

Координационная химия, 2019, T. 45, № 9, стр. 527-538
Комплексы олова(II) на основе N-алкилзамещенных о-амидофенолятных лигандов. Кислотно-основные и окислительно-восстановительные превращения
А. В. Пискунов 1, *, К. В. Цыс 1, М. Г. Чегерев 2, А. В. Черкасов 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Институт физической и органической химии Южного федерального университета
Ростов-на-Дону, Россия
* E-mail: pial@iomc.ras.ru
Поступила в редакцию 27.03.2019
После доработки 24.04.2019
Принята к публикации 25.04.2019
Аннотация
Синтезирован и структурно охарактеризован новый станнилен AdAPSn (I) на основе 4,6-ди-трет-бутил-N-адамантил-о-аминофенола. В кристаллическом состоянии станнилен I формирует бесконечные цепочки за счет межмолекулярных донорно-акцепторных Sn–N и металлофильных Sn···Sn взаимодействий. Реакционная способность I, а также синтезированного ранее t-BuAPSn (II) изучена на примере окислительно-восстановительных и кислотно-основных реакций. Установлено, что станнилены I и II внедряются по связи S–S тетраметилтиурамдисульфида с образованием соответствующих дитиокарбаматных комплексов олова(IV). При этом взаимодействие с мягкими одноэлектронными окислителями протекает с участием редокс-активного о-амидофенолятного лиганда и приводит к генерации лабильных парамагнитных станниленов, изученых методом спектроскопии ЭПР. Наличие неподеленной электронной пары у атома низковалентного олова обуславливает его основные свойства, что продемонстрировано на примере взаимодействия I c нонакарбонилдижелезом. Структуры некоторых синтезированных соединений установлены методом РСА (СIF files CCDC № 1905419–1905421).
Координационные и металлоорганические соединения, содержащие в своем составе редокс-активные лиганды, являются одной из перспективных точек развития современной химии и находят применение в целом ряде областей исследования, таких как фундаментальные вопросы теории химической связи, каталитические превращения органических субстратов и малых молекул, молекулярный магнетизм и многое другое [1–8]. Другим интенсивно развивающимся направлением координационной химии стали исследования соединений непереходных металлов в низких степенях окисления, обладающих уникальными химическими свойствами [9–13]. Среди них большое внимание уделяется двухвалентным производным элементов 14 группы – так называемым тяжелым аналогам карбенов. К настоящему времени в литературе накоплен большой массив информации о синтезе и химических свойствах стабильных гермиленов, станниленов и плюмбиленов [14–20]. Введение редокс-активных лигандов (о-хинонов [21, 22], о-иминохинонов [22–27] и альфа-дииминов [28–34]) в состав тяжелых аналогов карбенов позволило сформировать новую ветвь развития химии низковалентных производных металлов, для которых существенно расширены рамки окислительно-восстановительных превращений. Появилась возможность не только синтезировать принципиально новые парамагнитные аналоги карбенов [33, 34], но и использовать их в качестве донорных лигандов в дизайне координационных соединений переходных металлов [35]. Настоящая работа посвящена синтезу и исследованию химических свойств станниленов – комплексов олова(II) на основе редокс-активных 4,6-ди-трет-бутил-N-(алкил)-o-аминофенолятных лигандов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу и исследованию химических превращений комплексов олова проводили в условиях отсутствия кислорода и влаги воздуха. Использованные в работе растворители очищали и обезвоживали согласно рекомендациям [36]. Применяли коммерческие реактивы: тетраметилтиурамдисульфид (ТMTDS), галогениды ртути(II). 2-Этокси-3,6-ди-трет-бутилфеноксильный радикал [37], Fe2(CO)9 [38], Sn(N(SiMe3)2)2 [39] и комплекс t-BuAPSn (II) [26] получали по известным методикам. 4,6-Ди-трет-бутил-N-(адамантил)-o-аминофенол синтезировали по реакции 3,5-ди-трет-бутилпирокатехина с 1-аминоадамантаном [40, 41] по модифицированной методике, описанной в [42].
Синтез комплекса AdAPSn (I). Sn(N(SiMe3)2)2 (0.444 г, 1 ммоль) в Et2O (20 мл) добавляли к раствору 4,6-ди-трет-бутил-N-(адамантил)-o-аминофенола (1 ммоль) в том же растворителе (10 мл). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре. После окончания реакции растворитель удаляли при пониженном давлении. Сухой остаток перекристаллизовали из горячего толуола (20 мл). Выход комплексa I в виде диамагнитного кристаллического порошка желто-оранжевого цвета 0.38 г (80%).
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д. (J, Гц)): 7.23 (д., 1H, HAP, JH–H = 2.3); 6.83 (д., 1H, HAP, JH–H = = 2.3); 1.88 (с. 9H, (t-Bu)); 1.83 (с., 3H, –CH–); 1.54 (с., 6H, –CH2–); 1.45 (с., 6H, –CH2–); 1.35 (с., 9H, (t-Bu)). Спектр ЯМР 13C (C6D6; 20°С; δ, м.д.): 138.1, 137.6 (C–(t-Bu)); 135.0 (C–O); 125.5 (C–N); 121.0–104.1 (Cарил); 57.3 (N–CAd); 41.8, 36.0 (CH2Ad); 32.0–31.1 (Cчетв); 30.3 (CH3(t-Bu)); 29.7 (CHAd). Спектр ЯМР 119Sn (C6D6; 20°С; δ, м.д.): ‒295.2.
ИК-спектр (ν, см–1): 1583 сл, 1554 сл, 1533 с, 1409 сл, 1395 ср, 1323 сл, 1304 сл, 1270 ср, 1248 ср, 1239 ср, 1213 сл, 1183 сл, 1157 ср, 1136 сл, 1115 сл, 1096 ср, 1052 сл, 1026 сл, 1005 ср, 972 с, 944 сл, 916 сл, 902 ср, 865 сл, 851 ср, 828 ср, 806 сл, 757 ср, 732 с, 694 ср, 652 ср, 641 сл, 615 сл, 568 ср, 542 сл, 528 ср, 507 ср, 465 ср.
Синтез комплексов III и IV. К желтому раствору комплексов I или II (1 ммоль) в CH2Cl2 (20 мл) приливали раствор ТMTDS (1 ммоль) в гексане (10 мл). Окраска реакционной смеси мгновенно изменялась на красно-фиолетовую. Реакционную смесь выдерживали в течение 3 ч при комнатной температуре. После концентрирования раствора комплексы III и IV выделяли в виде диамагнитных кристаллических веществ насыщенного красно-фиолетового цвета, которые после фильтрования сушили при комнатной температуре в вакууме форвакуумного насоса.
Комплекс III: выход 0.489 г (70%).
ИК-спектр (ν, см–1): 1746 ср, 1718 сл, 1631 сл, 1599 с, 1563 сл, 1526 сл, 1482 с, 1416 ср, 1377 с, 1358 с, 1339 сл, 1299 с, 1264 с, 1241 ср, 1201 ср, 1185 ср, 1164 сл, 1145 сл, 1124 сл, 1103 сл, 1094 сл, 1080 ср, 1049 сл, 1024 сл, 996 сл, 968 ср, 928 ср, 917 ср, 877 с, 865 с, 830 с, 805 ср, 774 ср,743 с, 671 сл, 654 сл, 645 сл, 594 сл, 559 ср, 529 с, 477 ср.
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д. (J, Гц)): 7.56 (д., 1H, HAP, JH–H = 1.8); 7.09 (д., 1H, HAP, JH–H = = 1.7); 2.33 (с., 12 H, Me); 1.88 (с., 9H, N(t-Bu)), 1.53 (с., 9H, (t-Bu)), 1.65 (с., 3H, –CH–); 2.78 (с., 6H, –CH2–); 2.22 (с., 6H, –CH2–). Спектр ЯМР 13C (C6D6; 20°С; δ, м.д.): 198.6 (C (TMTDS)); 138.1, 137.6 (C–(t-Bu)); 35.0 (C–O); 128.9 (C–N); 115.0–110.3 (Cарил); 57.8 (N–CAd); 45.29 (CH3 (TMTDS)); 37.8, 35.7 (CH2Ad); 34.0 (CHAd); 32.2–30.7 (Cчетв); 30.5 (CH3(t-Bu)). Спектр ЯМР 119Sn (C6D6; 20°С; δ, м.д.): –656.5.
Комплекс IV: выход 0.524 г (84%).
ИК-спектр (ν, см–1): 1582 с, 1414 с, 1359 ср, 1329 с, 1286 ср, 1266 ср, 1256 ср, 1232 ср, 1211 с, 1111 сл, 1101 сл, 1051 сл, 1026 ср, 994 с, 914 сл, 870 с, 797 с, 775 сл, 693 сл, 656 ср, 609 ср, 534 сл, 446 сл.
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д. (J, Гц)): 7.35 (д., 1H, HAP, JH–H = 2.2); 7.20 (д., 1H, HAP, JH–H = = 2.2); 2.26 (с., 12 H, Me); 1.76 (с., 9H, N(t-Bu)), 1.43 (с., 9H, (t-Bu)), 1.19 (с., 9H, (t-Bu)). Спектр ЯМР 119Sn (C6D6; 20°С; δ, м.д.): –654.8.
Синтез комплекса V. К раствору станнилена II (0.280 г, 0.78 ммоль) в толуоле (10 мл) добавляли суспензию Fe2(CO)9 (0.28 г, 0.78 ммоль) в том же растворителе (10 мл). Реакционную смесь выдерживали в темноте при 20°C в течение 2 сут. За это время цвет раствора изменился на интенсивно коричневый. Выход комплекса V в виде желтого кристаллического порошка после концентрирования раствора до 10 мл составил 0.35 г (70%).
ИК-спектр (ν, см–1): 2043 с, 1982 ср, 1935 с, 1557 сл, 1412 ср, 1285 сл, 1267 сл, 1244 ср, 1202 сл, 1186 сл, 1130 сл, 1105 сл, 1043 ср, 972 ср, 939 ср, 939 ср, 914 ср, 872 ср, 829 ср, 804 ср, 766 сл, 750 ср, 692 ср, 665 ср, 617 с, 549 сл, 522 сл.
Спектр ЯМР 1Н (d8-THF; 20°С; δ, м.д. (J, Гц)): 6.84 (д., 2H, JH–H = 2.3, HAP); 6.70 (д., 2H, JH–H = = 2.3, HAP); 1.19 (с., 9H, t-Bu); 1.08 (с., 9H, t-Bu); 2.08 (с., 3H, –CH–); 2.05 (с., 6H, –CH2–); 1.66 (с., 6H, –CH2–). Спектр ЯМР 13C (d8-THF; 20°С; δ, м.д.): 137.6, 134.3 (C–(t-Bu)); 125.9 (C–O); 124.6 (C–N); 120.8, 123.9 (Cарил); 57.4 (N–Cчетв); 34.4, 33.3 (Cчетв); 31.6–29.6 (CH3(t-Bu)); 211.1 (C=O).
ИК-спектры регистрировали на ИК Фурье-спектрометре ФСМ-1201 в вазелиновом масле в кюветах из KBr. Спектры ЭПР фиксировали на спектрометре Bruker EMX. В качестве стандарта при определении g-фактора использовали 2,2-дифенил-1-пикрилгидразил (g = 2.0037). Для определения точных параметров спектр ЭПР симулировали с помощью программы WinEPR SimFonia (Bruker).
Квантово-химические расчеты выполняли с использованием программного пакета Gaussian09 [43] методом теории функционала плотности (DFT) с применением функционала B3LYP [44] и стандартного базисного набора def2svp для всех атомов.
РСА соединений I, III и IV проведен на дифрактометрах Agilent Xcalibur E (III), Bruker Smart Apex (IV) и Bruker D8 Quest (I) (ω-сканирование, МоКα-излучение, λ = 0.71073 Å). Измерение и интегрирование экспериментальных наборов интенсивностей, учет поглощения и уточнение структур проведены с использованием программных пакетов CrysAlis Pro [45], Smart, APEX2 [46], SADABS [47] и SHELX [48]. Структуры решены прямым методом и уточнены полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для неводородных атомов. Водородные атомы в I, III и IV помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв (U(H)изо = 1.5U(C)экв для метильных фрагментов).
Кристаллографические данные, параметры рентгеноструктурных экспериментов и уточнения структур приведены в табл. 1. Структуры зарегистрированы в Кембриджском банке структурных данных (CCDC № 1905419 (I), 1905420 (III), 1905421 (IV) и доступны по адресу: ccdc. cam.ac.uk/structures).
Таблица 1.
Кристаллографические данные и параметры уточнения соединений I, III и IV
| Параметр | Значение | ||
|---|---|---|---|
| I | III | IV | |
| Брутто-формула | C48H70N2O2Sn2 | C30H47N3OS4Sn · С7H8 | C24H41N3OS4Sn ∙ CH2Cl2 |
| М | 944.44 | 804.77 | 719.45 |
| Сингония | Триклинная | Моноклинная | Моноклинная |
| Пр. гр. | P1 | I2/a | I2/a |
| T, K | 100 | 298 | 100 |
| a, Å | 6.7263(4) | 18.6353(4) | 18.1573(16) |
| b, Å | 12.6897(6) | 12.9182(3) | 17.3440(8) |
| c, Å | 13.7217(7) | 36.6481(9) | 21.3163(10) |
| α, град | 99.1260(10) | 90 | 90 |
| β, град | 101.0710(10) | 104.272(2) | 91.8820(10) |
| γ, град | 101.3530(10) | 90 | 90 |
| V, Å3 | 1103.20(10) | 8550.2(4) | 6709.3(7) |
| Z | 1 | 8 | 8 |
| ρ(выч.), г/см3 | 1.422 | 1.250 | 1.425 |
| µ, мм–1 | 1.171 | 0.822 | 1.192 |
| Размер кристалла, мм | 0.52 × 0.22 × 0.18 | 0.76 × 0.15 × 0.11 | 0.40 × 0.14 × 0.14 |
| Область сканирования θ, град | 2.50–30.18 | 2.94–30.51 | 2.25–27.88 |
| Число измеренных/ независимых отражений |
16 202/6527 | 82 190/13 051 | 32 826/7983 |
| Rint | 0.0151 | 0.0818 | 0.0402 |
| Число независимых отражений с I > 2σ(I) | 6286 | 7744 | 6721 |
| Число уточняемых параметров/ ограничений | 250/0 | 452/160 | 396/21 |
| R (I > 2σ(I)) | R1 = 0.0167 wR2 = 0.0419 |
R1= 0.0474 wR2 = 0.0917 |
R1 = 0.0419 wR2 = 0.0932 |
| R (по всем данным) | R1 = 0.0177 wR2 = 0.0424 |
R1 = 0.1017 wR2 = 0.1066 |
R1 = 0.0529 wR2 = 0.1008 |
| S (F 2) | 1.049 | 0.987 | 1.033 |
| Остаточная электронная плотность (min/max), e Å–3 | –0.27/0.72 | –0.39/0.57 | –0.78/1.18 |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
о-Амидофенолятный комплекс олова(II) AdAPSn (I) получали согласно методике, описанной ранее для синтеза t-BuAPSn (II) [26]. Реакция элиминирования амина между соответствующим o-аминофенолом и бис-(триметилсилил)амидом олова(II) протекает с высоким выходом (~80%), в эфире при комнатной температуре (схема 1 ).
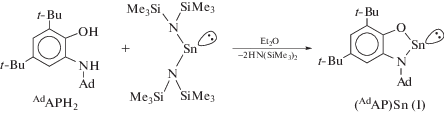
Схема 1 .
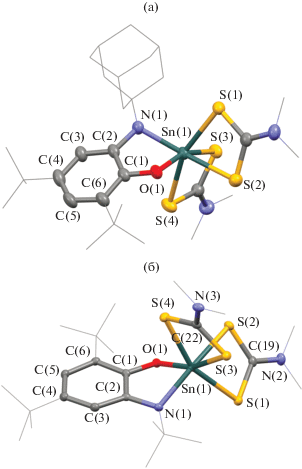
Станнилен I представляет собой желтый кристаллический продукт, чувствительный к влаге и кислороду воздуха. Молекулярное и кристаллическое строение I установлено методом РСА (рис. 1). Избранные длины связей и углы приведены в табл. 2. Длины связей Sn(1)–O(1) и Sn(1)–N(1) (2.0533(8) и 2.2427(9) соответственно) близки к аналогичным значениям у станилена II. Длины связей O(1)–C(1) 1.349(2), N(1)–C(2) 1.463(2) Å, а также расстояния C–C в шестичленном цикле, лежащие в узком интервале значений 1.393(2)–1.416(2) Å и характерные для ароматических систем, однозначно указывают [49, 50] на дианионную природу редокс-активного амидофенолятного лиганда. Станнилен I димеризуется в кристаллическом состоянии посредством сильных межмолекулярных контактов Sn(1)–N(1А) 2.3153(9) Å (сумма ван-дер-ваальсовых радиусов олова и азота равна 3.8 Å [51]). Вакантная p-орбиталь при атоме олова выступает в качестве сильной кислоты Льюиса по отношению к неподеленной паре атома азота соседней молекулы. Данное свойство определяет агрегационный характер станниленов [52–54]. В образующемся димере плоскости {SnNSnN} и {SnON} близки к ортогональности – двугранный угол между ними составляет 77.31(3)°. Аналогичный характер образования димерных молекул был обнаружен для опубликованных ранее амидофенолятных станниленов, содержащих фенильный [22] и трет-бутильный заместитель [26] у атома азота. Примечательно, что станнилен DippAPSn [23] на основе более стерически загруженного 4,6-ди-трет-бутил-N-(2,6-ди-изо-пропилфенил)-о-аминофенола образует димер посредством межмолекулярных Sn···O контактов. Можно заключить, что при переходе к менее объемистым заместителям при атоме азота происходит изменение характера димеризации комплексов с Sn···O на Sn···N.
Рис. 1.
Молекулярная структура комплекса I. Для ключевых атомов тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода не изображены для ясности.
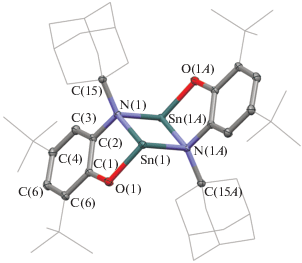
Таблица 2.
Основные длины связей (Å) и валентные углы (град) в комплексах I, III и IV
| Связь | I | III | IV | Угол | I | III | IV |
|---|---|---|---|---|---|---|---|
| Sn(1)–N(1) | 2.2427(9) | 2.089(2) | 2.092(3) | O(1)Sn(1)N(1) | 80.13(3) | 78.97(8) | 79.25(9) |
| Sn(1)–O(1) | 2.0533(8) | 2.035(2) | 2.034(2) | O(1)Sn(1)S(1) | 103.91(5) | 101.75(6) | |
| C(1)–O(1) | 1.349(2) | 1.362(3) | 1.359(4) | N(1)Sn(1)S(1) | 100.88(6) | 100.17(7) | |
| C(2)–N(1) | 1.463(2) | 1.405(4) | 1.404(4) | O(1)Sn(1)S(3) | 159.69(6) | 161.75(7) | |
| C(1)–C(2) | 1.414(2) | 1.424(4) | 1.428(4) | N(1)Sn(1)S(3) | 108.74(6) | 105.62(7) | |
| C(2)–C(3) | 1.398(2) | 1.388(4) | 1.400(4) | S(1)Sn(1)S(3) | 93.24(3) | 94.75(3) | |
| C(3)–C(4) | 1.393(2) | 1.395(4) | 1.393(5) | O(1)Sn(1)S(2) | 86.75(6) | 87.17(6) | |
| C(4)–C(5) | 1.398(2) | 1.377(5) | 1.394(4) | N(1)Sn(1)S(2) | 160.80(6) | 161.91(7) | |
| C(5)–C(6) | 1.399(2) | 1.394(5) | 1.402(4) | S(1)Sn(1)S(2) | 70.02(3) | 70.79(3) | |
| C(6)–C(1) | 1.416(2) | 1.395(4) | 1.403(4) | S(3)Sn(1)S(2) | 89.02(3) | 90.99(3) | |
| Sn(1)–N(1A) | 2.3153(9) | O(1)Sn(1)S(4) | 90.09(5) | 91.38(6) | |||
| Sn(1)–S(1) | 2.5228(8) | 2.5279(8) | N(1)Sn(1)S(4) | 98.01(6) | 100.74(7) | ||
| Sn(1)–S(2) | 2.6110(9) | 2.5818(9) | S(1)Sn(1)S(4) | 158.29(3) | 157.03(3) | ||
| Sn(1)–S(3) | 2.5550(7) | 2.5502(8) | S(3)Sn(1)S(4) | 70.47(3) | 70.50(3) | ||
| Sn(1)–S(4) | 2.5708(7) | 2.5930(8) | S(2)Sn(1)S(4) | 94.74(3) | 91.40(3) | ||
| S(1)–C | 1.730(3) | 1.735(3) | O(1)Sn(1)N(1А) | 100.82(3) | |||
| S(2)–C | 1.707(3) | 1.718(3) | N(1)Sn(1)N(1А) | 81.24(3) | |||
| S(3)–C | 1.729(3) | 1.732(3) | C(1)O(1)Sn(1) | 115.96(6) | |||
| S(4)–C | 1.711(3) | 1.726(3) | C(2)N(1)Sn(1) | 106.26(6) | |||
| Sn(1)N(1)Sn(1А) | 98.76(3) |
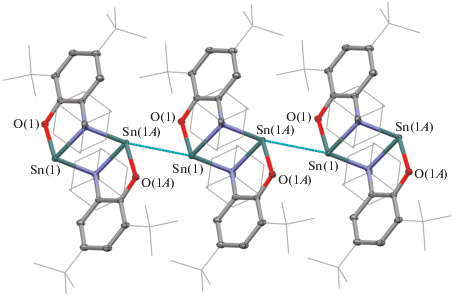
Димерные молекулы I формируют “бесконечные” цепочки за счет металлофильного взаимодействия Sn···Sn (3.5360(2) Å) (рис. 2), длина которого несколько больше, чем в стерически менее загруженном аналоге II (Sn···Sn 3.3523(2) Å [26]), и сопоставима с расстоянием Sn···Sn в PhApSn 3.5480(2) Å [22]. Тщательное изучение экспериментальной электронной плотности и анализа NBO (Natural bond orbital) для II показало ковалентный характер такого взаимодействия [26]. В результате формирования цепочек димерных молекул станнилена адамантильная группа сильно отклоняется от плоскости {SnOССN} (отклонение атома C(15) от плоскости металлоцикла составляет 1.12(2) Å). Таким образом, атом азота имеет пирамидальную конфигурацию.
Рис. 2.
Фрагмент кристаллической упаковки комплекса I. Для ключевых атомов тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода не изображены для ясности.
Представленные выше о-амидофенолятные комплексы олова(II) I и II, равно как и другие дииминовые и катехолатные [22, 25, 55] комплексы низковалентных металлов 14 группы, способны демонстрировать множественную разностороннюю реакционную способность. Низковалентный атом олова Sn(II) может вступать в реакции окислительного присоединения с образованием производных Sn(IV) (схема 2 , путь а). В то же время в окислительно-восстановительное взаимодействие может быть вовлечен редокс-активный лиганд с сохранением степени окисления металла (схема 2 , путь г). Благодаря наличию неподеленной электронной пары и вакантной p-орбитали станнилены проявляют амфотерность Льюиса – они способны выступать в роли как мягких кислот, так и мягких оснований (схема 2 , пути б и в).
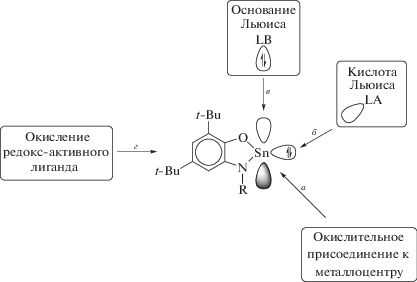
Схема 2 .
Дисульфиды являются удобными реагентами для изучения различных реакций окислительного присоединения [56–58]. Так, мы установили, что соединения I и II легко взаимодействуют с эквимолярным количеством TMTDS с образованием фиолетовых кристаллических диамагнитных продуктов (AdAP)Sn(S2R)2 (III) и (t-BuAP)Sn(S2R)2 (IV) (схема 3 ). Реакция заканчивается в течение нескольких часов при комнатной температуре. В данных условиях проведения реакции не следует ожидать гомолитической диссоциации связи S–S в дисульфиде, поскольку термическая диссоциация TMTDS происходит при T = 130–150° [59]. Следовательно, изучаемый процесс сопровождается внедрением атома низковалентного олова по связи S–S с последующим образованием новых шестикоординированных дитиокарбаматных комплексов олова(IV).
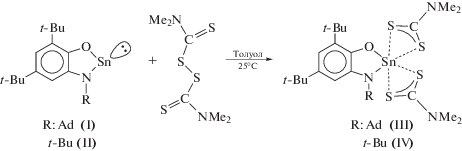
Схема 3 .
Молекулярное и кристаллическое строение комплексов III и IV (рис. 3) было определено методом РСА. Кристаллы, пригодные для РСА, получали из толуола(III) и смеси н-гексан–CH2Cl2 (IV). В независимой области кристаллических ячеек в III и IV помимо молекул комплексов содержатся по одной сольватной молекуле толуола(III) и хлористого метилена(IV). Геометрические характеристики III и IV близки между собой (табл. 2), поэтому ниже обсуждается лишь молекулярное строение комплекса IV.
Рис. 3.
Молекулярная структура комплексов: а – (AdAP)Sn(S2R)2 (III); б – (t-BuAP)Sn(S2R)2 (IV). Для ключевых атомов тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода не изображены для ясности.
Координационное окружение атома металла в IV – искаженный октаэдр. Длины связей Sn(1)–O(1) 2.034(2) и Sn(1)–N(1) 2.092(3) Å в соединении IV меньше суммы ковалентных радиусов соответствующих элементов (2.09 Å для Sn–O, 2.1 Å для Sn–N [51]) и близки к таковым в о-амидофенолятах олова(IV) [22, 42]. Данные связи существенно короче наблюдаемых в исходном станнилене I, что согласуется с изменением степени окисления центрального атома олова(II) до олова(IV).
Расстояния C(1)–O(1) 1.359(4) и C(2)–N(1) 1.404(4) Å лежат в интервале, характерном для дианионной формы о-иминохинонового лиганда [49, 50]. Связи C–C в шестичленном кольце близки к типично ароматическим значениям ~1.40 Å. Полученные метрические параметры указывают на дианионное состояние редокс-активного лиганда. Каждый дитиокарбаматный фрагмент бидентатно хелатирует металлоцентр, образуя четырехчленные циклы {CS2Sn}. Углы SSnS (70.50(3)°, 70.79(3)°) в циклах близки к 70°. Длины связей Sn–S лежат в интервале значений 2.5279(8)–2.5930(8) Å. Расстояния C–S в дитиокарбаматных фрагментах близки между собой (1.718(3)–1.735(3) Å, средн. C–S = 1.728(6) Å) и указывают на значительную делокализацию заряда в рассматриваемом фрагменте [60].
Способность станнилена I предоставлять вакантную р-орбиталь для донорно-акцепторного взаимодействия наглядно продемонстрированa на примере формирования димерных молекул в соответствующем кристалле. В литературе имеются обширные данные о комплексообразовании низковалентных производных олова с переходными металлами путем вовлечения во взаимодействие неподеленной электронной пары металлена [61]. Необходимо отметить, что в данном случае станнилены выступают в качестве оснований Льюиса. Реакция I c Fe2(CO)9 в растворе толуола завершается при комнатной температуре в течение 2 сут. и дает аддукт V в виде мелкокристаллического порошка желто-коричневого цвета (схема 4 ).
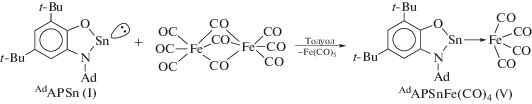
Схема 4 .
Образовавшийся продукт идентифицирован по данным спектроскопии ЯМР 1H, 13C, а также методом ИК-спектроскопии. В спектре ЯМР 1H соединения V наблюдается незначительное смещение всех сигналов по сравнению со спектром исходного станнилена I. Спектр ЯМР 13C характеризуется появлением нового сигнала при δC 210.7 м.д., принадлежащего карбонильным группам, связанным с атомом железа. Колебания карбонильных CO связей в комплексе V проявляются в ИК-спектре как сильные полосы поглощения в интервале 1980–2043 см–1.
Как было показано выше, станнилены I и II реагируют с TMTDS с образованием производных олова(IV) (схема 3 ). В ходе данной реакции редокс-активный лиганд не изменяет своей степени окисления. Введение в реакцию с I и II мягких одноэлектронных окислителей позволяет перенести реакционный центр с металла на лиганд, сохранив при этом степень окисления металла.
Мы исследовали реакции полученных о-амидофенолятов олова(II) с такими окислителями, как соли ртути HgHal2 (Hal = Cl, Br, I), а также со стабильным 2-этокси-3,6-ди-трет-бутил-2-этоксифеноксильным радикалом (далее феноксил) [39].
Известно, что парамагнитные производные элементов 14 группы на основе радикальных редокс-активных лигандов весьма лабильны [22, 55, 62, 63]. В настоящий момент известно лишь две работы, где удалось выделить в их индивидуальном состоянии [33, 34]. Нам удалось зарегистрировать сигналы, отвечающие парамагнитным производным олова(II) общего вида (RImSQ)SnOR в спектрах ЭПР в ходе реакции I и II с феноксилом (RImSQ = анион-радикальная форма лиганда). Так, в условиях ЭПР-эксперимента при окислении комплекса II феноксилом окраска реакционной смеси становится интенсивно зеленой, а в спектре наблюдается хорошо разрешeнный сигнал (рис. 4а), свидетельствующий об образовании соответствующего моно-о-иминосемихинолятного производного олова(II) (t-BuImSQ)SnOR (VI). Спектр полученного анион-радикального комплекса VI представляет собой триплет (1 : 1 : 1) дублетов (1 : 1). Сверхтонкая структура (CTC) спектра обусловлена сверхтонким взаимодействием (СТВ) неспаренного электрона с двумя неэквивалентными магнитными ядрами атома водорода 1Н (99.98%, I = 1/2, μN = 2.7928) [64] и атома азота 14N (99.63%, I = 1, μN = 0.4037) [64]; кроме того, наблюдается сателлитное расщепление на магнитных изотопах олова 117Sn (7.68%, I = 1/2, μN = 1.000) и 119Sn (8.58%, I = 1/2, μN = 1.046) [62]. Параметры спектра ЭПР (табл. 3), наблюдаемого в ходе реакции комплекса II с феноксилом, а именно большие значения констант СТВ аi(119Sn) = 145.0 Э, аi(119Sn) = 138.6 Э, существенно отличаются от полученных ранее для о-иминосемихинолятов олова(IV) [65, 66]. Для последних характерны более низкие величины констант СТВ аi(117, 119Sn) (20–50 Э). Соединения двухвалентного олова, напротив, обычно характеризуются большими значениями констант СТВ (ai117, 119Sn ≥ 90 Э) [22, 55, 67, 68].
Рис. 4.
Изотропный спектр ЭПР комплексов VI (а) и IХ (б) в ТГФ: экспериментальный (1), симулированный (2) при 298 К.
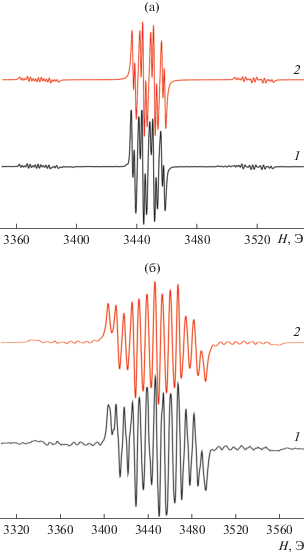
Таблица 3.
Параметры изотропных спектров ЭПР (Э) для комплексов VI–IX
| Комплекс | gi | аi(1H) | аi(1H) | аi(14N) | аi(119Sn) | аi(117Sn) | аi(Hal) |
|---|---|---|---|---|---|---|---|
| VI | 2.0008 | 2.0 | 5.1 | 7.2 | 145.0 | 138.6 | |
| VII | 2.0003 | 1.6 | 5.2 | 7.0 | 145.8 | 139.0 | |
| VII | 2.0003 | 1.2 | 5.5 | 7.3 | 138.0 | 132.1 | 4.3 (35Cl) 3.5 (37Cl) |
| IX | 2.0013 | 1.2 | 5.4 | 7.2 | 138.4 | 132.3 | 17.0 (79Br) 20.5 (81Br) |
Обнаруженные различия в значениях констант СТВ с магнитными изотопами металла в высоко- и низковалентном состояниях являются типичными как для олова, так и для других элементов [69, 70]. На основании данных спектроскопии ЭПР можно однозначно заключить, что в реакционной смеси присутствуют частицы, содержащие о-иминосемихиноновый лиганд и металлоцентр в низкой степени окисления.
Интенсивная зеленая окраска и соответствующий спектр ЭПР, присущие о-иминосемихиноновому комплексу VI, сохраняются ~1.5 ч. Выдерживание реакционной смеси в течение 2 ч при комнатной температуре приводит к интенсивному желтому окрашиванию и полному исчезновению сигнала в спектре ЭПР. Аналогичное поведение демонстрирует станнилен I, реакция которого с феноксилом приводит к образованию парамагнитного производного (AdImSQ)SnOR (VII). Параметры спектров ЭПР парамагнитных станниленов VI–IX приведены в табл. 3.
Важно отметить, что зарегистрированные парамагнитные станнилены VI и VII обладают существенно более высокой стабильностью по сравнению с их N-арилзамещенными аналогами. Схожие парамагнитные частицы (DippImSQ)SnOR удавалось наблюдать в растворе только при пониженных температурах (–18°С) [22]. Кроме того, нам удалось зафиксировать спектры ЭПР, отвечающие галогенсодержащим радикальным станниленам (AdImSQ)SnCl (VIII) и (AdImSQ)SnBr (IX) в ходе взаимодействия I с галогенидами ртути(II) (табл. 3). Спектр ЭПР комплекса IX представлен на рис. 4б. Продукты с иодид ионом у атома металла, а также N-трет-бутильным заместителем в редокс-активном лиганде зарегистрировать не удалось ввиду их быстрого превращения даже при низких температурах.
СТС спектра комплекса IX обусловлена взаимодействием неспаренного электрона с двумя неэквивалентными магнитными ядрами атома водорода 1Н, атомом азота 14N и с магнитными изотопами брома 79, 81Br. Также наблюдается сателлитное расщепление на магнитных изотопах олова 117, 119Sn. Высокое значение констант СТВ с магнитными изотопами галогеновых заместителей свидетельствует о том, что σ(σ*)-орбитали Sn–Hal вносят заметный вклад в π-МО, занятую неспаренным электроном.
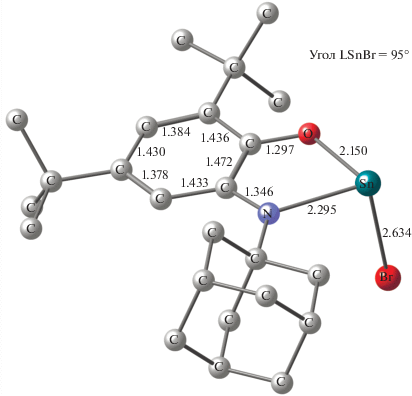
Наиболее вероятная геометрия образующегося промежуточного станнилена IХ установлена при помощи квантово-химического моделирования (DFT B3LYP/def2svp) (рис. 5). Атом олова в соединении IХ имеет тригонально-пирамидальную координацию. Атом галогена практически ортогонален плоскости редокс-активного лиганда (угол LSnBr равен 95°), что обеспечивает эффективное перекрывание орбитали неспаренного электрона и σ(σ*)-орбитали Sn–Hal.
Рис. 5.
Оптимизированная геометрия комплекса IХ (DFT B3LYP/def2svp). Атомы водорода опущены для ясности. Длины связей приведены в Å.
На основании полученных результатов по окислению рассматриваемых станниленов можно заключить, что образующиеся несимметричные производные олова(II) общего вида (RImSQ)SnX неустойчивы в растворе. По окончании взаимодействия соединения I из реакционных смесей были выделены только известные ранее диамагнитные бис-о-амидофеноляты олова(IV) [42]. На основании данных спектроскопии ЭПР и продуктов реакций можно предложить следующий механизм окисления комплексов I и II (схема 5 ):
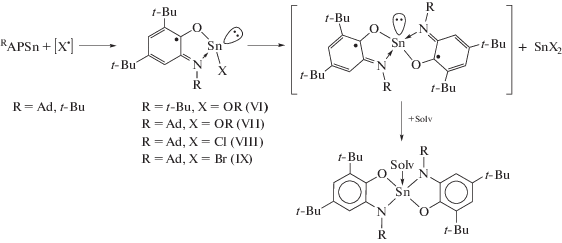
Схема 5 .
На первой стадии происходит одноэлектронное окисление исходного станнилена с образованием парамагнитного производного олова(II) общего вида (RImSQ)SnX. Комплексы такого рода неустойчивы и симметризуются с образованием бирадикальных продуктов типа (RImSQ)2SnII и SnX2. На последней стадии производные (RImSQ)2SnII претерпевают внутримолекулярный редокс-процесс, заключающийся в окислении металлоцентра до четырехвалентного состояния и переходе редокс-активных лигандов в дианионную форму. Возможность такого редокс-изомерного превращения описана в [42].
Таким образом, редокс-активные лиганды не только влияют на реакционную способность металлоцентра, но и сами активно участвуют в окислительно-восстановительных реакциях с различными субстратами, не теряя при этом связь с комплексообразователем.
В ходе проведенного исследования синтезирован новый комплекс низковалентного олова (AdAP)Sn. Показано, что насыщение координационной сферы металла в кристаллическом состоянии достигается за счет межмолекулярных донорно-акцепторных взаимодействий. Установлено, что подобные соединения способны реагировать с кислотами Льюиса, а также имеют двойственную природу в окислительно-восстановительных превращениях: за реакции с одноэлектронными окислителями отвечает редокс-активный лиганд, а за взаимодействие с двухэлектронными окислителями – ион двухвалентного металла.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Abakumov G.A., Piskunov A.V., Cherkasov V.K. et al. // Russ. Chem. Rev. 2018. V. 87. P. 393.
Luca O.R., Crabtree R.H. // Chem. Soc. Rev. 2013. V. 42. P. 1440.
Broere D.L.J., Plessius R., van der Vlugt J.I. // Chem. Soc. Rev. 2015. V. 44. P. 6886.
Kaim W., Paretzki A. // Coord. Chem. Rev. 2017. V. 344. P. 345.
Jacquet J., Murr M.D., Fensterbank L. // ChemCatChem. 2016. V. 8. P. 3310.
Kaim W. // Inorg. Chem. 2011. V. 50. P. 9752.
Lyaskovskyy V., de Bruin B. // ACS Catal. 2012. V. 2. P. 270.
Tezgerevska T., Alley K.G., Boskovic C. // Coord. Chem. Rev. 2014. V. 268. P. 23.
Schulz S. // Chem. Eur. J. 2010. V. 16. P. 6416.
Hadlington T.J., Driess M., Jones C. // Chem. Soc. Rev. 2018. V. 47. P. 4176.
Li Z., Thiery G., Lichtenthaler M.R., Guillot R. et al. // Adv. Synth. Catal. 2018. V. 360. P. 544.
Yadav S., Saha S., Sen S.S. // ChemCatChem. 2016. V. 8. P. 486.
Kazarina O.V., Gourlaouen C., Karmazin L. et al. // Dalton Trans. 2018. V. 47. P. 13800.
Benedek Z., Szilvási T. // Organometallics. 2017. V. 36. P. 1591.
Kühl O. // Coord. Chem. Rev. 2004. V. 248. P. 411.
Tokitoh N., Okazaki R. // Coord. Chem. Rev. 2000. V. 210. P. 251.
Asay M., Jones C., Driess M. // Chem. Rev. 2011. V. 111. P. 354.
Mizuhata Y., Sasamori T., Tokitoh N. // Chem. Rev. 2009. V. 109. P. 3479.
Lee V.Y., Sekiguchi A. Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds. Chichester: Wiley, 2010. 448 p.
Mandal S.K., Roesky H.W. // Acc. Chem. Res. 2012. V. 45. P. 298.
Абакумов Г.А., Черкасов В.К., Пискунов А.В. и др. // Изв. АН. Сер. хим. 2006. P. 1103.
Chegerev M.G., Piskunov A.V., Maleeva A.V. et al. // Eur. J. Inorg. Chem. 2016. P. 3813.
Piskunov A.V., Aivaz’yan I.A., Fukin G.K. et al. // Inorg. Chem. Commun. 2006. V. 9. P. 612.
Tsys K.V., Chegerev M.G., Fukin G.K., Piskunov A.V. // Mendeleev Commun. 2018. V. 28. P. 527.
Tsys K.V., Chegerev M.G., Pashanova K.I. et al. // Inorg. Chim. Acta. 2019. V. 490. P. 220.
Chegerev M.G., Piskunov A.V., Tsys K.V. et al. // Eur. J. Inorg. Chem. 2019. P. 875.
Чегерев М.Г., Пискунов А.В. // Коорд. химия. 2018. Т. 44. С. 109 (Chegerev M.G., Piskunov A.V. // Russ. J. Coord. Chem. 2018. V. 44. P. 258. https://doi.org/10.1134/S1070328418040036).
Zabula A.V., Hahn F.E. // Eur. J. Inorg. Chem. 2008. P. 5165.
Gans-Eichler T., Gudat D, Nättingen K., Nieger M. // Chem. Eur. J. 2006. V. 12. P. 1162.
Leites L.A., Bukalov S.S., Aysin R.R. et al. // Organometallics. 2015. V. 34. P. 2278.
Fedushkin I.L., Skatova A.A., Chudakova V.A. et al. // Organometallics. 2004. V. 23. P. 3714.
Druzhkov N.O., Kazakov G.G., Shavyrin A.S. et al. // Inorg. Chem. Commun. 2018. V. 90. P. 92.
Fedushkin I.L., Khvoinova N.M., Baurin A.Y. et al. // Inorg. Chem. 2004. V. 43. P. 7807.
Janes T., Zatsepin P., Song D.T. // Chem. Commun. 2017. V. 53. P. 3090.
Fedushkin I.L., Sokolov V.G., Piskunov A.V. et al. // Chem. Commun. 2014. V. 50. P. 10108.
Гордон А., Форд Р. Спутник химика. М.: Мир, 1976. 543 с.
Abakumov G.A., Cherkasov V.K., Nevodchikov V.I. // Tetrahedron Lett. 2005. V. 46. P. 4095.
Brauer G. Handbuch der Praparativen Anorganischen Chemie. Print book. German, 1981.
Harris D.H., Lappert M.F. // Chem. Commun. 1974. P. 895.
Matson E.M., Opperwall S.R., Fanwick P.E., Bart S.C. // Inorg. Chem. 2013. V. 52. P. 7295.
Shadyro O. I., Sorokin V.L., Ksendzova G.A. et al. // Pharm. Chem. J. 2012. V. 46. P. 27.
Chegerev M.G., Piskunov A.V., Starikova A.A. // Eur. J. Inorg. Chem. 2018. P. 1087.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09. Revision D.01. Wallingford (CT, USA): Gaussian, Inc., 2013.
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648.
Agilent. CrysAlis Pro. Yarnton (Oxfordshire, England): Agilent Technologies Ltd., 2014.
Smart. APEX2. Madison (WI, USA): Bruker AXS Inc., 2014.
Krause L., Herbst-Irmer R., Sheldrick G.M., Stalke D. // J. Appl. Cryst. 2015. V. 48. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Brown S.N. // Inorg. Chem. 2012. V. 51. P. 1251.
Poddel’sky A.I., Cherkasov V.K., Abakumov G.A. // Coord. Chem. Rev. 2009. V. 253. P. 291.
Бацанов С.С. // Журн. неорган. химии. 1991. Т. 36. C. 3015.
Mansell S.M., Russell C.A., Wass D.F. // Inorg. Chem. 2008. V. 47. P. 11367.
Aysin R.R., Leites L.A., Bukalov S.S. et al. // Inorg. Chem. 2016. V. 55. P. 4698.
Zabula, A.V., Rogachev A.Y., West R. // Chem. Eur. J. 2014. V. 20. P. 16652.
Piskunov A.V., Aivaz’yan I.A., Cherkasov V.K., Abakumov G.A. // J. Organomet. Chem. 2006. V. 691. P. 1531.
Broere D.L.J., Metz L.L., de Bruin B. et al. // Angew. Chem. Int. Ed. 2015. V. 54. P. 1516.
Fedushkin I.L., Maslova O.V., Hummert M., Schumann H. // Inorg. Chem. 2010. V. 49. P. 2901.
Fedushkin I.L., Nikipelov A.S., Skatova A.A. et al. // Eur. J. Inorg. Chem. 2009. P. 3742.
Steudel R., Steudel Y., Mak A.M., Wong M.W. // J. Org. Chem. 2006. V. 71. P. 9302.
Bouška M., Novák M., Dostál L. et al. // Eur. J. Inorg. Chem. 2014. P. 310.
Baumgartner J., Marschner C. // Rev. Inorg. Chem. 2014. V. 34. P. 119.
Tumanskii B., Pine P., Apeloig Y. et al. // J. Am. Chem. Soc. 2004. V. 126. P. 7786.
Tumanskii B., Pine P., Apeloig Y. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 8248.
Эмсли Д. Элементы. М.: Мир, 1993. С. 256.
Пискунов А.В., Мещерякова И.Н., Баранов Е.В. et al. // Изв. АН. Сер. хим. 2010. С. 354.
Пискунов А.В., Чегерев М.Г., Ваганова Л.Б., Фукин Г.К. // Коорд. химия. 2015. Т. 41. С. 394 (Piskunov A.V., Chegerev M.G., Vaganova L.B., Fukin G.K. // Russ. J. Coord. Chem. 2015. V. 41. P. 428. https://doi.org/10.1134/S1070328415070076).
Абакумов Г.А., Черкасов В.К., Пискунов А.В., Дружков Н.О. // Докл. РАН. 2004. Т. 399. С. 353.
Чегерев М.Г., Пискунов А.В., Старикова А.А. // Журн. общ. химии. 2017. Т. 76. С. 1841.
Абакумов Г.А., Неводчиков В.И., Черкасов В.К., Разуваев Г.А. // Докл. АН СССР. 1978. Т. 242. С. 609.
Климов Е.С., Абакумов Г.А., Гладышев Е.Н. и др. // Докл. АН СССР. 1974. Т. 218. С. 844.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия