

Координационная химия, 2019, T. 45, № 9, стр. 569-576
Стереохимия октаэдрических цис-тетрафторокомплексов титана c энантиомерами Ph2P(O)CH(Me)CH2C(O)Et В СН2Сl2
Е. Г. Ильин 1, *, А. С. Паршаков 1, В. И. Привалов 1, Е. И. Горюнов 2, И. Б. Горюнова 2, Э. Е. Нифантьев 2
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
2 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
* E-mail: eg_ilin@mail.ru
Поступила в редакцию 26.09.2018
После доработки 29.03.2019
Принята к публикации 10.04.2019
Аннотация
Методом ЯМР 19F{1Н} и 31Р{1Н} изучено комплексообразование TiF4 с фосфорилированным кетоном Ph2P(O)CH(Me)CH2C(O)Et (L), имеющим асимметрический атом углерода в алифатическом углеводородном радикале и представляющим собой рацемическую смесь энантиомеров. Определен состав образующихся в растворе комплексов и на основе анализа спектров ЯМР 19F с применением концепции гетеротропности проведено отнесение резонансных линий к двум хиральным оптически активным рацемическим и мезо-стереоизомеру цис-TiF4L2. Показано преимущественное влияние конфигурации сосуществующих в координационной сфере энантиомеров монодентатного лиганда на аксиальные атомы фтора смешанных октаэдрических цис-тетрафторокомплексов d0-переходных металлов. Разработан новый эффективный метод синтеза лиганда L.
Координационные соединения фторидов d0‑переходных металлов находят применение в ряде важных каталитических процессов как прекурсоры в синтезе новых классов координационных соединений, в медицине и др. [1–3]. В частности, для активации связей фтор–углерод фторорганических соединений – важной области органической химии из-за ее практических применений для решения проблем окружающей среды, в фармацевтике и катализе (например, для дефторирования фторалканов путем комбинации фторидного прекатализатора и силана) [4]. Показана перспективность применения комплексов фторидов металлов как новых переносчиков 18F радиоизотопа в медицине, первоначально основанного на использовании фторорганических соединений [5, 6]. Постоянный научный интерес привлекает развитие неплатиновой противоопухолевой терапии. Например, фторсодержащие соединения титана типа CpR2TiF2 (CpR = 4-MeO–C6H4(CH2)C5H4, 3‑MeO–C6H4(CH2)C5H4) обладают высокой противоопухолевой активностью и при меньшей токсичности только в 2 раза ниже “золотого стандарта” – цис-платины [7–9]. Комплексные соединения фторидов d0-переходных металлов активно исследовали [10] в качестве модельных систем для установления особенностей природы химической связи в комплексах этих элементов. Активными реагентами по отношению к фторидам – жестким кислотам Льюиса – являются фосфорилсодержащие основания. Для исследования новых классов координационных соединений фторидов d0-переходных металлов интересны функционализированные фосфиноксиды, различающиеся числом, природой (Р=О, С(О)R, С(О)NR2, С(О)OH, NH2, NRH и др.) и относительным расположением функциональных групп. Ранее методом ЯМР 19F{1Н} и 31Р{1Н} изучены реакции TiF4 с представителями нового класса лигандов – дифенилфосфорилалканонами Ph2P(O)CH2C(O)Me (L1) [11] и Ph2P(O)- (CH2)2C(O)NMe2 (L2) [12]. Показано, что при двукратном избытке лиганда последние координируются к TiF4 через группу Р=О с образованием цис-TiF4(L1,2)2. В эквимолярном растворе TiF4 : L2 образуются димерные и полиядерные комплексы с фторными мостиками. При двукратном избытке TiF4 в качестве мостиков, наряду с ионами F–, выступает L2, использующий при координации группы Р=О и С=О [12]. Замена метильного радикала в группе С=О на –NR2 приводит к возникновению хелатирующей способности, и лиганд Ph2P(O)CH2C(O)NBu2 (L3), в зависимости от соотношения реагентов, координируется к TiF4 хелатно с образованием комплекса (η2-L3)TiF4 или монодентатно через более сильную по основности группу Р=О в цис-Ti(L3)2F4 [12]. Наряду с аддуктами обнаружены стереоизомеры димерного катиона {(μ-F)[Ti(η2-L3)F3]2}+, в котором фрагменты [(η2-L3)TiF3]+ с хелатно координированным лигандом связаны фторным мостиком [13]. С увеличением длины углеводородного мостика в Ph2P(O)(CH2)2C(O)NМе2 (L4) высокая хелатирующая способность сохранялась, и методами ЯМР 19F{1Н} и 13Р{1Н} при низких температурах удалось наблюдать в растворе конформационную изомерию семичленного гетероцикла TiOPCCCO в комплексе (η2-L4)TiF4 [14]. В его монокристалле обнаружены четыре кристаллографически независимые молекулы (η2-L4)TiF4 [15], в которых геометрия хелатных циклов TiOPCCCO попарно практически совпадает и, следовательно, в кристалле, как и в растворе, существуют два конформационных изомера [15]. В реакции L4 с TaF5 лиганд не только координируется хелатно с замещением иона фтора и образованием катиона [(η2-L)TaF4]+, но и выступает в качестве мостиковой группы, связывая две молекулы пентафторида (µ-L)[TaF5]2 [16].
Цель настоящей работы – изучение методом ЯМР 19F{1Н} и 31Р{1Н} комплексообразования TiF4 с представителем дифенилфосфорилалканонов, имеющим асимметрический атом углерода, 5-дифенилфосфорилгексан-3-оном – Ph2P(O)CH(Me)CH2C(O)Et (L) и представляющим собой рацемическую смесь энантиомеров. Проявление диастереотопии атомов фтора в спектрах ЯМР 19F фторокомплексов с оптически-активными бидентатными хелатными лигандами показано на примере диоксо- [17] и оксофторокомплексов вольфрама [18, 19]. Представляло интерес установить возможность идентификации и оценки методом ЯМР 19F относительных концентраций рацемических и мезо-стереоизомеров смешанных фторокомплексов, содержащих во внутренней сфере два монодентатных оптически активных лиганда (R и R), (S и S) или (R и S). Это также открывает возможности, путем введения фторидной метки, определять оптическую чистоту некоторых лекарственных препаратов и биологически активных соединений.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Соединение TiF4 получали фторированием порошка металла элементарным фтором в кварцевой системе. Лиганд L синтезировали по специально разработанной нами методике из коммерчески доступных соответствующих хлорфосфина и енона в присутствии АсОН в среде ацетонитрила по схе-ме 1 :
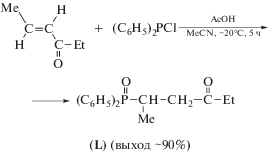
Схема 1 .
Реакция протекает с высокой скоростью при комнатной температуре, легко масштабируется и позволяет получать аналитически чистое целевое соединение с выходом, близким к количественному.
Синтез L. К раствору 3.0 г (30.5 ммоля) гекс-4-ен-3-она в 24 мл абсолютного МеСN в атмосфере аргона последовательно прибавляли по каплям раствор 1.94 г (32.3 ммоля) ледяной АсОН в 14 мл абсолютного MeCN и раствор 6.42 г (29 ммоля) свежеперегнанного дифенилхлорфосфина в 10 мл абсолютного MeCN. Смесь перемешивали на магнитной мешалке 5 ч при комнатной температуре. Затем отгоняли растворитель, остаток выдерживали 2 ч при 60°С в вакууме (~1 Торр) и растворяли его в 40 мл CH2Cl2. Полученный раствор фильтровали через основной носитель Al2O3 (5.0 г), промывали 20 мл CH2Cl2. Объединенные органические фильтраты упаривали в вакууме (~15 Торр), остаток растирали со смесью 25 мл Et2O и 12.5 мл гексана. Образовавшийся кристаллический осадок отделяли, промывали смесью Et2O–гексан (2 : 1) (2 × 37.5 мл) и сушили на воздухе. Получили 6.44 г лиганда L. Из промывных растворов дополнительно выделили 1.39 г продукта. Общий выход L 7.83 г (90%). Тпл = 97.5–98.5°С.
Спектр ПМР (δ, м.д.; J, Гц.): 0.72 т. (СН3СНAHB, 3Н, 3JHH = 7.3), 1.14 д.д. (СН3СН, 3H, 3JHH = 7.1, 2JHP = 16.2), 1.64 д. кв. (СН3СНАНВ, 1Н, 3JHH = 7.3, 2JHAHB = 17.8), 1.68 д. кв. (СН3СНАНВ, 1Н, 3JHH = 7.4, 2JHВHА = 17.8), 2.45–2.57 м. (СН2СН, 2Н), 3.16–3.25 м. (СН, 1Н), 6.98–7.07 м. (м- + р-С6Н5, 6Н), 7.78–7.85 м. (о-С6Н5, 2Н), 7.86–7.92 м. (о‑С6Н5, 2Н). Спектр ЯМР 13С (δ, м.д.; J, Гц.): 7.34 с. (СН3СН2), 12.76 д. (СН3СН, 2JCP = 2.7), 27.43 д. (СН, 1JCP = 74.5), 36.00 c. (CH3CH2), 41.46 c. (CHCH2), 128.42 д. (м-С6Н5, 3JCP = 10.9), 128.50 д. (м-С6Н5, 3JCP = 10.9), 130.92 д. (о-С6Н5, 2JCP = 8.2), 130.98 д. (р-С6Н5, 4JCP = 2.7), 131.11 д. (р-С6Н5, 4JCP = 2.7), 133.28 д. (ipso-C6H5, 1JCP = 94.5), 133.60 д. (ipso-C6H5, 1JCP = 93.6), 207.43 д. (С=О, 3JCP = = 12.7). Спектр ЯМР 31Р (δ, м.д.): 33.53 с.
Настоящая методика была успешно опробована в укрупненном варианте (трехкратное увеличение загрузок).
Элементный анализ L выполняли в Лаборатории микроанализа ИНЭОС РАН. Анализ С и Н проводили на анализаторе Elementar vario Micro cube, на фосфор – на спектрофотометре Agilent Cary 100 в виде фосфорномолибденовой гетерополикислоты после минерализации образцов концентрированной H2SO4 по Кьельдалю.
Температуру плавления определяли на приборе MPA 120 EZ-Melt automated melting point apparatus (Stanford Research Systems).
Спектры ЯМР 19F{1Н} и 31Р{1Н} растворов регистрировали на спектрометре Bruker АVANСE-300. Химические сдвиги ЯМР 19F и 31Р измеряли относительно CCl3F и 85%-ной H3PO4 соответственно. Для исследований готовили раствор с отношением L : TiF4 = 2.1 (несколько выше 2), чтобы получить основным продуктом комплекс TiF4L2. Расчетное количество TiF4 вводили в раствор L в предварительно осушенном CH2Cl2 и перемешивали на магнитной мешалке в течение 30 мин при комнатной температуре, TiF4 растворялся полностью. Все операции проводили в атмосфере сухого азота.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
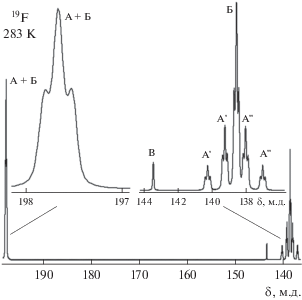
Была изучена зависимость спектров ЯМР 19F {1Н} и 31Р{1Н} раствора от температуры в интервале 263–313 К. При 313 К в спектре ЯМР 19F{1Н} (рис. 1) основными сигналами были группа широких перекрывающихся линий в области 138.0–142.5 м.д. и широкий сигнал при 200.5 м.д., что указывало на протекание процессов обмена лигандов. Положение наблюдаемых сигналов в областях химических сдвигов изученных ранее комплексов тетрафторида титана с дифенилфосфорилалканонами цис-TiF4(L1,2)2 [11, 12] и равенство суммарной относительной интенсивности линий А', Б', A" в сильном поле и сигнала при 200.5 м.д. указывало на образование тетрафторокомплекса титана цис-TiF4L2. Широкие линии в сильном поле мы отнесли к сигналам атомов фтора F1, расположенных в транс-положении друг к другу [11, 12]. Сигнал в слабом поле при 200.5 м.д. отнесен к атомам фтора F2, находящимся в транс-положении к группам Р=О на ординатах F–Ti–OP···L. Малоинтенсивный сигнал В свидетельствует о присутствии в растворе транс-TiF4L2 в незначительной концентрации.
Рис. 1.
Зависимость спектров ЯМР 19F{1Н} раствора TiF4 + 2L в CH2Cl2 от температуры. А, А', A" – F2, F1, F1'; Б, Б'– F2, F1 оптических стереоизомеров I и II комплекса цис-TiF4L2; В – F1 – транс-TiF4L2.
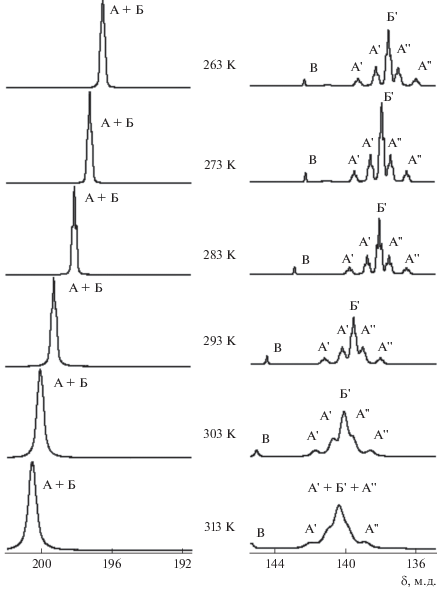
При снижении температуры до 303 К и ниже сигналы в области атомов фтора F1 начинают расходиться. При 293 К обнаружено пять резонансных линий, проявляющих тонкую структуру триплетов при 283 К (рис. 1). Суммарная интенсивность двух пар триплетов А' и А" (в пределах погрешности) равна интенсивности триплета Б'. Тонкую структуру триплета проявляет также линия в слабом поле, отнесенная к атомам фтора F2, находящимся в транс-положении к группе Р=О лиганда. Наилучшее разрешение спектра наблюдается при 283 К (рис. 2), а дальнейшее понижение температуры приводит к уширению сигналов.
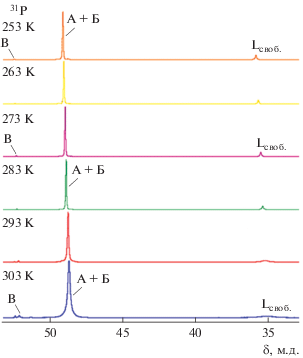
В спектре ЯМР 31Р{H} при 300 К атомам фосфора координированных лигандов цис-TiF4L2 отвечает интенсивный синглет при 48.5 м.д. (рис. 3).
Рис. 3.
Зависимость спектров ЯМР 31P{1Н} раствора TiF4 + 2L в CH2Cl2 от температуры. А + Б – 31P лиганда L оптических стереоизомеров I и II комплекса цис-TiF4L2; В – 31P лиганда в транс-TiF4L2.
Обычно спектры ЯМР 19F первого порядка цис-тетрафторокомплексов с монодентатно координированными лигандами TiF4Х2 представляют собой два триплета равной интенсивности, отвечающих двум парам неэквивалентных атомов фтора – на ординатах F1–Ti–F1 и F2–Ti–Х октаэдра [20–22]. Появление бόльшего числа мультиплетных резонансных линий в области сигналов атомов фтора F1, расположенных в транс-положении друг к другу, можно объяснить присутствием в растворе стереоизомеров цис-TiF4L2, поскольку лиганд L, имеющий асимметрический атом углерода, существует в виде рацемической смеси R- и S-энантиомеров. Нельзя также исключить образования конформационных изомеров комплекса вследствие заторможенного вращения координированных лигандов L. Для отнесения наблюдаемых резонансных линий и установления причины появления необычной группы резонансных линий в области химических сдвигов сигналов атомов фтора F1, рассмотрим зависимость спектров ЯМР 19F {1Н} раствора от температуры (рис. 1) более подробно.
Обращают на себя внимание особенности температурного поведения пар сигналов А' и А": сдвиг в противоположных направлениях с понижением температуры при сохранении величины разности резонансных частот (Δν = 284.4 Гц) между мультиплетами в парах А' и А" и уменьшение различий в их относительной интенсивности. Эти факторы, а также равенство суммарных интегральных интенсивностей пар линий А' и А" позволяют отнести их к резонансным линиям компонент ХY спектра ЯМР 19F второго порядка (А2ХY) октаэдрического тетрафторокомплекса цис-TiF4L2, в котором атомы фтора в транс-положении друг к другу неэквивалентны F1–Ti–F1'. Такое отнесение подтверждается также уменьшением различий (выравниванием) относительной интенсивности “внешних” и “внутренних” линий пар триплетов А' и А" с увеличением разности химических сдвигов между ними, т.е. приближение к спектру первого порядка – двум дублетам триплетов равной интенсивности. Сохраняющуюся при снижении температуры разность резонансных частот между мультиплетами в парах А' и А" мы отнесли к константе спин-спинового взаимодействия (КССВ) между ядрами неэквивалентных атомов фтора ${{J}_{{{{{\text{F}}}^{1}}{{{\text{F}}}^{{1'}}}}}}$ = 284.4 Гц на ординате F1–Ti–F1'.
Таким образом, две пары мультиплетов А' и А" мы отнесли к атомам фтора F1 в транс-положении друг к другу стереоизомера цис-TiF4L2 (I), в котором они неэквивалентны F1–Ti–F1', а триплет В' – к стереоизомеру цис-TiF4L2 (II), в котором атомы фтора в транс-положении друг к другу эквивалентны F1–Ti–F1'. Оба стереоизомера присутствуют в растворе в равных концентрациях. Разделить резонансные сигналы ЯМР 19F атомов фтора F2, расположенных в транс-положении к лиганду на ординатах F2–Ti–L стереоизомеров I и II (А и Б), при снижении температуры не удалось и в спектре им отвечает линия в слабом поле (А + Б), по форме напоминающая триплет (рис. 1). Его интенсивность равна суммарной интенсивности сигналов атомов фтора F1 изомеров I и II (А', А", Б'), химические сдвиги которых различаются. Значения КССВ между неэквивалентными концевыми атомами фтора, находящимися в цис-положении друг к другу (${{J}_{{{{{\text{F}}}^{{\text{2}}}}{{{\text{F}}}^{{\text{1}}}}}}}$ и ${{J}_{{{{{\text{F}}}^{{\text{2}}}}{{{\text{F}}}^{{\text{1}}}}'}}}$ ) стереоизомеров I и II в пределах ошибки совпадают (38.2 Гц). Их величина более чем в 7 раз ниже значения КССВ между транс-расположенными атомами фтора (${{J}_{{{{{\text{F}}}^{1}}{{{\text{F}}}^{{1'}}}}}}$ = 284.4 Гц) в стереоизомере I. Это подтверждает сделанный на основании обобщения экспериментальных данных по изучению стереохимии смешанных фторидов групп IV–VI в кристаллическом состоянии и в растворе [23] вывод о преимущественном характере межлигандных взаимодействий через центральный атом в октаэдрических комплексах d0-переходных металлов, между лигандами, находящимися в транс-положении друг к другу. Наблюдаемая величина ${{J}_{{{{{\text{F}}}^{1}}{{{\text{F}}}^{{1'}}}}}}$ существенно превышает КССВ между мостиковым и транс-концевыми атомами фтора в спектрах ЯМР 19F димерных комплексов [M2F11]– (M = Nb, Ta, Sb, Ti) [23–27] и M2F10L (M = Nb, Ta) [28], значения которых лежат в интервале 150–175 Гц.
Образование в растворе стереоизомеров I и II комплекса цис-TiF4L2, по нашему мнению, связано с существованием лиганда L в виде рацемической смеси двух оптических R- и S-изомеров.
Комплекс цис-TiF4L2 в растворе может образовывать три оптических стереоизомера: цис-TiF4LRLR, цис-TiF4LSLS и цис-TiF4LRLS (схема 2 ). В соответствии с теорией статистического распределения лигандов [29], вероятность образования стереоизомеров, содержащих две одноименные по оптической активности – правовращающие или левовращающие молекулы лиганда (цис-TiF4LRLR и цис-TiF4LSLS) в два раза ниже, чем содержащих противоположные по направлению оптической активности лиганды – цис-TiF4LRLS. Следовательно, суммарная концентрация первых двух должна быть равной концентрации последнего, что и наблюдается. Поскольку лиганд является рацематной смесью энантиомеров, это подтверждает возможность существования в растворе трех оптических стереоизомеров тетрафторокомплекса цис-TiF4L2 (схема 2 ):

Схема 2 .
Для отнесения групп линий А, А', А" и Б, Б' в спектрах ЯМР 19F к определенным оптическим стереоизомерам цис-TiF4L2 мы выбрали за основу фундаментальные работы по определению стереохимической конфигурации хиральных молекул органических соединений методами ЯМР спектроскопии [30, 31]. В частности, рассмотрение органических молекул, имеющих два асимметрических центра, разделенных одним углеродным атомом. Органические молекулы, содержащие симметрично расположенные хиральные группы (GR, GS) могут существовать в двух формах, называемых мезо или оптически неактивным изомером и оптически активными рацемическими изомерами, присутствующими в виде двух энантиомеров (схема 3 ) [30]. Для таких симметричных молекул предполагалось [32], что соотношения симметрии между протонами позволят различать мезо и рацемические стереоизомеры методом ЯМР (схема 3 ):
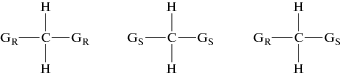
Энантиомеры рацемического изомера мезо-Изомер
Схема 3 .
В энантиомерах рацемического изомера две хиральные группы (GR или GS) имеют одинаковую абсолютную конфигурацию, тогда как в мезоформе они имеют противоположные конфигурации [30]. Из этого следует, что в энантиомерах рацемического изомера два протона мостикового атома углерода могут поменяться операцией симметрии С2, поэтому должны быть изохронными [30] и в спектрах ПМР не различаются. В мезо-изомере протоны у мостикового атома углерода не могут быть совмещены никакой операцией симметрии, поэтому они диастереотопны, должны быть анизохронными [30] и могут различаться в спектрах ПМР.
Мы применили эти выводы к возможным оптическим стереоизомерам I и II октаэдрического тетрафторокомплекса цис-TiF4L2 (схема 4 ), заменив атом углерода на атом титана, а атомы водорода – на аксиальные атомы фтора F1. В энантиомерах цис-TiF4LRLR и цис-TiF4LSLS рацемического стереоизомера октаэдрического комплекса TiF4L2, содержащих симметрично расположенные одноименные энантиомеры лиганда, последние могут быть совмещены путем вращения вокруг оси второго порядка С2 – биссектрисы угла OTiO (схема 2 ), лежащей в экваториальной плоскости октаэдра перпендикулярно ординате F1–Ti–F1. Из этого следует, что в энантиомерах рацемического изомера, при условии одинаковой абсолютной конфигурации координированных лигандов L, аксиальные атомы фтора могут поменяться операцией симметрии С2, должны быть изохронными, а их химические сдвиги не различаются.
В мезо-стереоизомере октаэдрического комплекса цис-TiF4LRLS лиганды, координированные к центральному иону, имеют противоположные конфигурации и не связаны никакой операцией симметрии, поэтому аксиальные атомы фтора анизохронны и могут различаться по химическим сдвигам.
Учитывая вышесказанное, стереоизомер I, в котором аксиальные атомы фтора находятся в транс-положении друг к другу, стерически неэквивалентны и различаются по химическим сдвигам F1 и F1' и которому в спектре ЯМР 19F отвечает группа линий А, А', А", по нашему мнению, является мезо-стереоизомером октаэдрического тетрафторокомплекса цис-TiF4LRLS.
Стереоизомер II, в котором атомы фтора в транс-положении эквивалентны и которому в спектре ЯМР 19F отвечают два триплета Б и Б' равной интенсивности, мы отнесли к сумме оптически активных энантиомеров рацемического стереоизомера цис-TiF4LRLR (IIа) и цис-TiFSLS (IIб), находящихся в равных концентрациях. Учитывая, что геометрическое расположение лигандов L относительно атомов фтора F1 симметрично, последние эквивалентны, изохронны и в спектре ЯМР 19F не различаются.
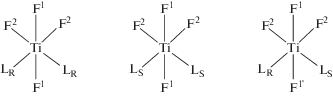
Энантиомеры IIа и IIб рацемического изомера мезо-Изомер I цис-TiF4L2
Схема 4 .
Суммарная концентрация оптически активных энантиомеров IIа и IIб рацемического изомера должна быть равна концентрации мезостереоизомера I цис-TiF4LRLS [18], что и наблюдается. Различить сигналы от ядер атомов фосфора оптически активных стереоизомеров IIа и IIб и мезостереоизомера I комплекса цис-TiF4L2 в спектрах ЯМР 31Р не удалось (рис. 2).
Для комплекса транс-TiF4L2 в растворе также возможно образование мезо-транс-TiF4LRLS и оптически активных энантиомеров транс-TiF4LRLR и транс-TiF4LSLS рацемического стереоизомера, однако наблюдать их отдельные сигналы не удалось. Это может быть связано с усреднением эффектов оптической стереоизомерии хиральных лигандов при их транс-расположении в координационной сфере, что обеспечивает им свободу вращения.
Таким образом, установлено образование оптических стереоизомеров октаэдрического комплекса цис-TiF4L2 с представителем фосфорилированных кетонов, имеющим асимметрический атом углерода в алифатическом радикале. На основе анализа спектров ЯМР 19F с применением концепции гетеротропности сделано заключение об относительной стереохимической конфигурации присутствующих в растворе энантиомерах хирального и мезо-оптически неактивного стереоизомеров. В мезо-стереоизомере цис-TiF4LRLS атомы фтора F1, расположенные на одной ординате октаэдра (в транс-положении друг к другу), стерически неэквивалентны, различаются по химическим сдвигам и между ними наблюдается КССВ ${{J}_{{{{{\text{F}}}^{1}}{{{\text{F}}}^{{1'}}}}}}$. В энантиомерах оптически активного рацемического стереоизомера цис-TiF4LRLR и цис-TiF4LSLS атомы фтора в транс-положении друг к другу эквивалентны и в спектре ЯМР 19F комплексу отвечают два триплета равной интенсивности. Разработана новая методика синтеза с высоким выходом фосфорилированных кетонов из коммерчески доступных реагентов.
Список литературы
Nikiforov G.B., Roesky H.W., Koley D. // Coord. Chem. Rev. 2014. V. 258–259. P. 16.
Benjamin S.L., Levason W., Reid G. // Chem. Soc. Rev. 2013. V. 42. P. 1460.
Haiges R., Deokar P., Christe K.O. // Angew. Chem. Int. Ed. 2014. V. 53. 5431.
Kühnel M.F., Holstein P., Kliche M. et al. // Chem. Eur. J. 2012. V. 18. P. 10701.
Shetty D., Choi S.Y., Jeong J.M. et al. // Chem. Commun. 2011. V. 47. P. 9732.
McBride W.J., Sharkey R.M., Karacay H. et al. // J. N-ucl. Med. 2009. V. 50. P. 991.
Garbutcheon-Singh K.B., Grant M.P., Harper B.W. et al. // Curr. Top. Med. Chem. 2011. V. 11. P. 521.
Buettner K.M., Valentine A.M. // Chem. Rev. 2012. V. 112. P. 1863.
Eger S., Immel T.A., Claffey J. et al. // Inorg. Chem. 2010. V. 49. P. 1292.
Beste A., Kramer O., Gerhard A. et al. // Eur. J. Inorg. Chem. 1999. V. 11. P. 2037.
Ильин Е.Г., Паршаков А.С., Яржемский В.Г. и др. // Докл. РАН. 2015. Т. 465. № 3. С. 314 (Il’in E.G., Parshakov A.S., Yarzhemskii V.G. et al. // Dokl. Chem. 2015. V. 465. Pt 1. P. 272). https://doi.org/10.1134/ S0012500815110075).
Ильин Е.Г., Паршаков А.С., Данилов В.В. и др. // До-кл. РАН. 2016. Т. 471. № 2. С. 163 (Il’in E.G., Parshakov A.S., Danilov V.V. et al. // Dokl. Chem. 2016. V. 471. Part 1. P. 314. https://doi.org/10.1134/S0012500816110045).
Ильин Е.Г., Ковалев В.В., Нестерова Н.П. и др. // Докл. РАН. 2008. Т. 423. № 4. С. 493.
Ильин Е.Г., Паршаков А.С., Привалов В.И. и др. // Докл. РАН. 2016. Т. 467. № 5. С. 547 (Il’in E.G., Parshakov A.S., Privalov V.I. et al. // Dokl. Chem. 2016. V. 467. Pt. 2. P. 122. https://doi.org/10.1134/S0012500816040054).
Ильин Е.Г., Паршаков А.С., Александров Г.Г. и др. // Докл. РАН. 2016. Т. 470. № 2. С. 176 (Il’in E.G., Parshakov A.S., Aleksandrov G.G. et al. // Dokl. Chem. 2016. V. 470. P. 255. https://doi.org/10.1134/S0012500816090068).
Ильин Е.Г., Паршаков А.С., Данилов В.В. и др. // Коорд. химия. 2018. Т. 44. № 5. С. 318 (Il’in E.G., Parshakov A.S., Danilov V.V. et. al. // Russ. J. Coord. Chem. 2018. V. 44. 10. P. 619. https://doi.org/10.1134/ S1070328418100068).
Calve I.J., Guerchais J.E., Kergoat R., Kheddar N. // Inorg. Chim. Acta. 1979. V. 33. P. 95.
Кокунов Ю.А., Бочкарева В.А., Чубар Ю.Д., Буслаев Ю.А. // Коoрд. химия. 1980. Т. 6. № 8. С. 1205.
Кокунов Ю.А., Бочкарева В.А. Буслаев Ю.А. // Коорд. химия. 1980. Т. 6. № 11. С. 1619.
Muetterties E.L. // J. Am. Chem. Soc. 1960. V. 82. P. 1082.
Ковалев В.В., Ильин Е.Г. // Журн. неорган. химии. 2016. Т. 61. № 1. С. 67 (Kovalev V.V., Il’in E.G. // Russ. J. Inorg. Chem. 2016. V. 61. № 1. P. 63. https://doi.org/10.1134/S0036023616010125).
Ковалев В.В., Ильин Е.Г. // Журн. неорган. химии. 2016. Т. 61. № 9. С. 1244 (Kovalev V.V., Il’in E.G. // Russ. J. Inorg. Chem. 2016. V. 61. № 9. P. 1191. https://doi.org/10.1134/S0036023616090114).
Buslaev Yu.A., Ilyin E.G. // Soviet Sci. Rev. B. 1987. V. 1. p. 90
Edwards A.J., Jones G.R. // J. Chem. Soc. A. 1970. P. 1491.
Brownstein S. // Inorg. Chem. 1973. V. 12. P. 584.
Dean P.A.W., Fergusson B.J. // Canad. J. Chem. 1974. V. 52. P. 667.
Ilyin E.G., Buslaev Yu.A. // J. Fluor. Chem. 1984. V. 25. № 1. P. 57.
Ilyin E.G., Buslaev Yu.A., Ignatov M.E. et al. // J. Fluor. Chem. 1978. V. 12. P. 381.
Calingaert G., Beatty H.A. // J. Am. Chem. Soc. 1939. V. 61. P. 2748.
Gaudemer A. // Stereochemistry: Fundamentals and Methods. V. 1. Determination of Configurations by Spectroscopic Methods / Ed. Kagan H.B. Stuttgart: Georg Thieme Publishers, 1977. P. 73.
Eliel E.L., Wilen S.H., Doile M.P. Basic Organic Stereochemistry. John. Wiley & Sons, Inc., 2001. 754 p.
Mislow K., Raban M. Topics in Stereochemistry. V. I. N.Y.: Interscience Puplishers, 1967. p. 73.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия