5086(14) | ||
| Угол | ω, град | Угол | ω, град |
| II | |||
| Re(1)–Br(4) | 2.5392(10) | Re(1)–O(1) | 2.035(7) |
| Re(1)–Br(5) | 2.5289(11) | Re(3)–O(2) | 1.956(6) |
| Re(1)–Br(7) | 2.6135(12) | Re(3)–O(4) | 2.053(6) |
| Re(2)–Br(5) | 2.5300(11) | Re(3)–O(4)ii | 2.205(7) |
| Re(2)–Br(6) | 2.5547(11) | Re(1)–P(3) | 2.409(3) |
| Re(2)–Br(8) | 2.5496(11) | Re(2)–P(1) | 2.339(3) |
| Re(3)–Br(4) | 2.5385(10) | Re(2)–P(2) | 2.477(3) |
| Re(3)–Br(6) | 2.5325(10) | ||
Координаты атомов и другие параметры структур депонированы в Кембриджском банке структурных данных (CCDC 1829565 (I) и 1829566 (II); deposit@ccdc.cam.ac.uk или http://www.ccdc.cam. ac.uk/data_request/cif).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
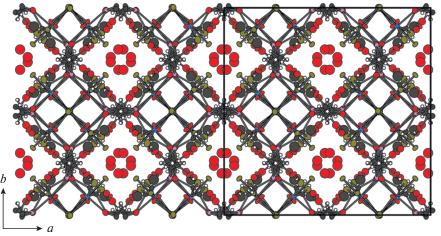
При обработке Re3Br9 избытком P(CH2OH)3 в метаноле без доступа воздуха наблюдается медленная реакция (2 нед.), сопровождающаяся изменением окраски раствора с темно-оранжевой на темно-зеленую. При этом в спектре ЯМР 31Р уменьшается интенсивность сигнала исходного THP (δ = –24.3 м.д.) с одновременным появлением сигнала при +36.8 м.д., отвечающего координированному ТНР. Кристаллизация приводит к образованию коричневых монокристаллов соединения I составa [Re3(µ2-Br)3Br3(µ3-P,O,O-P(CH2OH)(CH2O)2)2H] · 0.8H2O · 1.6MeOH (по данным РСА и элементного анализа). Растворы I в метаноле дают тот же сигнал в спектре ЯМР, что и реакционная смесь – один пик при 36.8 м.д. Таким образом, даже при большом мольном избытке ТНР (соотношение кластер : лиганд = 1 : 6) реакция протекает с образованием продукта I, в котором к кластерному ядру координированы только два лиганда ТНР. Структура I построена из нейтральных треугольных кластеров [Re3(µ2-Br)3Br3(µ3-P(CH2OH)(CH2O)2)2H] (рис. 1а), содержащих кластерное ядро {Re3(µ2-Br)3}6+ с атомами рения, образующими три эквивалентные двойные связи Re=Re 2.45–2.48 Å. Каждый атом рения имеет координационный полиэдр в виде пентагональной (с учетом связей металл–металл) или тригональной (без учета этих связей) бипирамиды. Помимо двух атомов рения, в координационную сферу металла входят в качестве экваториальных два мостиковых и один концевой атом брома, а в качестве апикальных – либо два атома кислорода (Re(2)), либо атом кислорода и атом фосфора (пара атомов Re(1)). При этом в кластере атомы Re(1) связаны поворотной осью второго порядка, проходящей через атомы Br(4), Re(2) и Br(2). Длины связей Re–(μ2‑Br), Re–Br, Re–P и Re–O (2.53, 2.58–2.67, 2.36 и 1.99–2.13 Å соответственно) близки к таковым, наблюдаемым в других фосфиновых комплексах [36–41]. Наблюдаемая стехиометрия продукта (отсутствие аниона) требует нейтрализации заряда за счет депротонирования трех координированных гидроксильных групп лигандов ТНР. По всей видимости, протон находится при одном из атомов кислорода, отвечающих более длинным связям Re(1)–O(2) 2.128(11) Å. Более коротким расстояниям Re(2)–O(1) (1.984(10) Å) отвечает координация депротонированного метилольного остатка. Полная кристаллическая структура I определяется упаковкой молекул кластера, воды и кристаллизационного метанола, образующих водородные связи. При этом сольватные молекулы занимают каналы, тянущиеся вдоль кристаллографического направления [001] (рис. 2).
Рис. 1.
Строение [Re3(μ2‑Br)3((P(CH2O)2(CH2OH))2H] в I (a) и [{Re3(µ2-Br)3Br2(µ4-P,P,O,O(OCH2)2PCH2OP-(CH2OH)2)2}2{µ2-P,O-(P(CH2OH)(CH2O)2)}2] в II.
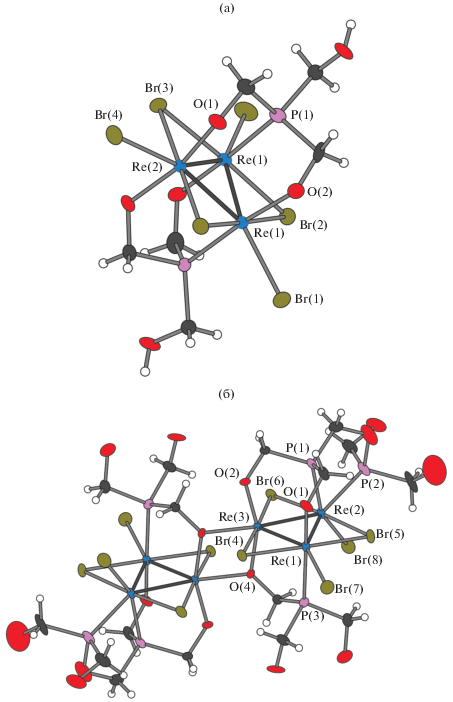
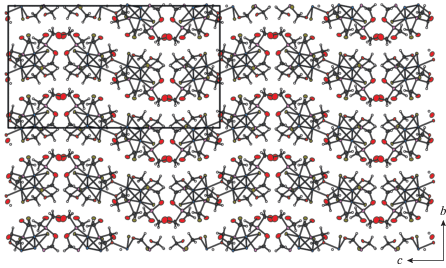
При доступе воздуха выделен другой продукт. При медленной кристаллизации (1 мес.) из аэрированного раствора, содержащего первоначально образовавшийся комплекс I, получены монокристаллы зеленого цвета II. По данным РСА, строение этого продукта описывается формулой [{Re3(µ2-Br)3Br2(µ4-P,P,O,O-(OCH2)2PCH2OP(CH2OH)2)2}2{µ2-P,O-(P(CH2OH)- (CH2O)2)}2] (рис. 1б). Шестиядерный молекулярный комплекс II построен на основе двух кластерных ядер {Re3(μ2-Br)3(P,P-PCH2OP)} ((P,P-PCH2OP)2– = = ((OCH2)2PCH2OP(CH2OH)2)2–), связанных в димеры двумя депротонированными лигандми THP. Каждый атом рения в треугольном кластерном остове {Re3(μ2-Br)3} связан с двумя другими атомами Re короткими двойными связями Re=Re 2.48–2.53 Å и посредством мостиковых атомов брома, лежащих в плоскости металлоостова (Re–(μ2‑Br) = 2.53–2.56 Å). Оставшиеся три координационных места занимают концевые атомы брома, а также фосфора и кислорода. Два треугольных кластера {Re3(μ2-Br)3}6+ связаны парой депротонированных лигандов ТНР через атомы Re(3), формируя тем самым структурный мотив {(O)2Re–(μ2-O)2–Re(O)2} (Re–(μ2‑O) 2.05–2.20 Å). Координационный полиэдр атома Re(3) можно описать {ReO3Br2}. Атом Re(2) координирован атомом брома (Re–Br 2.55 Å) и обоими атомами фосфора (фосфинитным и фосфиновым) депротонированного бидентатного лиганда ((OCH2)2PCH2OP(CH2OH)2)2–. Расстояние Re–P до фосфинитного атома существенно длиннее, чем до фосфинового (2.48 и 2.34 Å соответственно), что можно объяснить более слабыми донорными свойствами атома фосфора фосфинитного типа. Таким образом, координационный полиэдр атома Re(2) описывается как ReBr3P2. Атом Re(1) координирован атомом фосфора лиганда THP и алкоксидным атомом кислорода депротонированной группы OCH2 в аксиальных позициях (Re(1)–P 2.41, Re(1)–О 2.03 Å) и атомом брома в экваториальной позиции (Re(1)–Br 2.61 Å). Все эти расстояния типичны для комплексов Re3Br9 [36–41]. Молекула шестиядерного комплекса II электронейтральна, и заряд двух кластерных ядер уравновешивается депротонированием гидроксильных групп метилольных фрагментов –CH2OH. Отметим, что депротонирование лиганда (HOCH2)2PCH2OP(CH2OH)2 наблюдали впервые, хотя примеры обратимого депротонирования структурно близкого лиганда (HOCH2)2PCH2СН2P(CH2OH)2 известны [19]. В кристаллической структуре молекулы кластера образуют “извивающиеся” цепочки посредством слабых водородных связей H⋅⋅⋅Br (рис. 3).
Бидентатный фосфино-фосфинитный лиганд (HOCH2)2PCH2OP(CH2OH)2 можно рассматривать как продукт конденсации двух молекул P(CH2OH)3. К сожалению, низкая растворимость II в доступных растворителях не позволяет использовать спектроскопию ЯМР 31P и масс-спектрометрию для подробного изучения процесса образования этого лиганда. Мы можем лишь предположить механизм его формирования по аналогии с ранее описанными нами для комплекса [Ni3S2((HOCH2)2PCH2OP(CH2OH)2)3] [25]. Присутствие кислорода необходимо для одной из ключевых стадий, а именно (2):
(1)
${\text{HOC}}{{{\text{H}}}_{{\text{2}}}}{\text{P}}{{({\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}})}_{{\text{2}}}} = {\text{HP}}{{({\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}})}_{{\text{2}}}} + {\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}},$(2)
${\text{2HP}}{{\left( {{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}} \right)}_{{\text{2}}}} + {{{\text{O}}}_{{\text{2}}}} = {\text{2HOP}}{{\left( {{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}} \right)}_{{\text{2}}}},$(3)
$\begin{gathered} {\text{HOP}}{{\left( {{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}} \right)}_{{\text{2}}}} + {\text{HOC}}{{{\text{H}}}_{{\text{2}}}}{\text{P}}{{\left( {{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}} \right)}_{{\text{2}}}} = \\ = {{\left( {{\text{HOC}}{{{\text{H}}}_{{\text{2}}}}} \right)}_{{\text{2}}}}{\text{PC}}{{{\text{H}}}_{{\text{2}}}}{\text{OP}}{{\left( {{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OH}}} \right)}_{{\text{2}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}. \\ \end{gathered} $Также известно, что в реакции MCl2(P(CH2OH)3)2] (M = Pd, Pt) с избытком THP образуется [M{(CH2OH)2PCH2OP(CH2OH)2}2]2+, но попытки выделить из этих комплексов свободный лиганд приводят к его распаду на P(CH2OH)3 и HP(O)(CH2OH)2 [46]. Известны и другие примеры конденсации ТНР [47].
Таким образом, нам удалось показать, что реакция Re3Br9 c трис(гидроксиметил)фосфином не приводит, в отличие от нефункционализированных фосфинов, к образованию ожидаемого комплекса [Re3Br9(THP)3]. Отличия вызваны, во-первых, вовлечением в координацию гидроксильных групп ТНР, во-вторых, окислительной конденсацией двух молекул ТНР. При этом характерной особенностью координации обоих метилолфосфиновых лигандов (THP и продукта его конденсации) является тенденция к замыканию пятичленных циклов за счет (P,O)-мостиковой функции лиганда по отношению к связи рений–рений.
Конфликт интересов между авторами статьи отсутствует.
Вклад каждого автора: М.Н. Соколов – руководитель проекта, автор идеи, обсуждение результатов и написание статьи. А.В. Анюшин – ответственный исполнитель, проведение экспериментов и написание статьи. П.А. Абрамов – рентгеноструктурный анализ.
Список литературы
Pinault T., Bruce D.W. // Coord. Chem. Rev. 2003. V. 241. P. 1.
Herrmann W.A., Kohlpaintner C.W. // Angew. Chem. Int. Ed. 1993. V. 32. P. 1524.
Kothari K.K., Raghuraman K., Pillarsetty N.K. et al. // Appl. Radiat. Isot. 2003. V. 58. P. 543.
Dilworth J.R., Parrott S.J. // Chem. Soc. Rev. 1998. V. 27. P. 43.
Blower P.J. // Transition Met. Chem. 1998 V. 23. P. 109.
James B.R., Lorenzini F. // Coord. Chem. Rev. 2010. V. 254. P. 420.
Katti K.V., Gali H., Smith C.J. et al. // Acc. Chem. Res. 1999. V. 32. P. 9.
Ellis W., Harrison K.N., Hoye P.A.T. et al. // Inorg. Chem. 1992. V. 31. P. 3026.
Chatt J., Leigh J.G., Slade R.M. // Dalton Trans. 1973. P. 2021.
Berning D.E., Katti K.V., Barbour L.G. et al. // Inorg. Chem. 1998. V. 37. P. 334.
Driessen-Holscher B., Heinen J. // J. Organomet. Chem. 1998. V. 570. P. 141.
Fukoka A., Kosugi W., Morishita F. et al. // Chem. Commun. 1999. P. 489.
Moiseev D., James B.R., Patrick B.J. et al. // Inorg. Chem. 2006. V. 45. P. 2917.
Kalio R., Lönnecke P., Hey-Hawkins E. // J. Organomet. Chem. 2008. V. 693. P. 590.
Cadierno V., Crochet P., García-Garrido S.E. et al. // Dalton Trans. 2004. P. 3635.
Čubrilo J., Hartenbach I., Schleid T. et al. // Z. Anorg. Allg. Chem. 2006. V. 632. P. 400.
Casey C.P., Singer S.W., Powell D.R. et al. // J. Am. Chem. Soc. 2001. V. 123. P. 1090.
Sokolov, M.N., Anyushin, A.V., Hernandez-Molina R. et al. // Pure Appl. Chem. 2017. V. 89. P. 379.
Algarra A.G., Basallote M.G., Fernández-Trujillo M.J. et al. // Inorg. Chem. 2007. V. 46. P. 7668.
Basallote M.G., Fernández-Trujillo M.J., Pino-Chamorro J.Á. et al. // Inorg. Chem. 2012. V. 51. P. 6794.
Beltrán T.F., Llusar R., Sokolov M.N. et al. // Inorg. Chem. 2013. V. 52. P. 8713.
Sokolov M.N., Anyushin A.V., Virovets A.V. et al. // Inorg. Chem. Commun. 2011. V. 14. P. 1659.
Anyushin A.V., Sokolov M.N., Virovets A.V. et al. // Inorg. Chem. Commun. 2012. V. 24. P. 225.
Anyushin A.V., Sokolov M.N., Virovets A.V. et al. // J. Struct. Chem. 2013. V. 54. P. 638.
Anyushin A.V., Abramov P.A., Kompankov N.B. et al. // Russ. J. Coord. Chem. 2013. V. 39. P. 77. https://doi.org/10.1134/S1070328412090011
Anyushin A.V., Korotaev E.V., Andreeva A. Yu. et al. // Russ. Chem. Bull. 2016. P. 173.
Falcone D.D., Hack J.H., Klyushin A.Yu. // ACS Catal. 2015. V. 5. P. 5679.
Takeda Y., Tamura M., Nakagawa Y. et al. // Catal. Sci. Technol. 2016. V. 6. P. 5668.
Jothimurugesan K., Nayak A.K., Mehta G.K. et al. // AIChE J. 1985. V. 31. P. 1997.
Fung A.S., Kelley M.J., Gates B.C. // J. Mol. Catal. 1992. V. 71. P. 215.
Fung A.S., Tooley P.A., McDevitt M.R. et al // Polyhedron. 1988. V. 7. P. 2421.
Nashner M.S., Somerville D.M., Lane P.D. et al. // J. Am. Chem. Soc. 1996. V. 118. P. 12964.
Ma L., Wilson S.R., Sharpley J.R. // J. Am. Chem. Soc. 1994. V. 116. P. 787.
Adams R.D., Falloon S.B. // J. Am. Chem. Soc. 1994. V. 116. P. 10540.
Adams R.D., Falloon S.B. // Organometallics. 1995. V. 14. P. 1748.
Cotton F.A., Lippard S.J., Mague T.J. // Inorg. Chem. 1965. V. 4. P. 508.
Johnson N.P., Lock C.J.L., Wilkinson G. // J. Chem. Soc. 1964. P. 1054.
Cotton F.A., Walton R.A. // Inorg. Chem. 1966. V. 5. P. 1802.
Ferguson J.E., Hickford J.H. // Inorg. Chim. Acta. 1968. V. 2. P. 475.
Ebner J.R., Walton W. // Inorg. Chem. 1975. V. 14. P. 1987.
Esjornson S.M.V., Fanwick P.E., Walton R. // Polyhedron. 1990. V. 9. P. 1165.
Колбин Н.И., Овчинников К.В. // Журн. неорган. химии. 1968. Т. 13. С. 1190
Sheldrick G.M. SADABS. Göttingen (Germany): Univ. of Göttingen, 1996.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Hübschle C.B., Sheldrick G.M., Dittrich B. // J. Appl. Cryst. 2011. V. 44. P. 1281.
Hoye P.A.T., Pringle P.G., Smith M.B. et al. // Dalton Trans. 1993. P. 269.
Lorenzini F., Patrick B.O., James B.R. // Inorg. Chem. 2007. V. 46. P. 8998.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия