

Координационная химия, 2020, T. 46, № 10, стр. 579-648
Синтез, реакции и строение арильных соединений пятивалентной сурьмы
В. В. Шарутин 1, *, А. И. Поддельский 2, О. К. Шарутина 1
1 Южно-Уральский государственный университет
(национальный исследовательский университет)
Челябинск, Россия
2 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: vvsharutin@rambler.ru
Поступила в редакцию 26.03.2020
После доработки 27.04.2020
Принята к публикации 07.05.2020
Аннотация
На основе анализа литературы, опубликованной преимущественно с 2009 г. по настоящее время, систематизированы и описаны методы получения, некоторые реакции, особенности строения арильных соединений пятивалентной сурьмы и примеры их возможного использования. Некоторые более ранние работы учтены в обзоре из-за их особой важности. При обсуждении методов синтеза основное внимание уделено наиболее эффективным подходам к получению арильных соединений, например, реакциям перераспределения лигандов, замещения и окислительного присоединения. Рассмотрены реакции образования гетероциклических соединений сурьмы. Приведены сведения о биологической и каталитической активности некоторых производных сурьмы. Библиография – 318 ссылок.
ОГЛАВЛЕНИЕ
Введение 579
Синтез пентаарильных соединений сурьмы Ar5Sb 580
Производные сурьмы Ar4SbХ и их реакции 581
Получение соединений сурьмы Ar4SbХ деарилированием пентаарилсурьмы кислотами 581
Синтез соединений Ar4SbX из галогенидов тетраарилсурьмы 585
Синтез соединений пятивалентной сурьмы по реакции перераспределения лигандов 593
Синтез комплексных соединений сурьмы, содержащих тетраарилстибониевые катионы 595
Методы синтеза и некоторые реакции производных сурьмы Ar3SbХ2 598
Получение производных сурьмы из дигалогенидов триарилсурьмы 598
Окисление производных Ar3Sb органическими и неорганическими окислителями 609
Окислительный метод синтеза арильных производных сурьмы(V) из триарильных соединений сурьмы, кислоты НХ и пероксида 615
Окислительный метод синтеза органических производных сурьмы(V) из триорганилсурьмы и орто-хинонов 619
Производные сурьмы Ar2SbХ3, ArSbХ4 и их реакции 626
Окисление 627
Замещение 628
Практическое применение арильных соединений сурьмы(V) 636
Заключение 638
Список литературы 639
ВВЕДЕНИЕ
Возрастающий интерес к органическим соединениям сурьмы во многом определяется растущим потенциалом их применения в самых разнообразных областях практической деятельности: в качестве лекарственных препаратов, биоцидов, фунгицидов, в качестве реагентов и компонентов каталитических систем при полимеризации, в тонком органическом синтезе, в качестве антиоксидантов, и др. Сурьма в сурьмаорганических производных имеет две основные степени окисления: +3 и +5 и, соответственно, образует органические соединения сурьмы(III) и сурьмы(V). При этом производные сурьмы(III) во многих случаях имеют олигомерное или полимерное строение, что в определенной и иногда значительной степени затрудняет их изучение и применение, в то время как органические производные сурьмы(V) предлагают большее разнообразие структурных типов и, соответственно, реакционной способности. С точки зрения токсичности, соединения сурьмы(V) менее токсичны по сравнению с органическими производными сурьмы(III). Фундаментальные исследования строения и свойств сурьмаорганичеких соединений дали толчок к раcширению возможностей по их прикладному применению: установлены случаи нетривиального химического поведения, например обратимое связывание кислорода о-амидофенолятами и катехолатами сурьмы(V), способность к избирательной фиксации галогенид-анионов моно- и биядерными металлоорганическими производными сурьмы, высокая активность сурьмаорганических соединений в некоторых важных реакциях органического синтеза, значительная биохимическая активность при относительно невысокой токсичности и др. Эти обстоятельства в значительной степени определяют интерес к получению новых арильных соединений пятивалентной сурьмы и разработке методов их синтеза.
СИНТЕЗ ПЕНТААРИЛЬНЫХ СОЕДИНЕНИЙ СУРЬМЫ Ar5Sb
В основе наиболее эффективных методов синтеза пентаарильных производных сурьмы лежат реакции с магний- или литийорганическими реагентами. Так, пентаарильные соединения сурьмы с п-метилфенильными и п-трифторметилфенильными заместителями ArnTol(5 –n)Sb (n = 0–5: Ar = p-CF3C6H4, Tol = p-CH3C6H4): Tol5Sb (1), ArTol4Sb (2), Ar2Tol3Sb (3), Ar3Tol2Sb (4), Ar4TolSb (5), Ar5Sb (6), были синтезированы из соответствующих дигалогенидов триарилсурьмы или фторидов тетраарилсурьмы и реактивов Гриньяра или ариллития в эфире при −78°С [1]. Выбор дифторидов триарилсурьмы и фторидов тетраарилсурьмы в качестве исходных соединений обусловлен тем, что скорость их реакции с реактивом Гриньяра или литийорганическим производным значительно выше, чем у бромидов. По данным рентгеноструктурного анализа, атомы сурьмы в соединениях 2–6 имеют тригонально-бипирамидальное окружение, причем более электроотрицательные п-трифторметилфенильные лиганды в 2–5 избирательно занимают аксиальные положения. Найдено, что в растворе смеси соединений 1 и 6 в дейтеробензоле при 60°С имеет место реакция обмена лигандами и в равновесной смеси методом ЯМР 13С-спектроскопии были обнаружены все шесть соединений 1–6, из которых наиболее устойчивое соединение 3.
Другой представитель арильных производных пятиковалентной сурьмы − пента(перхлорфенил)сурьма (C6Cl5)5Sb (7) был синтезирован непосредственно из пятихлористой сурьмы и перхлорфениллития в смеси диэтилового эфира и гексана при −78°С с выходом до 30% [2]. Невысокий выход целевого продукта обусловлен параллельным протеканием реакции восстановления Sb(V) → Sb(III). По данным РСА, атом сурьмы в молекуле 7 имеет слегка искаженную тригонально-бипирамидальную координацию.
При мольном соотношении перфторфениллития и пентахлорида сурьмы 4 : 1 реакция останавливается на стадии образования соответствующего монохлорида тетраарилсурьмы (С6F5)4SbCl (8, 25%) [3].
Производные сурьмы несимметричного строения Ph4ArSb (9) или Ph3ArSbCl (10) могут быть получены из ариллития, содержащего в арильном заместителе группу i-Pr2P, и дихлорида трифенилсурьмы или бромида тетрафенилсурьмы (схема 1 ) [4].
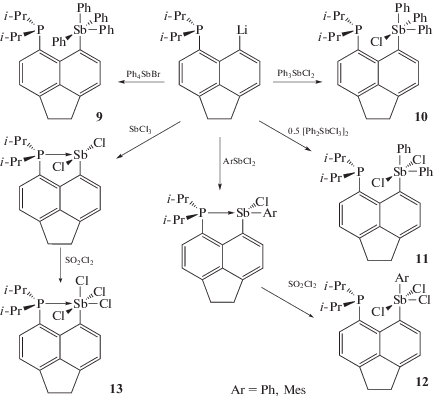
Схема 1 .
При изучении реакций литийорганического производного аценафтена была получена серия сурьмаорганических производных 9–13, содержащих группировку Ph(4 –n)SbCln.
ПРОИЗВОДНЫЕ СУРЬМЫ Ar4SbХ И ИХ РЕАКЦИИ
Получение соединений Ar4SbХ деарилированием пентаарилсурьмы кислотами. В основе эффективного получения производных сурьмы общей формулы Ar4SbX, где Х – электроотрицательный лиганд, лежат реакции пентаарилсурьмы с соединениями, содержащими подвижный атом водорода. В этом случае синтез целевого продукта происходит в одну стадию, а его выделение не является трудоемким. Таким способом получен ряд ароксидов тетрафенилсурьмы Ph4(ArO)Sb, в которых Ar = 2-NO2C6H4 (14) [5], 2,4,6-Cl3C6H2 (15) [6]; C6H7–C6H4 (4-циклогексадиенилфенил) (16) [7], 2,6-Br2-4-t-BuC6H2 (17) [7], 2,4-(NO2)2C6H3 (18) [7], 2,6-Br2-4-NO2C6H2 (19) [7], C6F5 (20) [8], C6Cl5 (21) [8], 2,6-Br2-4-MeC6H2 (22) [9] и тетра-п-толилсурьмы Tol4(ArO)Sb, где Ar – C6F5, (23) [8], C6Cl5 (24) [8], 2,6-Cl2C6H3 (25) [10], 2,4-(NO2)2C6H3 (26) [10], 2,4,6-(NO2)3C6H2 (27) [10].
Аналогично протекают реакции пентафенилсурьмы с такими карбоновыми кислотами, как 1‑адамантанкарбоновая (28) [11], N-бензоилглициновая (29) [12], фенилпропиоловая (30) [13], 4‑оксибензойная (31) [14] и фталевая (32) [15].
Взаимодействием эквимолярных количеств пентафенилсурьмы и малоновой кислоты в толуоле получен кислый малонат тетрафенилсурьмы (33) (схема 2 ) [16]. В то же время реакции двухосновных карбоновых кислот (янтарной, яблочной и винной) с двумя молями пентафенилсурьмы (толуол, 48 ч) приводят к образованию биядерных производных сурьмы (34–36) с выходом до 98% [17]. Продуктом реакции ацетилендикарбоновой кислоты с пентафенилсурьмой (толуол, 24 ч, 23°С, мольное соотношение 1 : 2) является ацетилендикарбоксилат бис(тетрафенилсурьмы|) (37) [18].
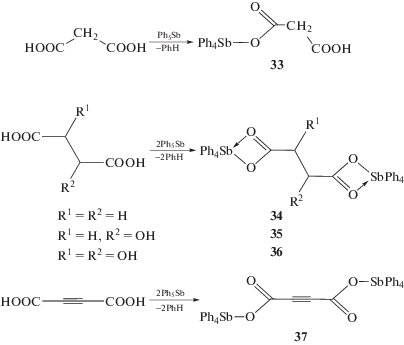
Схема 2 .
Отметим реакцию сукцината бис(тетрафенилсурьмы) с иодом в бензоле, в результате которой с выходом 72% был выделен сольват трииодида [(μ4-сукцинато)гексадекафенилтетрасурьмы] с бензолом (38) (схема 3 ) [19].
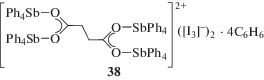
Схема 3 .
Неэквивалентные атомы Sb(1) и Sb(2) в центросимметричном катионе 38 имеют искаженную тригонально-бипирамидальную координацию с атомомами кислорода в аксиальных положениях (Sb(1,2)–O 2.347(4), 2.525(4), Sb(1,2)–Cэкв 2.109(7)–2.120(7) Å, 2.082(5)–2.106(7), Sb(1,2)–Cакс 2.158(7), 2.121(9) Å; углы OSb(1,2)Cакс 178.8(2)°, 174.5(3)°). Геометрия анионов [I3]– близка к линейной (угол I–I–I 179.41(4)°, расстояния I–I 2.880(1), 2.921(1) Å).
Особенность взаимодействия пентафенилсурьмы с тетрахлорфталевой кислотой заключается в том, что даже при соотношении реагентов 1 : 1 продуктом реакции является биядерное соединение 39 (схема 4 ) [20].
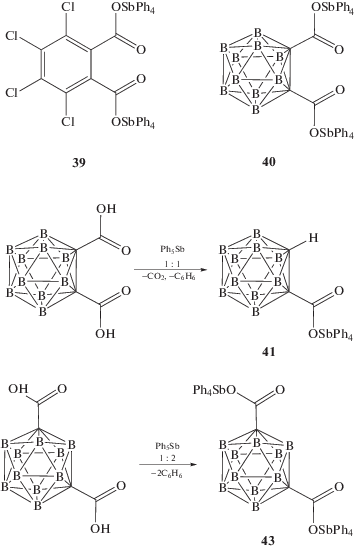
Схема 4 .
Если продуктом реакции пентафенилсурьмы с карборандикарбоновой кислотой при мольном соотношении исходных реагентов 2 : 1 (толуол, 24°С, 18 ч) является биядерное производное сурьмы (40) (94%), то взаимодействие эквимолярных количеств протекает с элиминированием углекислого газа и образованием монокарбоксилата тетрафенилсурьмы (41) (схема 4 ) [21].
Отметим, что взаимодействие эквимолярных количеств ацетилендикарбоновой кислоты и пентафенилсурьмы также сопровождалось элиминированием углекислого газа, при этом имело место образование пропиолата тетрафенилсурьмы (42), который синтезировали также из пентафенилсурьмы и пропиоловой кислоты [22].
В то же время мета-карборандикарбоновая кислота реагирует в растворе бензола с пентафенилсурьмой независимо от соотношения исходных реагентов с образованием только биядерного дикарбоксилата (43) (схема 4 ) [23].
Показано, что взаимодействие пентафенилсурьмы с 2,4-дигидроксибензойной кислотой независимо от соотношения реагентов протекает с участием карбоксильной и пара-гидроксильной групп и приводит к образованию 2-гидрокси-4-тетрафенилстибоксибензоата тетрафенилсурьмы(V) (44) (схема 5 ), в молекуле которого тригонально-бипирамидальная координация двух атомов сурьмы искажена в разной степени [24]. Установлено, что в реакции пентафенилсурьмы с 2,5- и 2,6-дигидроксибензойными кислотами участвуют только карбоксильные группы, продуктами являются 2,5-дигидроксибензоат тетрафенилсурьмы (45) и 2,6-дигидроксибензоат тетрафенилсурьмы (46) соответственно (схема 5 ) [25]:
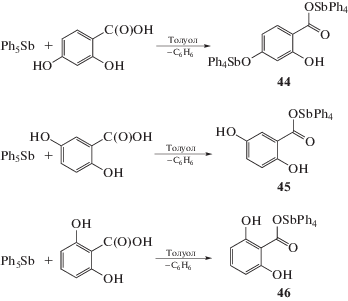
Схема 5 .
2,3-Дигидроксибензойная кислота при взаимодействии с пентафенилсурьмой проявляет свойства дигидроксибензола, при этом карбоксильная группа остается инертной даже при нагревании реакционной смеси, содержащей избыток пентафенилсурьмы. Единственным продуктом реакции является ионный комплекс (47) – 2-карбоксикатехолатотетрафенилстиботата(V) тетрафенилстибония, – в анионе которого присутствует пятичленный металлоцикл (схема 6 ).
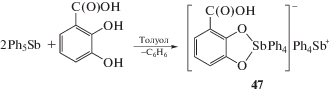
Схема 6 .
Оксиматы тетрафенилсурьмы Ph4SbON=CHR (R = C6H4(OH-2) (48) [26], C6H4(Br-2) (49) [27], C6H4(NO2-2) (50) [27], C4H3S (51) [27]), в которых координационное число (КЧ) центрального атома увеличивается до шести вследствие дополнительной координации атома азота оксимного лиганда с атомом Sb, синтезированы из эквимолярных количеств пентафенилсурьмы и оксима в растворе ароматического углеводорода (24 ч, 20°С) [26, 27].
В аналогичных условиях пента-пара-толилсурьма реагирует с 4-диметиламинобензальдоксимом с образованием оксимата тетра-пара-толилсурьмы (4‑MeC6H4)4SbON=CHC6H4NMe2-4 (52) [28].
Взаимодействием пентафенилсурьмы с 1,2-дифенилэтандиоксимом или 1,2-дифенил(1-окси)этаноксимом-2 независимо от соотношения исходных реагентов в толуоле синтезированы 1,2-дифенилэтандиоксимат бис(тетрафенилсурьмы) (53) в виде сольвата с толуолом и 1,2-дифенил(1-окси)-этаноксимат-2 тетрафенилсурьмы (54) (схема 7 ) [29].
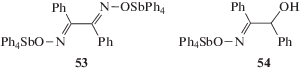
Схема 7 .
Показано, что взаимодействие диоксима метилендициклопентанона-2,2' с пентафенилсурьмой в жестких условиях (90°С, 5 ч) при мольном соотношении исходных реагентов 1 : 2 соответственно приводит к образованию макроциклического сурьмаорганического соединения – бис-μ-[(метилендициклопентанон-2,2'-диоксимато)трифенилсурьмы] (55), в молекулах которого симметричные диоксимные радикалы чередуются со структурными блоками трифенилсурьмы (схема 8 ) [30].
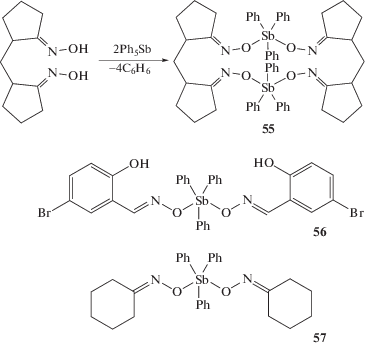
Схема 8 .
В центросимметричном соединении 55 атомы сурьмы имеют тригонально-бипирамидальную координацию с атомами кислорода в аксиальных положениях.
Взаимодействие пентафенилсурьмы с 2-гидрокси-5-бромбензальдоксимом и циклогексаноноксимом (толуол, 90–100°С, 1–5 ч) приводит к образованию соответствующих диоксиматов трифенилсурьмы (56) и (57) (схема 8 ) наряду с оксиматами тетрафенилсурьмы (до 10%) [31].
Реакция эквимолярных количеств пента-пара-толилсурьмы с бромистоводородной кислотой приводит к образованию бромида тетра(пара-толил)сурьмы (58) [32]. В некоторых случаях галогениды тетраарилсурьмы синтезируют из пентахлорида сурьмы и ариллития в растворе эфира, например хлорид тетракис(пентафторфенил)сурьмы (C6F5)4SbCl (59) был получен с выходом 25% [3].
Соли тетраарилсурьмы могут реагировать с избыточным количеством кислоты в реакционной смеси. Так, ацетат и нитрат тетрафенилсурьмы с эквимолярными количествами уксусной и азотной кислот образуют соответственно аддукты Ph4SbOC(O)CH3 ⋅ CH3C(O)OH (60) и Ph4SbONO2 ⋅ ⋅ HNO3 (61) [33].
Реакции пентафенилсурьмы с комплексными кислотами изучены на единичных примерах. Так, взаимодействием пентафенилсурьмы с золотохлористоводородной и гексахлороплатиноводородной кислотами в бензоле получены соответственно тетрахлороаурат тетрафенилсурьмы (62) [34, 35] и гексахлороплатинат бис(тетрафенилсурьмы) (63) [34]. При проведении последней реакции в растворе диметилсульфоксида (DMSO) из реакционной смеси были выделены желтые кристаллы комплекса [Ph4Sb(DMSO-O)][PtCl5(DMSO-S)] (64) [36].
Первые γ-алкилацетилацетонаты тетрафенилсурьмы (65–68), в которых атомы сурьмы имеют октаэдрическую координацию с бидентатным ацетилацетонатным лигандом, получили взаимодействием пентафенилсурьмы с γ-этилацетилацетоном, γ-аллилацетилацетоном, γ-фенилацетилацетоном и γ-тиобутилацетилацетоном (толуол, 90°С, 10 ч) (cхема 9) [37, 38].
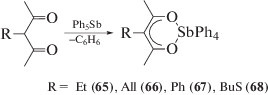
Схема 9 .
В гетероциклах SbO2C3 длины связей Sb−O близки к сумме ковалентных радиусов атомов сурьмы и кислорода.
Взаимодействием пентафенилсурьмы с октантетраоном-2,4,5,7 (мольное соотношение 2 : 1) в толуоле синтезирован биядерный хелатный комплекс Ph4Sb[OC(Ме)CHC(O)–(O)CCH(Ме)CO]SbPh4 (69) (cхема 10) [39].
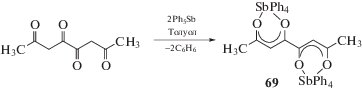
Схема 10 .
Синтез соединений Ar4SbX из галогенидов тетраарилсурьмы. Эффективный способ синтеза производных сурьмы общей формулы Ar4SbX основан на реакции замещения атома галогена в галогениде тетраарилсурьмы на электроотрицательную группу Х. Так, обработка хлорида тетракис(пентафторфенил)сурьмы трифторметансульфоновой кислотой в ацетонитриле приводит к образованию трифлата тетракис(пентафторфенил)сурьмы (70) (97%) [3]. Продуктом взаимодействия золотохлористоводородной кислоты с хлоридом тетрафенилстибония в ацетоне является [Ph4Sb]+[AuCl4]– (62) [34]. Если взаимодействие хлорида тетра-пара-толилстибония с гидратом золотохлористоводородной кислоты в ацетоне приводило к образованию тетрахлораурата тетра-пара-толилсурьмы [(p-Tol)4Sb]+[AuCl4]– (71), то в ацетоно-толуольном растворе направление реакции менялось. После испарения растворителя наблюдалось образование кристаллов темно-коричневого цвета, состояших, по данным РСА, из тетраэдрических тетра-п-толилстибониевых катионов и плоскоквадратных анионов [(p-Tol)AuCl3]− (72) (cхема 11) [35].

Схема 11 .
Анионы [(p-Tol)AuCl3]− имеют обычное плоскоквадратное строение, длины связей (Au−Cl 2.286(2)−2.389(2) Å, Au−C 2.028(7) Å, углы ClAuCl 89.92(9)°, 92.42(9)°, 176.68(8)°, CAuCl 88.27(18)°, 89.57(18)°, 175.59(19)°). Следует отметить, что реакции аурирования протекали только в присутствии хлорида тетра-пара-толилстибония.
Для замещения галогена в галогенидах тетраарилсурьмы часто используют соли натрия или калия. Например, взаимодействие хлорида тетрафенилсурьмы с перренататом натрия или хлор-атом калия приводит к образованию с высокими выходами перрената тетрафенилсурьмы (73) и хлората тетрафенилсурьмы (74) соответственно [40].
Подобным образом были синтезированы трифлаты тетраарилсурьмы – эффективные катализаторы реакции циклоприсоединения оксиранов к изоцианатам [41].
Продуктом взаимодействия динатриевой соли 4-гидроксибензойной кислоты с бромидом тетрафенилсурьмы (мольное соотношение 1 : 2 соответственно) в толуоле является сольват (µ2-оксибензоато-O,O',O")-бис-(тетрафенилсурьмы) (75) с толуолом (схема 12 ) [42]. Перемешивание смеси бромида тетрафенилсурьмы и метоксида натрия в метаноле в течение 72 ч в атмосфере воздуха приводит к образованию карбоната бис-(тетрафенилсурьмы) (76), кристаллизующегося из раствора в виде триклинной модификации.
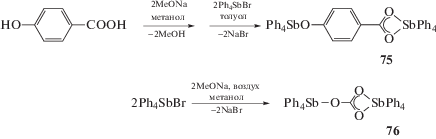
Схема 12 .
Аналогично получали ряд других бис-карбоксилатов трифенилсурьмы (77–80), карбоксилатов трифенилхлорсурьмы (81–84) и тетрафенилсурьмы (85–88) (схема 13 ) [43].
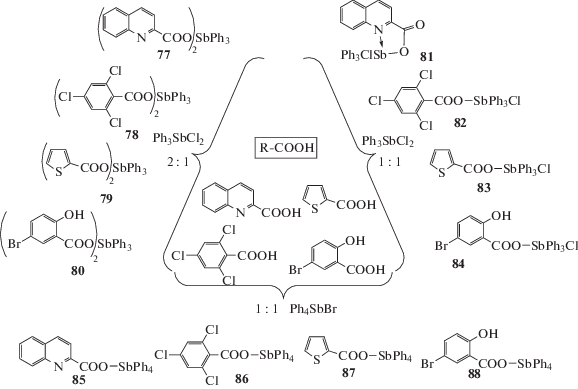
Схема 13 .
Производные бис-(феноксипропионато)триарилсурьмы (89–92) с выходом 74–80% синтезировали из соответствующего дихлорида триарилсурьмы и калиевой соли карбоновой кислоты в метаноле (кипячение, 8 ч) (схема 14 ) [44].
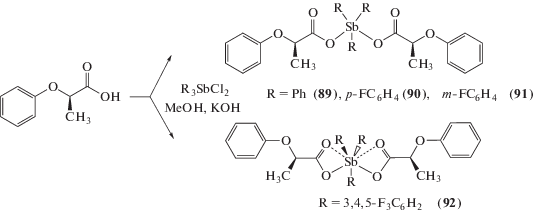
Схема 14 .
Первые производные тетраарилсурьмы с О,О-диалкилдитиофосфатными заместителями, способными проявлять структурную неэквивалентность, были получены из хлорида тетрафенилсурьмы и соответствующих О,О-диалкилдитиофосфатов калия (схема 15 ) [45–48].
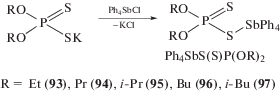
Схема 15 .
Показано, что атомы сурьмы в молекулах Ph4SbS(S)P(OR)2 (93–97) имеют координацию искаженной тригональной бипирамиды с аксиально расположенными О,О-диалкилдитиофосфатными лигандами, при этом основные структурные характеристики молекул близки между собой.
В (94) атом сурьмы имеет координацию искаженного октаэдра с атомами серы и углерода в экваториальной плоскости [48]. В молекулах комплекса дипропилдитиофосфатный лиганд обнаруживает анизобидентатный характер координации, так как одна из связей Sb–S (3.006 Å) заметно короче другой (3.557 Å), поэтому можно сделать вывод, что геометрия координационного полиэдра атома сурьмы в указанных комплексах чувствительна даже к небольшим изменениям в структуре серосодержащих лигандов.
Реакция бромида тетрафенилсурьмы с натриевыми солями тетракетонов независимо от мольного соотношения реагентов приводит к образованию с выходом до 76% биядерных хелатных комплексов общей формулы Ph4Sb[OC(R)CHC(O)- (O)CCH(R)CO]SbPh4 (R = Et (98); R = tBu (99) – аналогов комплекса 69 [49].
Карбоксилаты тетрафенилсурьмы общей формулы ArC(O)OSbPh4 [Ar = 3-F-4-CH3C6H3 (100), 4-F-2-CH3C6H3 (101), 5-F-2-CH3C6H3 (102)] синтезированы кипячением эквивалентных количеств бромида тетрафенилсурьмы, карбоновой кислоты и этилата натрия в метаноле в течение 12 ч [50].
Взаимодействие бромида тетрафенилсурьмы с трифлатом серебра в хлористом метилене приводит к образованию трифлата тетрафенилсурьмы, обработка которого такими донорами (donor) как оксид триметилфосфина или 4-метилпиридиноксид сопровождается образованием продуктов присоединения [Ph4Sb(donor)]+[OSO2CF3]− (103 и 104) [51].
Замещением атома хлора арильной группой в хлороксистиборане (105), содержащем C,O,C'-тридентатный лиганд, была получена смесь двух стереоизомеров комплексов сурьмы (106 и 107) (схема 16 ) [52].
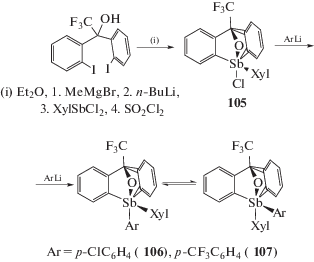
Схема 16 .
Взаимодействие 1,8-дилитийнафталина с дибромидом трифенилсурьмы приводит к отщеплению одного фенильного лиганда от атома сурьмы и образованию четырехядерного комплекса (108), который при действии гексафторфосфата таллия превращается в гексафторфосфат бис-(µ2-нафталин-1,8-диилсурьма(V)-ртуть(II)) (109); продуктом реакции комплекса 108 с иодидом тетрабутиламмония является бис-(µ2-нафталин-1,8-диилиодосурьма(V)-ртуть(II)) (110) (схема 17 ) [53].
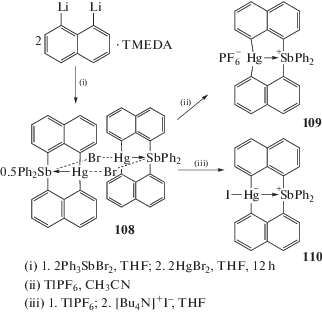
Схема 17 .
Бромиды тетраарилсурьмы общей формулы [ArSbPh3]+Br−, в которых Ar = 9-фенантрил (111), 1-пиренил (112), 3-периленил (113), описаны в [54]. Соединения (111–113) получены из дибромида трифенилсурьмы и литиевого производного соответствующего конденсированного арена. Установлено, что (111) не является стабильным в воде, но (112 и 113) можно использовать в качестве датчиков для обнаружения фторид-анионов в водных растворах DMSO (10% объемн.) (рН 4.8) (схема 18 ) [54].
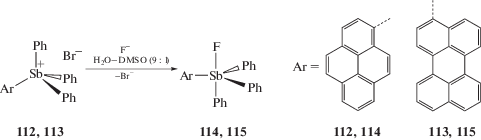
Схема 18 .
Хлорид алкокситриарилсурьмы (105) при действии ариллития, содержащего потенциальный координирующий центр в арильном кольце, образует соединения шестикоординированной сурьмы (116–118) (схема 19 ), в которых атом кислорода и объемный 3,5-диметилфенильный лиганд расположены напротив друг друга [55].
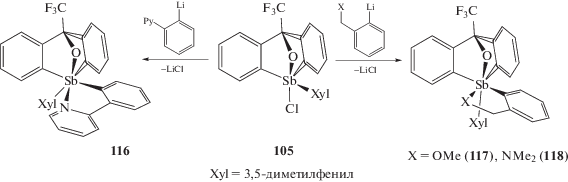
Схема 19 .
В реакциях замещения могут участвовать не только галогениды тетраарилсурьмы. Так, например, при взаимодействии водорастворимого трифлата антрацен-9-ил-трифенилсурьмы (119) с растворами, содержащими фторид-анионы, образуется фторид антрацен-9-ил-трифенилсурьмы (120) (схема 20 ) [56].
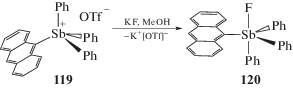
Схема 20 .
Тетрафенилбораты триарил(L2PdCl)стибония (121 и 122) ионного строения, содержащие фрагмент L2PdCl, связанный с центральным атомом сурьмы посредством связи Sb–Pd, при обработке фторидом тетрабутиламмония в хлористом метилене превращаются в ковалентные фториды триарил(L2PdCl)сурьмы (123 и 124) (схема 21 ) [57].
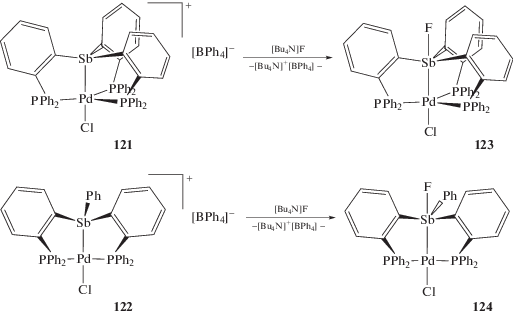
Схема 21 .
Весьма эффективным является способ синтеза производных тетраарилсурьмы Ar4SbХ, основанный на реакции галогенида тетраарилсурьмы с соединениями, содержащими активный атом водорода, в присутствии амина. Так были получены производные сурьмы Ar4SbL и Ar3SbL2 (125–139), где LH – N-гидроксигексагидро-4,7-эпоксиизоиндол-1,3-дион, N-гидрокси-3',4,7,7'-тетрагидро-4,7-эпоксиизоиндол-1,3-дион (схема 22 ) [58].
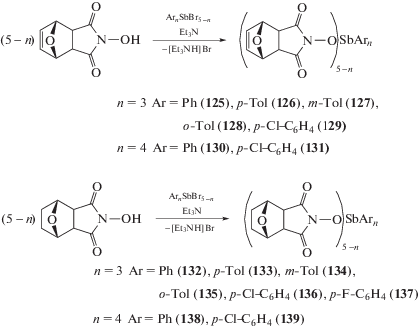
Схема 22 .
По аналогичной схеме (схема 23 ) были синтезированы сурьмаорганические производные арилгидроксамовых кислот (Ar = p-Tol (140), Ph (141)) [59].
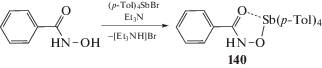
Схема 23 .
Серия ферроценилакрилатов тетраарилсурьмы C5H5FeC5H4CHCHC(O)ОSbAr4 (Ar = Ph (142), p-Tol (143), m-Tol (144), o-Tol (145), p-F-C6H4 (146) была синтезирована добавлением раствора 3-ферроценилакриловой кислоты в триэтиламине к суспензии бромида тетраарилсурьмы в толуоле (схема 24 ) [60].
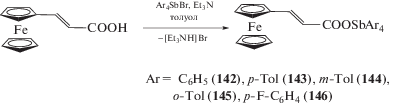
Схема 24 .
В молекуле фенильного производного (142) отношение d(Sb···O=C)/d(Sb–О) близко к 1 (2.400 Å/ 2.269 Å = 1.06), т.е. карбоксилатный лиганд является бидентатным. Вследствие этого координация атома сурьмы в (142) становится искаженной октаэдрической. В экваториальной плоскости находятся два атома углерода фенильных групп и два атома кислорода карбоксигруппы, аксиальные положения занимают атомы углерода двух других фенильных заместителей.
Найдено, что хлорид или бромид тетрафенилсурьмы реагируют при 180°С с карбамидом с образованием цианамида тетрафенилсурьмы Ph4Sb-NCN (147) с выходом 52%. По данным РСА, центральный атома сурьмы находится в искаженном тригонально-бипирамидальном окружении с цианамидной и фенильной группами в аксиальных положениях (СSbN 177.76(7)°, Sb–С 2.107(2)–2.167.2, Sb–N 2.3383(18) Å) [61]. Подобная реакция дибромида трифенилсурьмы с мочевиной при 155°C приводила к образованию соединения сурьмы (OCN)Ph3SbOSbPh3(NCO) (148), который был закристаллизован из диоксана (угол SbOSb 146.2°).
Сурьмасодержащие соединения ионного типа [Ar4Sb]+[X]− могут быть синтезированы из галогенида тетраарилсурьмы и комплексной соли переходного металла. Например, комплекс [p-Tol4Sb]+ [Au(CN)2]− (149) был получен из хлорида тетра(пара-толил)сурьмы и дицианоаурата калия в воде с выходом 83% [62]. Длины связей Au−C в (149) составляют 1.94(7)−2.00(6) Å, что меньше суммы ковалентных радиусов атомов золота и углерода (2.11 Å) [63].
Установлено, что при растворении дигидрата гексахлороосмата(IV) натрия и хлоридов тетрафенил- или тетра(пара-толил)стибония (мольное соотношение 2 : 1) в диметилсульфоксиде и последующем медленном испарении растворителя образуются устойчивые на воздухе зеленые кристаллы комплексов $[{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{2}}}^{ + }{{{\text{[OsC}}{{{\text{l}}}_{{\text{6}}}}]}^{{{\text{2}} - }}}$ (150) и $[p{\text{ - To}}{{{\text{l}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{2}}}^{ + }{{{\text{[OsC}}{{{\text{l}}}_{{\text{6}}}}]}^{{{\text{2}} - }}}$ (151) (схема 25 ) [64].
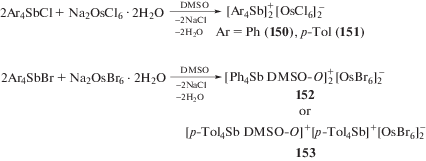
Схема 25 .
По данным РСА, кристаллы комплексов (150) и (151) состоят из тетраэдрических катионов тетраарилстибония и октаэдрических гексахлороосмат-анионов. Подобные реакции между бромидами тетрафенил- и тетра-п-толилсурьмы и гексабромоосматом натрия приводили к образованию комплексов (152 и 153) ионного типа (схема 25 ), однако в этих случаях молекулы растворителя входят в координационную сферу тетраарилстибониевого катиона, в результате чего КЧ атома сурьмы увеличивается до 5 с учетом О-связанной молекулы диметилсульфоксида [65]. Анионы [OsHal6]2− (Hal = Cl, Br) в реакциях с галогенидами тетраарилстибония в растворе DMSO кинетически инертны и не вступают в дальнейшие реакции внутрисферного лигандного обмена.
Хлорид тетрафенилсурьмы в растворе диметилсульфоксида реагирует с гидратом трихлорида рутения с образованием транс-бис-(диметилсульфоксидо)тетрахлорорутената (диметилсульфоксидо)тетрафенилсурьмы [Ph4Sb(DMSO-O)$]_{{}}^{ + }$ [RuCl4(DMSO-S)2]− (154) [66]. Атомы сурьмы в катионе имеют искаженную тригонально-бипирамидальную координацию с атомом кислорода DMSO в аксиальном положении (Sb–O 2.633(15), Sb–С 2.094(15)–2.146(15) Å, СSbO 178.54(16)°). В октаэдрических анионах комплекса [транс-RuCl4(DMSO-S)2]– лиганды DMSO координируются на атом металла атомом серы (Ru–S 2.349(3), Ru–Cl 2.353(5), 2.355(3) и 2.332(3), 2.344(6), 2.336(4)–2.353(3) Å соответственно), углы SRuS и транс-ClRuCl составляют 180°.
Комплексы иридия аналогичного строения [Ph4Sb(DMSO-O)$]_{{}}^{ + }$[IrCl4(DMSO-S)2]− (155) и [Ph4Sb]+[IrX4(DMSO-S)2-транс]– (X = Br (156); X = Cl, (157)) были получены из галогенида тетрафенилсурьмы и гексахлоро-, гексабромоиридата натрия в диметилсульфоксиде [67]. В продуктах реакции бромида тетра(пара-толил)сурьмы с гексабромоиридатом натрия в DMSO был также обнаружен комплекс [p-Tol4Sb(DMSO-O)]+[p-Tol4Sb]+ [IrBr6]2– (158). При быстром удалении растворителя образуются темно-синие кристаллы комплекса (158), анионы которого не содержат координированных молекул диметилсульфоксида (3 ч, 50°) [68]. Однако при медленном испарении растворителя (480 ч) наблюдается образование желто-коричневых кристаллов комплекса [p-Tol4Sb(DMSO-O)]+ [IrBr4(DMSO-S)2-транс]– (159) (схема 26 ).
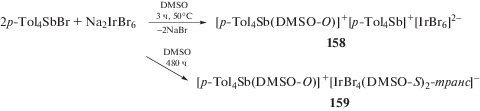
Схема 26 .
Отметим, что анион [IrBr6]2– менее устойчив в растворе DMSO, чем [OsBr6]2–, так как в случае комплексов осмия даже медленное испарение растворителя не приводило к реакциям восстановления и внутрисферного замещения в анионах.
В то же время взаимодействие бромида тетра-пара-толилстибония и гексабромородиата(III) натрия в DMSO приводило к образованию красно-коричневых кристаллов комплекса [p-Tol4Sb(DMSO-O)$]_{2}^{ + }$ [RhBr4(DMSO-S)2-транс]–[RhBr4(DMSO-S)2-цис]– (160) (cхема 27) [69].
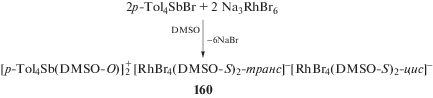
Схема 27 .
Изомерные октаэдрические анионы [RhBr4-(DMSO-S)2-транс]− и [RhBr4(DMSO-S)2-цис]− в 160 различаются расположением S-координированных молекул DMSO.
Первые диметилсульфоксидные комплексы палладия были получены путем растворения Pd-Cl2 в DMSO [70]. Ионные комплексы с сурьмасодержащими катионами и моноядерными анионами [PdHal3(DMSO-S)]– (161 и 162) могут быть синтезированы непосредственным взаимодействием галогенидов тетраарилсурьмы с галогенидами палладия в DMSO (cхема 28) [71, 72].
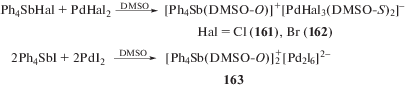
Схема 28 .
По данным РСА, координационный полиэдр атома палладия в анионах [PdHal3(DMSO–S)]– представляет собой искаженный квадрат, молекулы DMSO координируются на атом палладия атомом серы, валентные углы незначительно отличаются от теоретических значений.
Комплекс с биядерным анионом [Ph4Sb(DMSO-O)]$]_{2}^{ + }$[Pd2I6]2– (163) является продуктом реакции дииодида палладия с иодидом тетрафенилстибония (cхема 28) [73]. По данным РСА, в биядерных анионах [Pd2I6]2– координация атомов палладия плоско-квадратная, атомы палладия связаны друг с другом посредством двух μ2-мостиковых атомов иода. Связи Pd–Iтерм. (2.5836(8)–2.6093(4) Å) практически совпадают с расстояниями Pd–Iмост. (2.5875(5)–2.6129(3) Å).
Известно, что термическое разложение галогенидов тетрафенилстибония в отсутствии растворителя протекает при 250°С по схеме реакции восстановительного элиминирования с образованием трифенилстибина и галоидбензола [74]. Однако в присутствии K2[PtCl4] восстановление Sb(V) → Sb(III) осуществляется в растворе DMSO при комнатной температуре, при этом имеет место образование нейтрального смешанолигандного комплекса (Ph3Sb)(DMSO-S)Pt(Cl2-цис) (164) (cхема 29) [75].
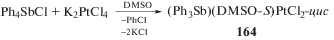
Схема 29 .
Как следует из данных РСА, комплекс 164 имеет характерное для соединений Pt(II) плоскоквадратное строение, в координационной сфере центрального атома, наряду с двумя цис-хлоро-лигандами, присутствуют S-координированная молекула DMSO и молекула трифенилстибина [75]. Транс-углы ClPtS 177.96(4)°–178.01(6)°, цис-SPtSb 90.75(4)°, SPtCl(1) 90.24(6)°, SbPtCl 88.36(4)°, ClPtCl 90.71(6)°. Длина связи Pt−Sb (2.5118(4) Å) меньше суммы ковалентных радиусов атомов платины и сурьмы (2.78 Å) [63]. Подобные соединения фосфора (Ph3P)(DMSO-S)PtCl2-цис (165) и (Ph3P)(SOEt2-S)PtCl2-цис (166) образуются из хлорида 2-бутенил-бис-(трифенилфосфония) и гексахлороплатиновой кислоты в присутствии диметил- или диэтилсульфоксида в ацетонитриле [76].
Для получения гексабромоплатината тетрафенилсурьмы $[{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{2}}}^{ + }{{{\text{[PtB}}{{{\text{r}}}_{{\text{6}}}}]}^{{{\text{2}}--}}}$ (167) в качестве исходных соединений были использованы гексабромоплатинат калия и бромид тетрафенилсурьмы [77]. По данным РСА, кристалл комплекса состоит из тетраэдрических стибониевых катионов и октаэдрических анионов [PtBr6]2–. При перекристаллизации полученного комплекса 167 из диметилсульфоксида наблюдалось внутрисферное замещение одного из атомов брома на молекулу DMSO с образованием комплекса 168 (cхема 30).
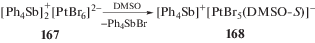
Схема 30 .
По данным РСА, октаэдрические анионы [PtHal5(DMSO-S)]– практически не искажены.
Взаимодействием тетрахлорида циркония с хлоридом тетрафенилстибония в ацетонитриле с выходом 86% синтезирован и структурно охарактеризован гексахлороцирконат тетрафенилстибония $[{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{2}}}^{ + }{{{\text{[ZrC}}{{{\text{l}}}_{{\text{6}}}}]}^{{{\text{2}}--}}}$ (169) [78].
Синтез соединений пятивалентной сурьмы по реакции перераспределения лигандов. Начало изучения реакций перераспределения лигандов в ряду арильных соединений пятивалентной сурьмы относится к 1974 г., когда Фергюсон и Хавли при перекристаллизации дигидроксида трифенилсурьмы из хлороформа в атмосфере воздуха наблюдали образование карбоната бис-(тетрафенилсурьмы) (76) [79]. Очевидно, что формирование фрагмента Ph4Sb можно объяснить только обменом фенильных заместителей между атомами сурьмы.
Изучая строение производного сурьмы, полученного из дихлорида трифенилсурьмы и оксалата серебра, Соверби с Миллингоном показали, что данное соединение имеет солеобразное строение [Ph4Sb][Ph2Sb(Ox)2] (170), где Ох = О2ССО2, и предположили, что оно может быть получено из дихлорида трифенилсурьмы по следующей схеме (cхема 31) [80].
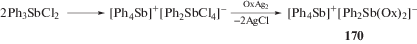
Схема 31 .
Решив исследовать взаимодействие пентаарилсурьмы с производными общей формулы Ar3SbX2, где Х − электроотрицательный лиганд, авторы [81] полагали, что будет иметь место либо образование комплексов ионного типа, либо перераспределение органических радикалов и синтез производных несимметричного строения. Действительно было обнаружено, что пентафенилсурьма реагирует с дигалогенидами, дибензоатом и дироданидом трифенилсурьмы с образованием продуктов несимметричного строения (171–174) – галогенида, бензоата и роданида тетрафенилсурьмы соответственно, с выходом до 99% (схема 32 ).
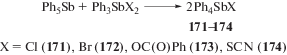
Схема 32 .
Взаимодействие реагентов протекает в ароматическом углеводороде при комнатной температуре в течение суток.
Возможность подобных реакций пентаарилсурьмы с другими производными пятивалентной сурьмы общей формулы Ar3SbХ2, где Х − электроотрицательный лиганд, установлена для диоксиматов [26], дисульфонатов [82], диароксидов [9, 83] и дикарбоксилатов [11–15, 81, 84] триарилсурьмы. В зависимости от природы лиганда Х исчезновение пентаарилсурьмы из реакционной смеси происходило при нагревании (90°С) через 0.25−2 ч. После удаления растворителя и перекристаллизации твердого остатка из смеси гептан−бензол выделяли единственный продукт реакции.
В продолжение этого направления исследований была изучена возможность синтеза производных сурьмы несимметричного строения общей формулы Ph3SbXY из соединений Ph3SbX2 и Ph3SbY2. Установлено, что хлорооксиматы трифенилсурьмы Ph3Sb(Cl)ONCMePh (175) [85] и Ph3Sb(Cl)ONCHC6H4(OH-2) (176) [86], в которых присутствуют различные электроотрицательные заместители, также могут быть получены по реакции перераспределения лигандов (cхема 33). Отметим, что хлороксиматы трифенилсурьмы (175 и 176) гладко фенилируются пентафенилсурьмой до оксимата тетрафенилсурьмы; вторым продуктом реакции является хлорид тетрафенилсурьмы. Подобная реакция синтеза хлорароксидов трифенилсурьмы осуществима и для бис-(4-нитрофеноксо)трифенилсурьмы [87], бис-(2,6-дихлорфеноксо)трифенилсурьмы [88] и бис-(бензолсульфоната) три(мета-толил)сурьмы (177–179, cхема 33) [89].
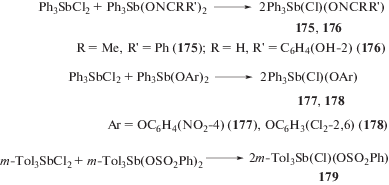
Схема 33 .
Реакции перераспределения радикалов между дифторидом триарилсурьмы и бис-(трифторметансульфонатом) триарилсурьмы в растворе хлористого метилена протекает с образованием производных сурьмы (180 и 181) несимметричного строения (cхема 34). Аналогичное перераспределение радикалов с образованием производных (182 и 183) имеет место в реакции дихлорида триарилсурьмы с пятихлористой сурьмой [90].
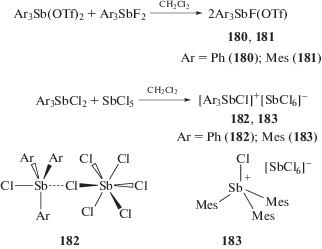
Схема 34 .
По аналогичной схеме реагирует дихлорид трифенилсурьмы с бис-(пентахлор-) и бис-(пентафторароксо)трифенилсурьмой [91].
В случае взаимодействия дихлорида три(пара-толил)сурьмы с бис-(4-нитрофенокси)-три-пара-толилсурьмой реакция перераспределения останавливается на образовании аддукта p-Tol3SbCl2 ⋅ ⋅ p-Tol3Sb(Cl)OC6H4(NO2-4) (184) [92].
Синтез комплексных соединений сурьмы, содержащих тетраарилстибониевые катионы. Известно, что хлорид тетрафенилсурьмы реагирует с тетрахлоридом олова в бензоле с образованием ионного комплекса (185) (cхема 35) [34]:
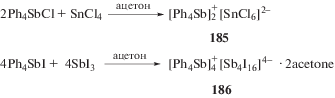
Схема 35 .
В реакциях перераспределения лигандов с участием производных пятивалентной сурьмы также предполагалось промежуточное образование комплексных соединений сурьмы, например [Ph4Sb]+[Ph4SbХ2]−, которые далее превращались в производные сурьмы общей формулы Ph4SbХ. В связи с этим авторы [93], изучая взаимодействие иодида тетрафенилсурьмы с трииодидом сурьмы в ацетоне, предполагали образование подобного комплекса, однако единственным продуктом реакции являлся комплекс $[{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{4}}}^{ + }{{{\text{[S}}{{{\text{b}}}_{{\text{4}}}}{{{\text{I}}}_{{{\text{16}}}}}]}^{{{\text{4}} - }}}$ · 2(CH3)2CO (схема 35 ) (186). Реакцию проводили при комнатной температуре в растворе ацетона, при этом цвет менялся с коричневого, характерного для растворов трехиодистой сурьмы, на красно-вишневый. По данным РСА, в комплексе (186) анионы [Sb4I16]4− имеют циклическое центросимметричное строение (схема 36 ).
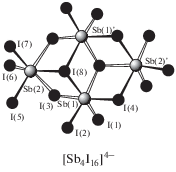
Схема 36 .
По аналогичной схеме был синтезирован изоструктурный комплекс $[{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{4}}}^{ + }{{{\text{[B}}{{{\text{i}}}_{{\text{4}}}}{{{\text{I}}}_{{{\text{16}}}}}]}^{{{\text{4}} - }}}$ · · 2(CH3)2CO (187) [94].
Несколько иначе протекают реакции 4-метилбензолсульфоната и 2,4-диметилбензолсульфоната тетрафенилсурьмы с трииодидом сурьмы в ацетоне (схема 37 ) [93].
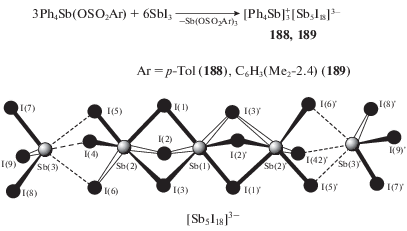
Схема 37 .
В этом случае из реакционной смеси были выделены комплексы(188 и 189), состоящие из тетрафенилстибониевых катионов и анионов [Sb5I18]3− (схема 37 ).
Реакции аренсульфонатов тетрафенилсурьмы с трииодидом висмута в диметилсульфоксиде приводят к образованию смешаннолигандного комплекса фенилтрииодовисмутата(III) тетрафенилстибония $[{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{2}}}^{ + }{{{\text{[P}}{{{\text{h}}}_{{\text{2}}}}{\text{B}}{{{\text{i}}}_{{\text{2}}}}{{{\text{I}}}_{{\text{6}}}}]}^{{{\text{2}} - }}}$ ⋅ 2DMSO (190), образованного из катионов [Ph4Sb]+, анионов [Ph2Bi2I6]2− и молекул кристаллизационного DM-SO (схема 38 ) [95].
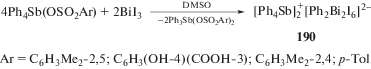
Схема 38 .
При взаимодействии эквимолярных количеств иодида тетра(пара-толил)сурьмы с трииодидом висмута в 2-этоксиэтаноле (схема 39 ) образуется сольватный комплекс (191) ионного типа [96].
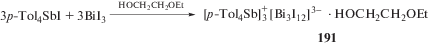
Схема 39 .
Продуктом взаимодействия эквимолярных количеств иодида тетра(пара-толил)сурьмы и трииодида висмута в растворе тетрагидрофурана являлся ионный комплекс $[p{\text{ - To}}{{{\text{l}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{2}}}^{ + }{{{\text{[B}}{{{\text{i}}}_{{\text{2}}}}{{{\text{I}}}_{{\text{8}}}}({\text{THF}})]}^{{{\text{2}} - }}}$ (192); увеличение количества иодида висмута в два раза и замена растворителя на ацетон приводит к образованию комплекса $[p{\text{ - To}}{{{\text{l}}}_{{\text{4}}}}{\text{Sb}}]_{n}^{ + }{{[{{({\text{B}}{{{\text{i}}}_{{\text{2}}}}{{{\text{I}}}_{{\text{7}}}})}_{n}}]}^{{n - }}}$ (193) с полимерным анионом [97].
Аналогично с образованием комплексов ионного типа реагируют с иодидом тетрафенилсурьмы дииодиды ртути или кадмия. Так, взаимодействие между указанными реагентами при комнатной температуре в растворе ацетона (12 ч) сопровождается образованием сурьмаорганических производных общей формулы $[{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{2}}}^{ + }{{{\text{[}}{{{\text{E}}}_{{\text{2}}}}{{{\text{I}}}_{{\text{6}}}}]}^{{{\text{2}} - }}}$ (E = Cd (194), Hg (195)) (схема 40 ) [98].
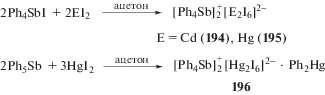
Схема 40 .
Полученные соединения имеют высокую температуру плавления и хорошо растворимы в органических растворителях. Установлено, что независимо от соотношения исходных реагентов пентафенилсурьма фенилирует иодид ртути в ацетоне до комплекса $[{\text{P}}{{{\text{h}}}_{{\text{4}}}}{\text{Sb]}}_{{\text{2}}}^{ + }{{{\text{[H}}{{{\text{g}}}_{{\text{2}}}}{{{\text{I}}}_{{\text{6}}}}]}^{{{\text{2}} - }}}$ ⋅ Ph2Hg (196), максимальный выход которого достигается при мольном соотношении исходных реагентов 2 : 3 (схема 40 ).
Из продуктов реакции 2,4-диметилбензолсульфоната тетрафенилсурьмы с дииодиом ртути в ацетоне при комнатной температуре из реакционной смеси дробной перекристаллизацией были выделены кристаллы (197) желтого цвета (схема 41 ).
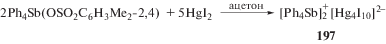
Схема 41 .
Показано, что изменение мольного соотношения исходных реагентов в реакции иодида тетра(пара-толил)сурьмы с иодидом ртути приводит к изменению структуры ртутьсодержащего аниона комплекса присоединения [99]. Так, при проведении реакции эквимолярных количеств иодида тетра(пара-толил)сурьмы и иодида ртути был получен с выходом 92% (ди-μ2-иодо)тетраиододимеркурат бис[(тетра-пара-толил)сурьмы] (198) (схема 42 ).
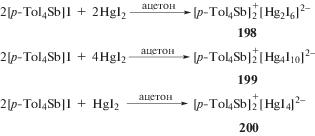
Схема 42 .
Из данных РСА следует, что кристалл комплекса (198) состоит из тетра(пара-толил)стибониевых катионов и центросимметричных биядерных анионов [Hg2I6]2−. При увеличении концентрации иодида ртути в реакционной смеси (мольное соотношение 1 : 2 соответственно) строение ртутьсодержащего аниона в целевом продукте усложняется. Реакция протекает с образованием комплекса (199), представляющего собой прозрачные кристаллы желтого цвета (схема 42 ). Наоборот, увеличение концентрации иодида тетра(пара-толил)сурьмы в реакционной смеси (2 : 1 мольн.) приводит к единственному продукту (200) (схема 42 ) с моноядерным анионом − тетрайодомеркурату бис[тетра(пара-толил)сурьмы]. В состав кристалла комплекса (200), кроме двух типов кристаллографически независимых тетраэдрических катионов тетра(пара-толил)стибония, входят тетраиодомеркуратные анионы [HgI4]2−.
Бромид дифенил[бис-(µ2-1,8-нафталиндиилртуть)]сурьмы (108), в котором два нафтильных лиганда связаны между собой через атомы ртути, при действии гексафторфосфата таллия в тетрагидрофуране или фторида тетрабутиламмония превращается в комплекс (201) с координированной молекулой THF или во фторид (202) соответственно [100]. Последующее прибавление к раствору (201) в THF 4-диметиламинопиридина (DMAP) приводит к образованию аддукта (203) с тремя молекулами DMAP (схема 43 ).
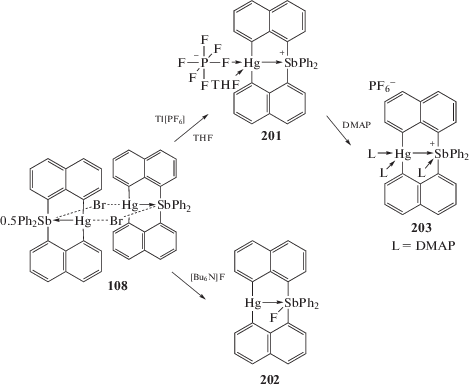
Схема 43 .
МЕТОДЫ СИНТЕЗА И НЕКОТОРЫЕ РЕАКЦИИ ПРОИЗВОДНЫХ СУРЬМЫ Ar3SbХ2
Одними из наиболее изученных арильных производных пятивалентной сурьмы являются соединения Ar3SbХ2, где Х – электроотрицательный лиганд. Существует несколько эффективных способов их получения, среди которых следует выделить реакции замещения атомов галогена на иные группы. Это прежде всего реакции дигалогенидов триарилсурьмы с натриевыми, калиевыми или серебряными солями кислот и подобных им соединений.
Получение производных сурьмы из дигалогенидов триарилсурьмы. Дихлорид трис(4-N,N-диметиламинофенил)сурьмы при действии фторида натрия в водно-ацетоновом растворе практически количественно превращается в дифторид трис(4-N,N-диметиламинофенил)сурьмы (204), представляющий собой бесцветные темнеющие на свету кристаллы (схема 44 ) [101]].
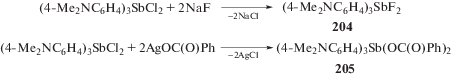
Схема 44 .
Дибензоат трис(4-N,N-диметиламинофенил) сурьмы (205) был также синтезирован по реакции обмена из бензоата серебра и дихлорида трис(4-N,N-диметиламинофенил)сурьмы (схема 44 ).
Взаимодействием дибромида трифенилсурьмы с азидом натрия (метанол, 10 мин) получен диазид трифенилсурьмы (206) (72%) [102].
Показано, что реакция дихлорида трифенилсурьмы с оксимом ацетилферроцена (мольное соотношение 1 : 2 соответственно) в присутствии метилата натрия в толуоле (4 ч, 110°С) приводит к образованию бис-(ферроценилэтаноноксимата) трифе-нилсурьмы (207). Координационный полиэдр атома сурьмы в (207) представляет собой тригональную бипирамиду с фенильными заместителями в экваториальных положениях и оксиматными лигандами − в аксиальных положениях (схема 45 ) [103].
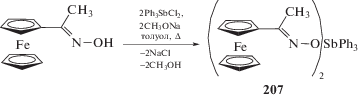
Схема 45 .
К образованию этого же соединения с выходом 72% приводит реакция дибромида трифенилсурьмы с ферроценилэтаноноксимом (12 ч, тетрагидрофуран) [104].
По аналогичной схеме получали дикарбоксилаты триарилсурьмы (208−210) из дибромида триарилсурьмы и натриевой соли мета- или пара-ферроценилбензойной кислоты в хлороформе (схема 46 ) [105] и дикарбоксилаты трифенилсурьмы (211, 212) из дихлорида трифенилсурьмы и натриевой соли 2-трифторметил- и 3-трифторметилбензойной кислоты соответственно с выходом до 84% [106].
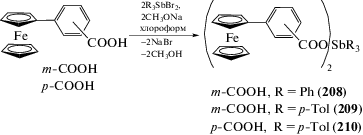
Схема 46 .
В реакции дихлорида трифенилсурьмы с N-оксифтальимидом и N-оксисукцинимидом (мольное соотношение 1 : 2 соответственно) присутствия основания не требуется (схема 47 ) [107]. Было обнаружено, что оба полученных соединения Ph3SbX2 (213, 214) являются перспективными противораковыми агентами, более активными, чем цисплатин.
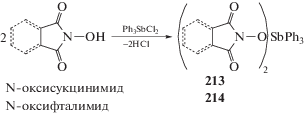
Схема 47 .
Взаимодействием дихлорида трифенилсурьмы с лапахолом (LpH) получено сурьмаорганическое производное лапахола (Lp)Ph3SbOH (215) (схема 48 ), которое ингибировало рост клеток хронической миелогенной лейкемии [108].
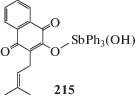
Схема 48 .
Из натриевой соли 2-гидроксибензальдоксима и дихлорида трифенилсурьмы была получена с выходом 86% бис-(2-гидроксибензальдоксимато)трифенилсурьма (216) [109].
Из натриевых солей 2-амино-4-хлоробензойной кислоты, 2-ацетилбензойной, 4-ацетилбензойной и дихлорида трифенилсурьмы в метаноле (0.5–3 ч, перемешивание) получены бис-(2-амино-4-хлоробензоато)трифенилсурьма (217) [110], бис-(2-ацетилбензоато)трифенилсурьма (218) и бис-(4-ацетилбензоато)трифенилсурьма (219), обладающие противоопухолевой активностью (схема 49 ) [111]. Подобным образом были синтезированы из 2-хлорфенилуксусной и 4-хлорфенилуксусной кислот в спирте аналогичные соединения сурьмы (220 и 221), однако в случае 3-хлорфенилуксусной кислоты из реакционной смеси было выделено лишь шестиядерное производное пятивалентной сурьмы (222) (схема 50 ) [112].
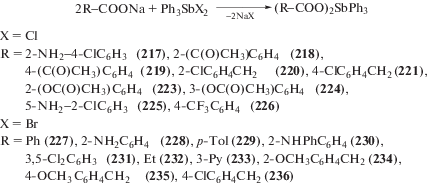
Схема 49 .
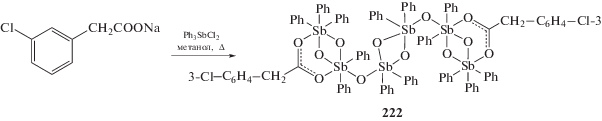
Схема 50 .
Обладающие антилейшманиoзной и антибактериальной активностью in vivo- и цитотоксичностью против макрофагов сурьмаорганические производные ацетилсалициловой (223) и 3-ацетоксибензойной (224) кислот [113] были получены по этой же схеме (в метаноле или толуоле), как и бис-(5-амино-2-хлоробензоато)трифенилсурьма (225) [114] с бис-(4-трифторметилбензоато)трифенилсурьмой (226) [115] (схема 49 ) и другие дикарбоксилаты триарилсурьмы (227–236) [116].
Дикарбоксилаты триарилсурьмы (4-ClC6H4CH= СHCOO)2SbPh3 (237) и (4-OMeC6H4CH= СHOO)2Sb(p-Tol)3 (238), обладающие антиопухолевой активностью, и их аналоги (239–241) были синтезированы с выходом до 82% из дибромида триарилсурьмы и натриевых солей соответствующих карбоновых кислот в толуоле (схема 51 ) [117].
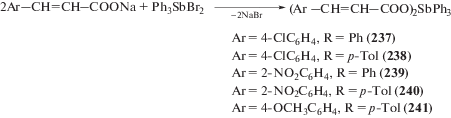
Схема 51 .
Взаимодействие эквимолярных количеств калиевых солей бензоилгидроксамовой и N-фенилбензоилгидроксамовой кислот с дихлоридом трифенилсурьмы в смеси растворителей (метанол–толуол, 1 : 1, перемешивание 0.5 ч) приводит к образованию биологически активных производных пятивалентной сурьмы – метоксида бензоилгидроксаматотрифенилсурьмы (242) и хлорида N-фенилбензоилгидроксамато-трифенилсурьмы (243) (схема 52 ) [118].
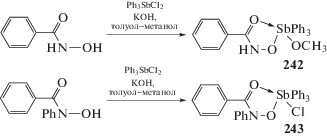
Схема 52 .
Реакция 3-метокси-2,4,5-трифторбензойной кислоты и этоксида натрия в метаноле с дихлоридом трифенилсурьмы при перемешивании в течение 12 ч при комнатной температуре после перекристаллизации из смеси дихлорметан-метанол (1 : 1) позволила получить целевой продукт (3-OCH3-2,4,5-F3C6H1COO)2SbPh3 (244) с выходом 86% [119]. По аналогичной методике синтезировали другие дикарбоксилаты трифенилсурьмы (4‑NH2C6H4COO)2SbPh3 (245) [120] и (3-F-4-CH3C6H3COO)2SbPh3 (246), (4-F-2-CH3C6H3COO)2SbPh3 (247), (5-F-2-CH3C6H3COO)2SbPh3 (248) [121]. В кристаллах (246 и 247) за счет межмолекулярных C–H···O, C–H···F и π–π-стекинг-взаимодействий реализуются супрамолекулярные структуры.
Супрамолекулярные структуры, обусловленные наличием нескольких типов невалентных взаимодействий (C–H…π, C–Cl…π, C–H…Cl), присутствуют и в кристаллах двух соединений [Ph3SbL1]2 (249) и [Ph3SbL2]2 (250) (H2L1 = 5-{[(2-карбоксиметил)метилен]амино}-4-хлорбензойная кислота, H2L2 = 5-{[(2-карбоксиметил)метилен]амино}-2-хлорбензойная кислота), синтезированные из дихлорида трифенилсурьмы, оснований Шиффа и этилата натрия и представляющие собой 24-членные макроциклы симметричного строения, содержащие по два атома сурьмы, связанных между собой мостиковыми основаниями Шиффа (схема 53 ) [122]:
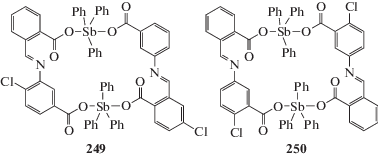
Схема 53 .
Взаимодействие 2,3-дибромбутандиовой кислоты и метоксида натрия с дихлоридом трифенилсурьмы в метаноле приводит к образованию соединения сурьмы 251, в котором два фрагмента Ph3Sb, связанные через мостиковый остаток 2,3-дибромбутандиовой кислоты, содержат еще и терминальные метоксильные заместители (схема 54 ) [123].
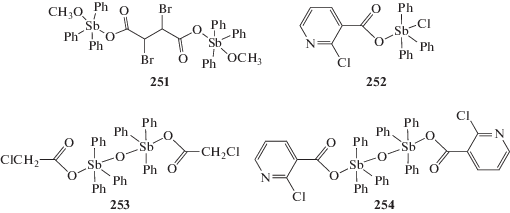
Схема 54 .
Производное сурьмы несимметричного строения – хлоридо(2-хлорникотинато)трифенилсурьму (252) можно получить кипячением (8 ч) смеси эквимолярных количеств 2-хлорникотиновой кислоты, метоксида натрия и дихлорида трифенилсурьмы в толуоле [124]. Атом сурьмы в кристалле (252) имеет идеальную тригонально-бипирамидальную координацию с атомом хлора и карбоксилатным лигандом в аксиальных положениях. Серия бромооксиматов трифенилсурьмы Ph3Sb(Br)(ON=CRR') была получена по аналогичной схеме с использованием дибромида трифенилсурьмы, оксима и метилата натрия, смесь которых перемешивали в растворе хлористого метилена 10–12 ч [125]. По аналогичной реакции из оксо-бис[(хлоро)трифенилсурьмы], метоксида натрия и хлоруксусной или 2-хлорпиридил-3-карбоновой кислоты в толуоле (24 ч, Tкомн) синтезированы с выходом до 85% оксо-бис[(хлорацето)трифенилсурьма] (253) [126] и оксо-бис[(2-хлорпиридинкарбоксилато)трифенилсурьма] (254) соответственно (схема 54 ) [127]. Атомы сурьмы в (253 и 254) имеют тригонально-бипирамидальную конфигурацию с карбоксильным лигандом и мостиковым атомом кислорода в аксиальных положениях.
По этой же схеме из дихлоридов триарилсурьмы, α-оксифенилуксусной кислоты и этоксида натрия в метаноле были синтезированы четырехъядерные производные пятивалентной сурьмы (255–258) (схема 55 ), обладающие противоопухолевыми свойствами [128].
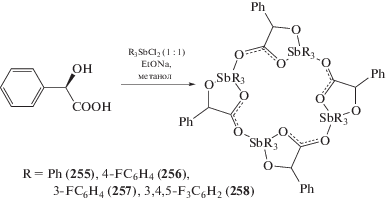
Схема 55 .
Аналогично с помощью калиевых или натриевых солей карбоновых кислот и дибромида триарилсурьмы был получен ряд биологически активных дикарбоксилатов три-п-толилсурьмы (259–266) (схема 56 ) [129].
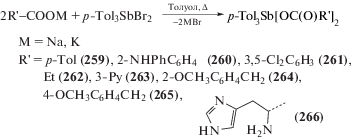
Схема 56 .
Показано, что взаимодействие дигалогенида триарилсурьмы с соединениями, содержащими активный атом водорода, в присутствии амина приводит к замещению атомов галогена и образованию производных сурьмы Ar3SbX2 c высоким выходом. Так, последовательное прибавление к суспензии дибромида триарилсурьмы Ar3SbBr2 (Ar = C6H5, p-Tol, m-Tol, o-Tol, 4-F-C6H4) в толуоле 3-ферроценилакриловой кислоты и триэтиламина с последующим перемешиванием реакционной смеси в течение 24 ч, фильтрованием и удалением растворителя из фильтрата приводило к образованию с высоким выходом дикарбоксилатов триарилсурьмы (267–271) (схема 57 ) [60].
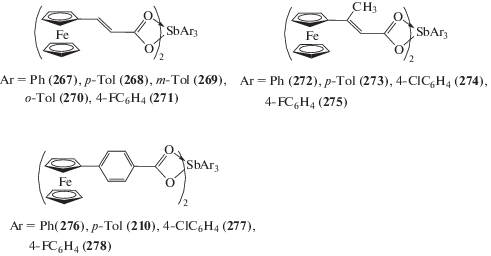
Схема 57 .
По аналогичной методике получены другие дикарбоксилаты триарилсурьмы: (272–275) с ферроценилметилакриловыми и (210, 276–278) с 4-ферроценилбензоатными заместителями [130].
Гетероциклические диакрилаты трифенил- и три(пара-толил)сурьмы (279, 280), синтезированные по указанной схеме, проявляют себя как эффективные средства в борьбе с лейшманиозом и стафилоккоком [131]. По аналогичной схеме, но в растворе тетрагидрофурана, проводили реакцию дибромида трифенилсурьмы с N-фенилглицином, при этом выделяли бис-(N-фенилглицинат) трифенилсурьмы (281) [132]. Молекулы перечисленных дикарбоксилатов триарилсурьмы имеют ожидаемую тригонально-бипирамидальную конфигурацию с несимметричной координацией на атом сурьмы двух атомов кислорода карбоксильных групп. Указанные выше дикарбоксилаты триарилсурьмы проявляют противоопухолевую активность in vitro.
Перемешивание толуольного раствора эквимолярных количеств дихлорида трифенилсурьмы с 8-оксихинолином или 2-пиридинэтанолом в присутствии триэтиламина приводит к образованию октаэдрических производных сурьмы (282 и 283) с выходом до 37%, в которых бидентатные N,O-донорные лиганды занимают у центрального атома металла (КЧ 6) аксиальное и экваториальное положения [133].
Несколько дикарбоксилатов триарилсурьмы [Ar3Sb(OOCR)2] (Ar = Ph, p-Tol; R = 4-NO2C6H4, 2-BrC6H4, CHPh2, CH2CH(i-Pr)NHSO2Ph) (284–291) были получены по реакции замещения с использованием дибромидов трифенил- или три(пара-толил)сурьмы, соответствующей карбоновой кислоты и триэтиламина в толуоле с выходом 78–81% (схема 58 ). Исследована биологическая активность полученных соединений [134]. Показано, что производные трис(пара-толил)сурьмы имеют более выраженную антилейшманиозную активность.
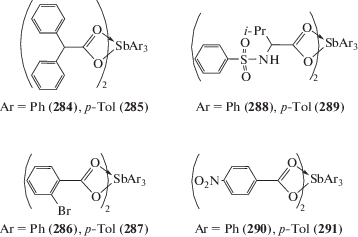
Схема 58 .
Взаимодействие оксидов триарилсурьмы (Ph3SbO)2 и [(2,6-(Me2NCH2)2C6H3)3SbO]2 с кислотой Льюиса B(C6F5)3 сопровождается образованием продуктов присоединения Ph3SbOB(C6F5)3 (292) (cхема 59) и 2,6-(Me2NCH2)2C6H3SbOB(C6F5)3 (293), имеющих чрезвычайно короткие связи Sb–O [135].
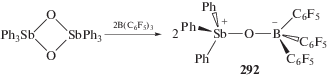
Схема 59 .
Реакция оксида трифенилсурьмы с метанолом приводит к образованию биядерного сольвата (294) (cхема 60) [136]. Взаимодействие полимерной формы оксида трифенилсурьмы с фенилфосфиновой, дифенилфосфиновой, трет-бутилфосфониевой и фенилселеновой кислотами приводит к образованию моно- и биядерных органических производных сурьмы (295–299) (cхема 60) [137].
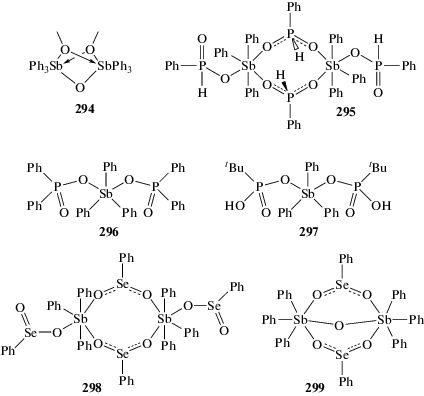
Схема 60 .
Гидролизом дибромида трифенилсурьмы или дибромида три(о-толил)сурьмы в бензоле получены сольваты (Ph3SbBr)2O ⋅ 2PhH (триклинная модификация) (300) и [(2-MeC6H4)3SbBr]2O ⋅ 1/2PhH (301), в которых атомы Sb имеют искаженную тригонально-бипирамидальную координацию с арильными лигандами в экваториальных положениях, мостиковым атомом кислорода и концевым лигандом Br в аксиальных положениях [138]. Из дибромида трис(4-фторфенил)сурьмы и роданида калия в водно-ацетоновом растворе получен оксид [(4-F-C6H4)3SbCNS]2O (302) [139]. Атомы сурьмы в его молекулах имеют искаженную тригонально-бипирамидальную координацию с электроноакцепторными лигандами в аксиальных положениях.
Дихлорид трицимантренилсурьмы при действии гидроксида калия в спирте превращается в дигидроксид трицимантренилсурьмы (303), в котором атомы сурьмы имеют искаженную тригонально-бипирамидальную координацию с гидроксильными группами в аксиальных положениях [140].
Соединения сурьмы мостикового типа μ-оксо-бис[(нитрато)трифенилсурьма] (304) и аддукт Ph2(2-PhC6H4)(NO3)SbOSbPh3(NO3) · (Ph3SbNO3)2O (305) синтезировали из дибромида трифенилсурьмы и азотнокислого серебра в растворах бензола и пара-ксилола, соответственно [141]. Если первый комплекс (304) выделяли сразу из реакционной смеси, то (305) перекристаллизовывали из бензола. Образование в пара-ксилоле комплекса (305), в котором присутствуют два типа молекул: μ-оксо-бис[(нитрато)трифенилсурьма] и μ-оксо[(нитрато)(2-фенилфениленил)дифенилсурьма]-[(нитрато)трифенилсурьма], несомненно вызывает интерес, поскольку в этой реакции имеет место орто-фенилирование фенильного заместителя при атоме Sb(V) (cхема 61).
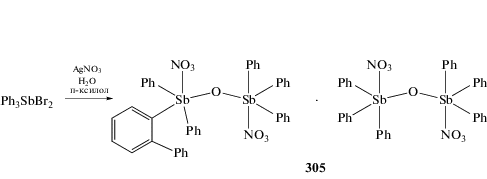
Схема 61 .
Образование последнего комплекса с выходом 48% можно объяснить лишь фенилированием молекул оксонитрата трифенилсурьмы дибромидом трифенилсурьмы в условиях реакции. Возможно, реакция идет через образование активированного комплекса, в состав которого входят две молекулы сурьмаорганического соединения и сольватные молекулы растворителя, превращающегося затем в фенилированный оксонитрат.
Последовательные реакции дихлорида трифенилсурьмы с водой и органосульфонатами серебра в ацетонитриле или тетрагидрофуране приводят к образованию термически устойчивых комплексов сурьмы (306 и 307) ионного строения (схема 62 ) [142].
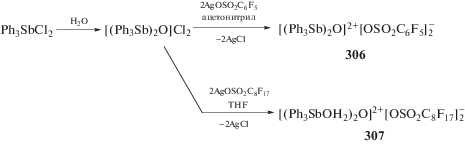
Схема 62 .
Взаимодействие дихлорида трифенилсурьмы с трифлатом серебра приводит к образованию ковалентного бис-(трифторметансульфоната) трифенилсурьмы (308) [143], который при добавлении таких нейтральных донорных лигандов как бипиридил или оксид трифенилфосфина, превращается в ионные продукты (309–311) с катионами [Ph3SbL2]2+ (схема 63 ).

Схема 63 .
Гетероциклическое соединение сурьмы (312), содержащее пятичленный цикл с чередующимися атомами азота, серы и сурьмы, получено из тиотритиазилхлорида и дихлорида трифенилсурьмы в жидком аммиаке (схема 64 ) [144].
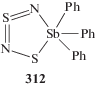
Схема 64 .
Связи атомов серы с атомом азота во фрагменте –S–N=S=N– неравноценны (две более короткие, одна более длинная).
Последовательная обработка дибромида трифенилсурьмы 1,8-дилитийнафталином, комплексом золота AuCl(Тht) (Тht = тетрагидротиофен) и трифенилфосфином в тетрагидрофуране приводило к образованию с выходом 37% золотоорганического соединения сурьмы (313) [145], в котором координационная связь Au⋅⋅⋅Sb (2.77 Å) близка к сумме ковалентных радиусов атомов золота и сурьмы (2.65 Å [63]). При действии фторида тетрабутиламмония на комплекс (313) в хлороформе образуется ионный комплекс (314) с анионом, имеющим две резонансные формы (схема 65 ).
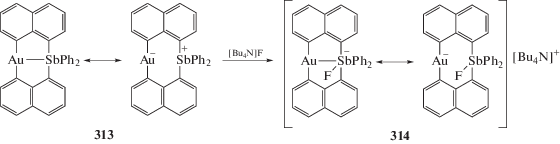
Схема 65 .
Из трис(2-дифенилфосфинофенил)стибина и комплекса AuCl(Тht) было получено другое биядерное Sb,Au-содержащее соединение (315), которое при действии дихлорида фенилиодония превращается в соответствующий дихлорид (316) (схема 66 ). Взаимодействие последнего комплекса с иодистым натрием сопровождается восстановлением сурьмы и образованием Au-содержащего моноиодида (317) [146].
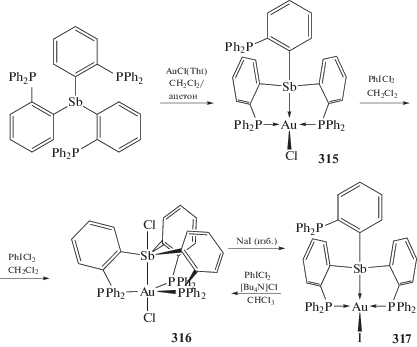
Схема 66 .
Весьма интересны реакции литийорганических производных первичных аминов с дихлоридом трифенилсурьмы и трихлоридом дифенилсурьмы (cхема 67) [147]. Взаимодействие амида лития с дихлоридом трифенилсурьмы (2 : 1, тетрагидрофуран) приводило к образованию циклодистибазанов (318), в которых два фрагмента Ph3Sb связаны между собой через два мостиковых атома азота NR-группы. Продуктами подобных реакций амида лития с трихлоридом дифенилсурьмы (3 : 1, тетрагидрофуран) являлись аналогичные димеры (319) с двумя терминальными фрагментами Ph2SbNHR, но при изменении мольного соотношения исходных реагентов на 2 : 1 образуется димер (320) с терминальными группами Ph2SbCl.
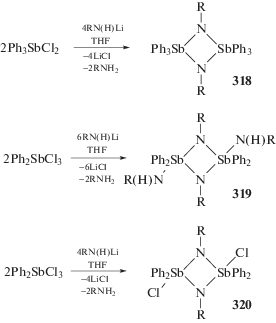
Схема 67 .
Действие ариллития, содержащего в арильном заместителе группировку i-Pr2P, на дихлорид трифенилсурьмы приводит к образованию хлорида трифениларилсурьмы Ph3SbArCl (10) (cхема 1) [4]. Продуктом реакции эквимолярных количеств ариллития и трихлорида дифенилсурьмы является дихлорид дифениларилсурьмы (11) (cхема 1).
Трифлат 1-дифенилфосфинонафтил-8-трифенилстибония (321) получен из 1-литий-8-дифенилфосфинонафталина и дибромида трифенилсурьмы с последующим отщеплением атома брома трифлатом серебра (cхема 68). Подобным способом получен и аналог (322) без фосфиновой группы.
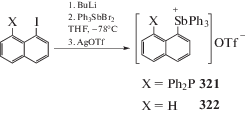
Схема 68 .
Cоединение 321, стабильное к действию кислорода и воды, является катализатором реакции альдегидов с триэтилсиланом, приводящей к синтезу симметричных эфиров (cхема 69). В то же время трифлат 321 катализирует реакцию альдольной конденсации. Понижение температуры реакции до −10°С приводит к образованию 1,3,5-триоксанов с выходом 50–90% [148].
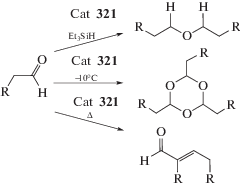
Схема 69 .
Случай орто-металлирования одного из фенильных колец исходного литийорганического соединения, полученного из фениллития и бензонитрила, наблюдали при взаимодействии дихлорида трифенилсурьмы с имидом лития LiN=CPh2 в тетрагидрофуране (cхема 70) [149].
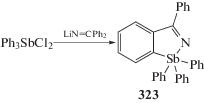
Схема 70 .
Из продукта гидролиза дихлорида трифенилсурьмы и 1,1,2,3,3-пентаметилтриметиленфосфиновой кислоты в бензоле получено биядерное производное сурьмы[(Ph3Sb)2(μ-O)(μ-cycPO2)2] (324) (cycPO2 = 1,1,2,3,3-пентаметилтриметиленфосфинат), которое при перекристаллизации из водного ацетонитрила превращается в девятиядерный комплекс сурьмы [(Ph2Sb)2(PhSb)7(μ-O)11(μ3-O)3(μ-OH)2(μ-cycPO2)2(cycPO2)2(H2O)2] · 2CH3CN · · H2O (325) (cхема 71) [150].
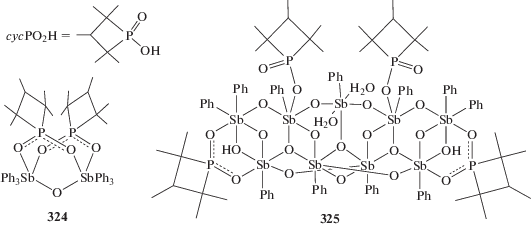
Схема 71 .
Стабилизирующий эффект дихлорида трифенилсурьмы проявляется в кристалле его аддукта (326) с бис-[4-(5-бромпиридин-2-ил)-1,2,3,5-дитиадиазолил-N,N')-трис(гексафторацетилацетонато-O,O')церием], включающим невалентные взаимодействия: контакты S⋯S, Cl⋯S и Br⋯Br (cхема 72) [151].
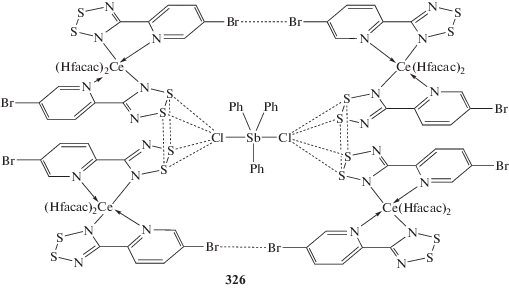
Схема 72 .
Окисление производных Ar3Sb органическими и неорганическими окислителями. Важным способом синтеза соединений сурьмы(V) является их получение по реакции окислительного присоединения. Так, стибины при действии хлористого сульфурила превращаются в производные пятивалентной сурьмы (327–329) (схема 73 ) [152].
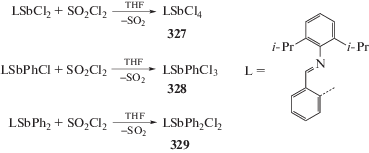
Схема 73 .
Известная реакция окислительного присоединения, когда из триарилстибина и алкилгалогенида образуется галогенид алкилтриарилстибония [74], в некоторых случаях протекает с образованием иных продуктов присоединения, например при метилировании трис(2-диметилстибинфенилметил)амина иодистым метилом (схема 74 ), когда продуктом взаимодействия является гетероциклическое соединение пятикоординированной сурьмы (330), в котором присутствует аномально короткая связь Sb···N (2.565(4) Å) [153].
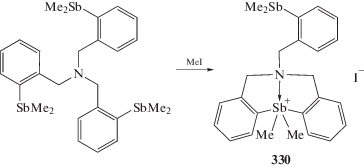
Схема 74 .
Реакция квартенизации триарилстибина, содержащего в одном из фенильных заместителей в положении 2 Mes2B-группу, протекает с образованием ионного трифторметансульфоната дифениларилметилстибония (331) (схема 75 ) [154].
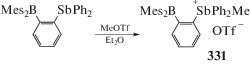
Схема 75 .
Исследуя реакцию (331) c фтор-(2-димезитилборилфенил)-метилдифенилфосфораном, авторы установили образование более стабильного производного (332) (схема 76 ), в котором наблюдается аномально короткая связь Sb−F (2.450(2) Å), что значительно меньше связи Р−F (2.666(2) Å) в аналогичном исходном фосфоране несмотря на меньший размер атома фосфора.
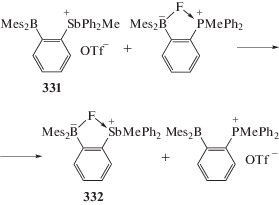
Схема 76 .
Обычная схема реакции квартенизации наблюдается при обработке соединения( 333) метилтрифлатом (схема 77 ) [155]. Образующийся продукт присоединения (334) при действии KF превращается во фторид метилдифениларилсурьмы (335).
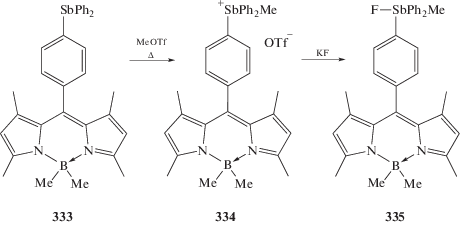
Схема 77 .
Дистибониевые соли (336 и 337) могут быть синтезированы обработкой o-фенилен-бис-(дифенилстибина) метилтрифторметансульфонатом (MeOTf) и тетрафторборатом триметилоксония ([Me3O]+[BF4]–) соответственно (схема 78 ) [156].
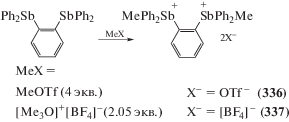
Схема 78 .
Еще в начале 1900-х гг. было показано, что триарилстибины легко и количественно окисляются галогенами, галогенидами двухвалентной меди и другими производными галогенов в органических растворителях [74]. В настоящее время дигалогениды триарилсурьмы получают этим же способом.
Установлено, что 2,3,6,7,14,15-гексаметил-9-фосфа-10-стибатриптицен и 2,3,6,7,14,15-гексаметил-9,10-дистибатриптицен окисляются бромом до соответствующих тетрабромидов (338 и 339) (схема 79 ), последний при перекристаллизации из тетрагидрофурана превращается в сольват (340), в котором две молекулы THF координируются на атомы сурьмы [157].
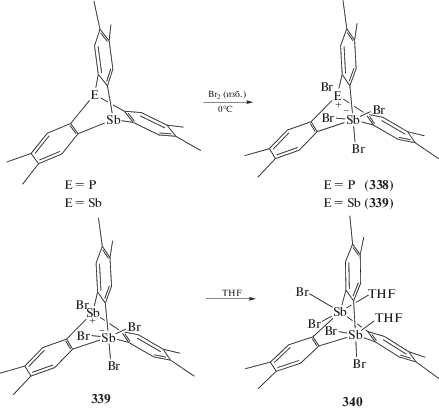
Схема 79 .
Показано, что трис(4-N,N-диметиламинофенил)сурьма окисляется дихлоридом меди в спиртово-ацетоновом растворе при комнатной температуре до дихлорида трис(4-N,N-диметиламинофенил)сурьмы (341) (схема 80 ), выделенного из реакционной смеси с выходом 60% в виде бесцветных чувствительных к свету кристаллов [101].
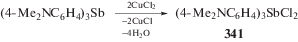
Схема 80 .
Взаимодействием трис(4-фторфенил)сурьмы с дибромидом меди в ацетоне синтезирован дибромид трис(4-фторфенил)сурьмы (4-F-C6H4)3SbBr2 (342), перекристаллизация которого из этанола привела к образованию аддукта (4-FC6H4)3SbBr2 ⋅ ⋅ [(4-FC6H4)3SbBr]2O (343) [139].
Аналогичное бромирование трицимантренилсурьмы в хлороформе приводило к образованию дибромида трицимантренилсурьмы (344) [140].
Дихлорид трис(4-бромфенил)сурьмы (345) был получен последовательным хлорированием триарилсурьмы в петролейном эфире, удалением растворителя и перекристаллизацией остатка из хлористого метилена с выходом 43% [158].
Комплекс платины, содержащий в лиганде 2,2'-Вipy группу Ph2Sb, при обработке дихлоридом иодбензола в растворе DMSO превращается в соответствующий платина-содержащий дихлорид триарилсурьмы (346) (схема 81 ), катализирующий реакцию присоединения мезитилена с этиловым эфиром ацетиленкарбоновой кислоты [159].
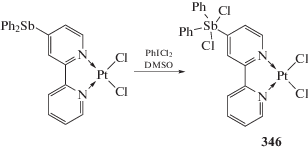
Схема 81 .
Отметим, что известны и другие реакции алкинов, катализируемые соединениями платины, например окисление толана кислородом воздуха, в результате чего образуется дифенилэтандион-1,2 с выходом 91% [160].
Взаимодействие комплекса (o-(Ph2P)−C6H4)3SbCl)Pt(Cl) (347), полученного из стибина [(o-(Ph2P)C6H4)3Sb и (Et2S)2PtCl2], с фторидами приводит к образованию фторстиборанильного комплекса ((o-(Ph2P)C6H4)3SbF)Pt(Cl) (348) (схема 82 ) – аналога комплекса палладия (123). Из (347) по реакциям обмена получены [((o‑(Ph2P)C6H4)3Sb)Pt(CyNC)]2+$[{\text{Sb}}{{{\text{F}}}_{{\text{6}}}}]_{{\text{2}}}^{--}$ (349), [((o-(Ph2P)C6H4)3SbF)Pt(CyNC)]+[SbF6]– (350) и ((o-(Ph2P)C6H4)3SbF2)Pt(CyNC) (351) (Cy = циклогексил), в которых фторидные лиганды связаны с атомом сурьмы. Структурные исследования этой серии показывают, что связь Sb–Pt удлиняется при последовательной координации фторидов у атома сурьмы, что согласуется с ослаблением взаимодействия Sb–Pt [161].
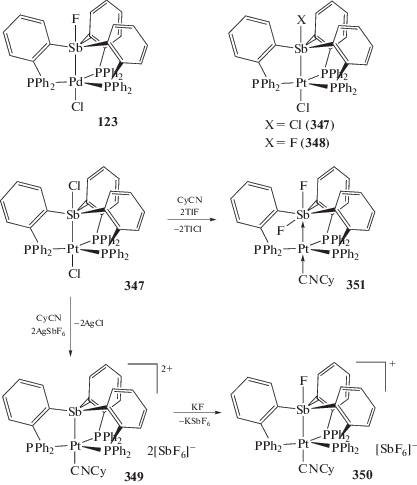
Схема 82 .
О контролируемом перемещении фенильной группы в биядерных Au–Sb комплексах (352–357) (схема 83 ), осуществляемом при действии на них различных реагентов, сообщали авторы работы [162]. Исходное соединение ((o-(i-Pr2P)C6H4)2SbPhCl2)Au(Cl) (352) для всей серии превращений синтезировали по реакции окислительного присоединения, обрабатывая Au–Sb биядерный комплекс ((o-(i-Pr2P)−C6H4)2SbPh)Au(Cl) хлористым фенилиодонием.
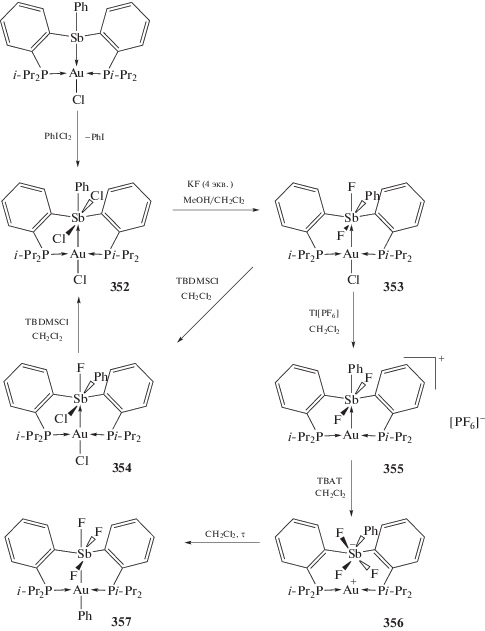
Схема 83 .
Cинтез и реакционная способность аналогичного Au–Sb биядерного комплекса ((o-(Ph2P)–C6H4)2SbPh)Au(Cl) с дифенилфосфиновыми заместителями в орто-положениях двух арильных групп исследована в [163, 164]. Найдено, что производное трехвалентной сурьмы реагирует с орто-хлоранилом при комнатной температуре в растворе хлористого метилена, при этом с выходом 60% образуется комплекс шестикоординированной сурьмы (358), который, в свою очередь, при действии гексафторфосфата таллия в ацетонитриле превращается в ионный комплекс (359), содержащий трехкоординированный атом золота (схема 84 ).
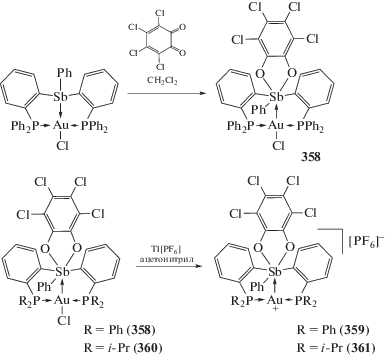
Схема 84 .
К образованию подобного ионного комплекса (361) приводит и аналогичная реакция диизопропилфосфинового производного (360) с гексафторфосфатом таллия (схема 84 ).
Комплекс ((o-(Ph2P)C6H4)2SbCl3)Au(Cl) (362), полученный аналогично комплексу (352), но при действии на прекурсор ((o-(Ph2P)C6H4)2SbCl)Au(Cl) фторида тетрабутиламмония вместо хлористого фенилиодония, превращается в соответствующий трифторид ((o-(Ph2P)C6H4)2SbF3)Au(Cl) (363), схожий по строению с комплексом ((o-(i-Pr2P)–C6H4)2SbF3)Au(Ph) (357) [165].
Окисление комплекса ((o-(i-Pr2P)C6H4)2SbCl)- Au(Cl) – производного хлордиарилстибина c хлоридом золота, содержащего в орто-положении арильного заместителя i-Pr2P-группировки, – орто-хлоранилом также приводит к образованию производного пятивалентной сурьмы (364) – аналога комплекса (358) (схема 85 ) [166].
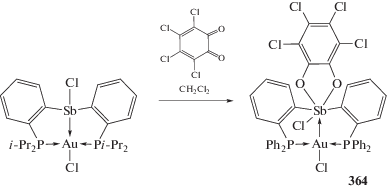
Схема 85 .
Аналогично окислением соответствующего стибина 3,5-ди-трет-бутил-о-бензохиноном (3,5-DBBQ) и о-хлоранилом (Cl4Q) были получены производные пятивалентной сурьмы (365 и 366) (схема 86 ) (перемешивание 15–30 мин, хлористый метилен) [167].
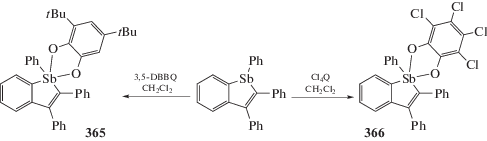
Схема 86 .
Окисление трифенилсурьмы N-хлоротрифенилфосфоранимином в растворе дихлорметана приводит к образованию продукта присоединения − хлорида трифенилфосфиниминотрифенилстибония [Ph3PNSbPh3]Cl (367), в котором атомы фосфора и сурьмы имеют тетраэдрическую и тригонально-бипирамидальную координацию соответственно [168].
При изучении реакций три-пара-толилсурьмы с трифторуксусной, трихлоруксусной, иодуксусной, толуолсульфоновой кислотами и 2,4,6-тринитрофенолом (НХ) в растворе толуола в присутствии или отсутствие кислорода воздуха было показано, что взаимодействие указанных реагентов протекает по двум конкурирующим между собой направлениям (схема 87 ) с образованием производных пятивалентной сурьмы общей формулы p-Tol3SbХ2, где Х – остаток карбоновой, толуолсульфоновой кислот или 2,4,6-тринитрофенола [169].
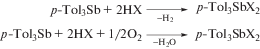
Схема 87 .
Бис-(трифторметансульфонат) трифенилсурьмы, полученный по реакции обмена из трифлата серебра и дихлорида трифенилсурьмы в хлористом метилене, при действии таких электронодонорных лигандов, как фосфины, превращается в ионный комплекс с элиминированием трифенилстибина (схема 88 ) [51].
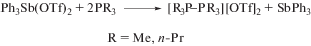
Схема 88 .
Аддукт трифенилсурьмы с азотнокислым серебром использовали для получения дикарбоксилатов трифенилсурьмы (схема 89 ) [170].
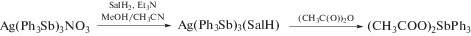
Схема 89 .
Окислительный метод синтеза арильных производных сурьмы(V) из триарильных соединений сурьмы, кислоты НХ и пероксида. В основе одного из эффективных способов синтеза соединений пятивалентной сурьмы лежит реакция окислительного присоединения, когда из триарильных соединений сурьмы, кислоты НХ и пероксида получают арильные производные пятивалентной сурьмы Ar3SbX2. Эта реакция впервые была осуществлена на примере синтеза диацетата трифенилсурьмы из трифенилсурьмы, уксусной кислоты и пероксида водорода [171]. Именно по этой схеме были синтезированы дикротонат трифенилсурьмы Ph3Sb(O2CCH=CHCH3)2 (368) [172], бис(1-адамантанкарбоксилат) трифенилсурьмы (369) [173], бис(бромацетат) трис(2-метокси-5-бромфенил)сурьмы (370) [174], бис(2-нитробензоат) трифенилсурьмы (371) [175], бис(1-адамантанкарбоксилат) три-м-толилсурьмы (372) [176], бис(фенилкарборанилкарбоксилат) три-р-толилсурьмы (373) [177], бис(2-метилкарборанилкарбоксилат) трифенилсурьмы (374) [178], бис(2-метоксибензоат) трифенилсурьмы (375) [179], бис(циклопропанкарбоксилат) трис(5-бром-2-метоксифенил)сурьмы (376) [180], [Ph3Sb(O2CCH=CHC6H4NO2-m)2 · · C6H6] (377) [181], бис(салицилальдоксимат) трифенилсурьмы (378) [26], бис(2-гидрокси-5-бромбензальдоксимат) трифенилсурьмы (56) [31], бис(циклогексаноксимат) три-пара-толилсурьмы (379) [31], дибензоат трис(4-N,N-диметиламинофенил)сурьмы (205) [101], бис(2-метилбензоат) трис(4-N,N-диметиламинофенил)сурьмы (380) [182], бис(4-метилбензоат) трис(4-N,N-диметиламинофенил)сурьмы (381) [183], бис(2,4,6-трибромфеноксид) трис(4-N,N-диметиламинофенил)сурьмы (382) [182], бис(2-нитробензоат) трис(5-бром-2-метоксифенил)сурьмы (383) [184], бис(хлорацетат) трис(5-бром-2-метоксифенил)сурьмы (384), бис(бромацетат) трис(5-бром-2-метоксифенил)сурьмы (385) и бис(иодацетат) трис-(5-бром-2-метоксифенил)сурьмы (386) [185], бис-(4-нитрофенилацетат) трис(5-бром-2-метоксифенил)сурьмы (387), бис(2-метоксибензоат) трис(5-бром-2-метоксифенил)сурьмы (388) и бис(фенилпропиолат) трис(5-бром-2-метоксифенил)сурьмы (389) [186], бис(4-оксибензолсульфонат) трифенилсурьмы (390) [187]. Сольват бис[(E)-3-(4-метоксифенил)про-2-еноато]трифенилсурьмы (391) с бензолом получали окислением трифенилсурьмы пероксидом водорода в присутствии фенилкоричной кислоты в тетрагидрофуране с последующей перекристаллизацией из смеси бензол–гексан [188]. Реже в реакции окислительного присоединения применяют смесь растворителей, например окисление трифенилсурьмы пероксидом водорода в присутствии метакриловой кислоты проводят в смеси изопропанол–диэтиловый эфир (4 : 1 объем.), при этом выход диметакрилата трифенилсурьмы достигает 79% [189].
Серия α-гидроксикарбоксилатных комплексов трифенилсурьмы (392–395) синтезирована по реакции окислительного присоединения из трифенилсурьмы, карбоновой кислоты и гидроперекиси третичного бутила (схема 90 ) [190].
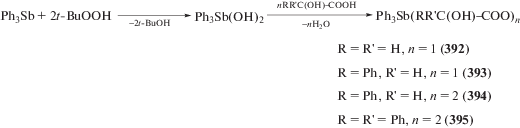
Схема 90 .
При растворении в таком полярном растворителе, как диметилсульфоксид, дикарбоксилат трифенилсурьмы (394) превращается в более устойчивый комплекс (396) (cхема 91).
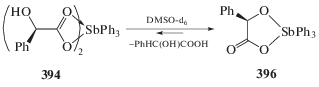
Схема 91 .
Отметим, что, несмотря на наличие двух карбоксильных групп в орто-фталевой кислоте, ее взаимодействие с трифенилсурьмой в присутствии пероксида водорода протекает по классической схеме реакции окислительного присоединения [191]. Особенностью молекулярной структуры дифталата трифенилсурьмы является отсутствие внутримолекулярной водородной связи, характерной для орто-фталевой кислоты.
Бис-μ-[(метилендициклопентанон-2,2'-диоксимат) трифенилсурьмы] (55), ранее полученный из пентафенилсурьмы и диоксима при нагревании [30], по реакции окислительного присоединения образуется практически с количественным выходом [192], как и бис(фенилметансульфонат) трифенилсурьмы (397) [193]. Eсли образование (397) по указанной схеме протекало с выходом 91%, то выход бис(2-нафталинсульфоната) трифенилсурьмы (398) и бис(1-нафталинсульфоната) трифенилсурьмы (399) достигал 46 и 25% соответственно. Bозможно, это связано с увеличением объема органического фрагмента в аренсульфонатном лиганде. Реакция между трифенилсурьмой и хлорангидридом фенилметансульфоновой кислоты в присутствии пероксида водорода протекает с образованием сурьмаорганического цвиттер-иона (400), который после перекристаллизации из толуола выделяли в виде бесцветных кристаллов с выходом 91% (схема 92 ) [193].
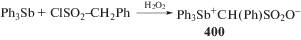
Схема 92 .
По данным РСА, атомы сурьмы в (400) имеют искаженную тетраэдрическую координацию, причем две молекулы (400) связаны между собой водородными связями посредством атома водорода метиновой группы и атома кислорода алкансульфонатного заместителя, который координирован с атомом кислорода второй молекулы (расстояние Sb–O(1') составляет 3.019(2) Å). Кроме того, в каждой молекуле (400) расстояние между атомом кислорода О(3) и атомом Sb равно 2.988(2) Å. Длина одной из связей S–O в сульфогруппе (S–O(2) 1.433(2) Å) значительно отличается от двух других (S–O(1,3) 1.446(2), 1.454(2) Å соответственно, что можно объяснить дополнительным взаимодействием атомов O(1,3) с атомами Sb и Н метиновой группы.
Бис(4-иодфенокси)трифенилсурьма (401) гладко образуется по схеме реакции окислительного присоединения из трифенилстибина, фенола и пероксида водорода в эфире [83].
При взаимодействии эквимолярных количеств триарилсурьмы, кислоты НХ и пероксида водорода в эфире образуются соединения сурьмы мостикового типа (Ar3SbX)2O (402), что, например, наблюдается в реакциях три(орто-толил)сурьмы с 2-гидрокси-5-бромбензальоксимом и фурфуральоксимом [194]. В отсутствие оксима в реакционной смеси имеет место окисление исходной триарилсурьмы до димерной формы оксида триарилсурьмы.
В случае подобной реакции трифенилстибина с салициловой кислотой с выходом 40% образуется мостиковое соединение сурьмы (403), обладающее противоопухолевой активностью [195].
Взаимодействием трифенилсурьмы с 6,6,6-трифтор-2,2-диметилгександионом-3,5 в присутствии пероксида водорода в эфире синтезирован комплекс (6,6,6-трифтор-3-гексанон-5,5-диолато)трифенилсурьма (404) (схема 93 ) [196].
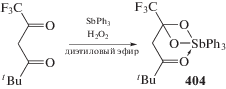
Схема 93 .
По данным РСА, атом Sb в комплексе имеет искаженную октаэдрическую координацию. Расстояния Sb–C изменяются в интервале 2.123(4)–2.131(4) A, длины связей Sb–О(1,2,3) составляют 2.052(2), 2.047(3), 2.669(2) A соответственно.
Установлено, что реакции эквимоляных количеств 2-гидроксибензальдоксима с трис(пара-толил)сурьмой, трис(3-фторфенил)сурьмой и трис(4-фторфенил)сурьмой в присутствии пероксида водорода в эфире приводят к образованию биядерных гетероциклических соединений сурьмы (405–407) [197].
Эффективным методом синтеза арильных производных пятивалентной сурьмы можно считать взаимодействие триарилсурьмы с органическими пероксидами в присутствии кислоты НХ. Показано, что в качестве кислот могут выступать спирты, фенолы, оксимы, карбоновые, сульфоновые, неорганические и другие ОН- и СН-кислоты. Роль окислителя в реакциях выполняет, как правило, гидропероксид третичного бутила. Указанный метод обладает рядом неоспоримых преимуществ по сравнению с другими способами: протекает в одну стадию при комнатной температуре с высоким выходом целевых продуктов. Так, из три-орто-толилсурьмы и циклогексаноноксима в присутствии трет-бутилгидропероксида (мольное соотношение 1 : 2 : 1) был синтезирован бис(циклогексаноноксимат) три-о-толилсурьмы (o-Tol)3Sb(ON=C6H10-цикло)2 (408). При мольном соотношении реагентов 1 : 1 : 1 из реакционной смеси выделяли аддукт (o-Tol)3Sb(ON=C6H10-цикло)2 · · [(o-Tol)3SbO]2 (409) [198].
Аналогично получены бис(2-бромбензальдоксимат) трифенилсурьмы (410), бис(3-нитробензальдоксимат) трифенилсурьмы (411), бис(3-бромбензальдоксимат) трифенилсурьмы (412), бис-(5-нитрофурфуральдоксимат) трифенилсурьмы (413) [199]. Другие диоксиматы триарилсурьмы, содержащие заместители в арильных кольцах (бис(2-оксибензальдоксимат) трис(пара-толил)сурьмы (414), бис(2-нитробензальдоксимат) трис(пара-толил)сурьмы (415), бис(2-бромбензальдоксимат) трис(пара-толил)сурьмы (416), бис(2-оксибензальдоксимат) трис(3-фторфенил)сурьмы (417), бис(2-бромбензальдоксимат) трис(4-фторфенил)сурьмы (418) и бис(2-нитробензальдоксимат) трис(4-фторфенил)сурьмы (419) [200]; бис(5-нитрофурфуральдоксимат) триc-(орто-толил)сурьмы (420) и бис(тиофен-2-карбальдоксимат) триc(орто-толил)сурьмы (421) [201]; бис[4-N,N-диметиламино)бензальдоксимат] триc(орто-толил)сурьмы (422), бис-(ацетофеноноксимат) триc-орто-толил)сурьмы (423), бис(фурфуральдоксимат) трис-(мета-толил)сурьмы (424) [202]; бис(2-нитробензальдоксимат) трис-(мета-толил)сурьмы (425) и бис(2-нитробензальдоксимат) трис(орто-толил)сурьмы (426) [203], бис(5-нитрофурфуральдоксимат) трис(мета-толил)сурьмы (427) [204]), были получены аналогично.
Взаимодействием трис(пара-толил)сурьмы с фенолами в присутствии трет-бутилгидропероксида (мольное соотношение 1 : 2 : 1) в диэтиловом эфире синтезированы диароксиды трис(пара-толил)сурьмы (p-Tol3Sb(OAr)2, Ar = С6Н3Br2-2,4 (428), С6Н2Br3-2,4,6 (429) [205], С6Н4Br-4 (430) [206], С6Н4(NO2-4) (431) [206].
Диароксиды трис(4-фторфенил)сурьмы (4-F-C6H4)3Sb(OAr)2 (Ar = С6H3Br2-2,4 (432), C6H4Cl-4 (433), C6H4Br-4 (434)) [207], С6Н4(NO2-4) (435), С6F5 (436), С6Cl5 (437) [206] получены из триарилсурьмы, фенолов и гидропероксида третичного бутила в эфире с выходом 70–91%. По этой же схеме были синтезированы бис(2,6-дибром-4-метилфеноксид) трифенилсурьмы (438) [9], бис-(4-бромфеноксид) трифенилсурьмы (439) [208] и диароксиды трис(3-фторфенил)сурьмы (3-F-C6H4)3Sb(OAr)2 (Ar = C6H3Br2-2,4 (440), OC6Cl5 (441)) [209]. По данным РСА, атомы сурьмы в полученных соединениях имеют искаженную координацию тригональной бипирамиды с кислородсодержащими заместителями в аксиальных положениях. Однако трис(пара-толил)сурьма в подобных условиях реагирует с 2,4-динитрофенолом, независимо от соотношения исходных реагентов, с образованием только соединения мостикового типа (p-Tol3SbOC6H3(NO2)2-2,5]2O (442) [210], что, видимо, обусловлено стерическими затруднениями.
Схема реакции окислительного присоединения не изменяется при добавлении трет-бутилгидропероксида в раствор трифенилсурьмы и 4-нитрофенола в гептане [87]. Взаимодействие триарилсурьмы с карбоновыми кислотами в эфире в присутствии трет-бутилгидропероксида приводит к образованию дикарбоксилатов трифенилсурьмы: бис(фенилпропиолат) трифенилсурьмы (443) [13], бис(4-оксибензоат) трифенилсурьмы (444) [14], бис(пропиолат) трифенилсурьмы (445) [22], бис(иодацетат) трис(4-фторфенил)сурьмы (446) и бис-(пентафторбензоат) трис(4-фторфенил)сурьмы (447) [211], бис(1-адамантанкарбоксилат) трис(4-фторфенил)сурьмы (448) и бис(циклопропанкарбоксилат) трис(4-фторфенил)сурьмы (449) [212], бис(хлорацетат) трис(4-фторфенил)сурьмы (450), бис-(4-нитрофенилацетат) трис-(4-фторфенил)сурьмы (451) и дибензоат трис(4-фторфенил)сурьмы (452) [213], дикарбоксилаты трис(3-фторфенил)сурьмы (453) [214, 215], диакрилаты трифенилсурьмы (454) [216, 217].
По аналогичной схеме протекают реакции три(мета-толил)сурьмы с бензолсульфоновой кислотой [218], бензойной кислотой [219] и три(пара-толил)сурьмы с 3,4-диметилбензолсульфоновой кислотой [220].
При использовании в подобных реакциях донорных лигандов возможно образование донорно-акцепторных комплексов сурьмы, например при взаимодействии триарилсурьмы с этиленгликолем или пирокатехином в бензоле в присутствии диметилсульфоксида [221].
Отметим, что в случае взаимодействия эквимолярных количеств исходных реагентов реакции окислительного присоединения протекают с образованием соединений мостикового типа. Так, взаимодействие трифенилсурьмы с пропиоловой кислотой в присутствии пероксида водорода (мольное соотношение 1 : 1 : 1) в диэтиловом эфире приводит к образованию μ2-оксо-бис[(пропиолато)трифенилсурьмы] [Ph3SbOC(O)C≡СН]2O (455) [22].
Производные μ2-оксо-бис[(арокси)триарилсурьмы] общей формулы [Ar3Sb(OAr’)]2O (Ar = = Ph, Ar' = C6H2Cl3-2,4,6 (456), C6H2Br2-2,6-(t-Bu)-4 (457); Ar = p-Tol, Ar’ = C6H2(NO2)3-2,4,6 (458) [222] и Ar = p-Tol, Ar’ = C6H2Br3-2,4,6 (459), C6Cl5 (460), C6H3(NO2)2-2,4 (461) [223] были также получены по аналогичной схеме.
Окисление трис-(5-бром-2-метоксифенил)сурьмы эквимолярным количеством трет-бутилгидропероксида в растворе тетрагидрофурана приводит к образованию бис[μ2-оксо-трис-(5-бром-2-метоксифенил)сурьмы] (462), в котором атомы металла имеют тригонально-бипирамидальную координацию с атомами кислорода в аксиальных положениях [224].
В некоторых случаях вместо целевого вещества реакции окислительного присоединения из реакционной смеси выделяли неожидаемые продукты. Так, взаимодействием трис-(5-бром-2-метоксифенил)сурьмы с 2-оксибензальдоксимом в присутствии пероксида водорода с выходом 94% синтезирована бис-(µ3-2-оксибензальдоксимато-О,О',N)-(µ2-оксо)-бис-(5-бром-2-метоксифенил)сурьма (463) (схема 94 ), в которой реализуются различные способы координации двух донорных атомов (кислорода и азота) оксимного лиганда [225].
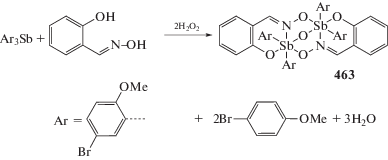
Схема 94 .
В молекуле комплекса связь атома сурьмы с лигандом осуществляется через атом кислорода (Sb–О 2.0768(11) Å), внутримолекулярные расстояния Sb···N равны 2.882(14) Å, гидрокси-группы в ароматическом кольце оксиматного лиганда участвуют в образовании внутримолекулярных водородных связей.
Реакции окислительного присоединения с участием триарилсурьмы, пероксида водорода и ацетоксима или ацетофеноноксима в диоксане сопровождались образованием сольватов тетраядерных сурьмаорганических перекисей (464, 465) с диоксаном (схема 95 ), кристаллизующиеся с различным содержанием молекул растворителя в кристаллической ячейке (1.5 и 6) [226].
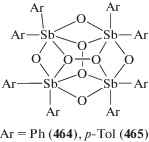
Схема 95 .
Отметим, что строение подобного соединения (464) в виде сольвата с хлороформом ранее было описано в [227].
Окислительный метод синтеза органических производных сурьмы(V) из триорганилсурьмы и орто-хинонов. Впервые о реакции окислительного присоединения между триарилсурьмой и орто-бензохиноном, приводящей к образованию 3,4,5,6-тетрахлоркатехолата трифенилсурьмы(V) (466), сообщалось в работе Холмса с соавторами [228]. Несколько позже было показано, что по аналогичной схеме реагируют с триорганилсурьмой различные замещенные о-бензохиноны и о-иминобензохиноны [229–243]. Показано, что некоторые катехолаты общей формулы (Cat)SbAr3 (467), содержащие электронодонорные группы, и о-амидофеноляты трифенилсурьмы(V) (AP)SbAr3 (468) обратимо реагируют с молекулярным кислородом с образованием циклических эндопероксидных комплексов типов LCat(OO)SbAr3 (469) и LAP(OO)SbPh3 (470), содержащих пятичленный триоксастиболановый цикл (cхема 96) [224–252 ].
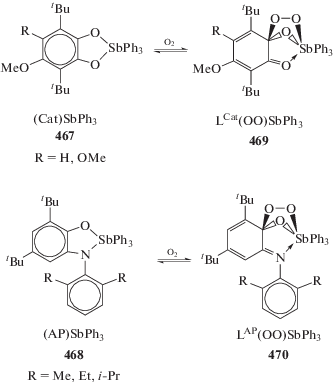
Схема 96 .
Предложен механизм взаимодействия комплексов с молекулярным кислородом [244, 246, 247]. Установлено, что одной из ключевых стадий процесса является одноэлектронное окисление дианионного лиганда в анион-радикальный (cхема 97).
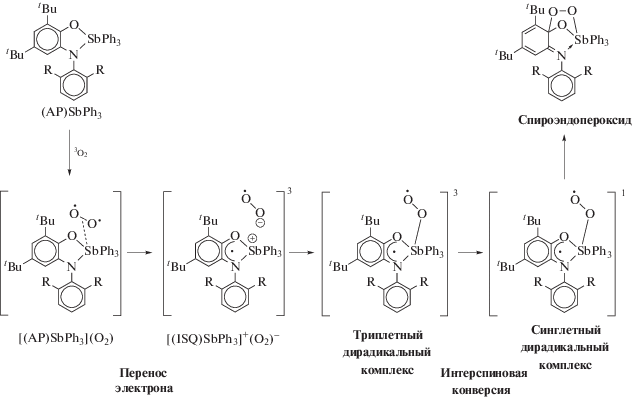
Схема 97 .
На данной стадии предполагается процесс одноэлектронного окисления лиганда (o-амидофенолятного, катехолатного или фенантрен-9,10-диолатного) молекулярным кислородом, при этом образуется ионная пара, состоящая из молекулярного сурьма-содержащего катиона [(ISQ)SbPh3]+ и супероксид-аниона. Далее ионная пара рекомбинирует с образованием триплетного дирадикального комплекса, имеющего o-иминобензосемихиноновый (или o-семихиноновый) и суперпероксидный лиганды. Последующий процесс – интерспиновая конверсия триплетного дирадикального комплекса в синглетный. Известно, что процесс интерспиновой конверсии триплетного состояния в синглетное облегчается в присутствии тяжелых атомов. В данном случае таким атомом является атом сурьмы, имеющий большое значение константы спин-орбитального взаимодействия. Последующая рекомбинация синглетной дирадикальной пары приводит к образованию эндопероксида.
Установлены и детально исследованы факторы, влияющие на способность данного класса комплексов к обратимому присоединению молекулярного кислорода [253–257]. Показано, что введение акцепторных заместителей в редокс-активный катехолатный лиганд повышает окислительный потенциал комплекса и ведет к его инертности по отношению к кислороду. Электронодонорные заместители, наоборот, понижают окислительный потенциал и увеличивают активность соответствующего комплекса в реакции с молекулярным кислородом.
Установлено, что варьирование заместителей при центральном атоме сурьмы, несмотря на удаленность от редокс-активного лиганда, позволяет точно задавать окислительно-восстановительные потенциалы производных комплексов и управлять их активностью в реакции с молекулярным кислородом [258–263].
Обнаружено влияние стерических факторов на активность комплексов сурьмы в данной реакции. Показана возможность управления строением образующихся эндопероксидов с помощью стерических факторов [247, 249].
Катехолатные и о-амидофенолятные комплексы были исследованы на противорадикальную активность, в качестве ингибиторов перекисного окисления липидов [264–271].
Взаимодействие тридентатных N-2-гидроксифенилзамещенных о-иминобензохиноновых лигандов с триорганилстибинами происходит по механизму окислительного присоединения, но сопровождается миграцией протона с гидрокси-группы на атом азота и приводит к получению амино-бис-о-фенолятных комплексов сурьмы(V) типа (LONHO)SbR3 (471) [272–274]. Данные соединения также проявляют заметную антирадикальную активность и могут быть применены в качестве перехватчиков радикалов. Комплексы типа (471) на основе триалкилсурьмы легко отщепляют алкан и превращаются в амидо-бис-фенолятные производные диалкилсурьмы типа (LONO)SbR2 (472). Этот процесс ускоряется в присутствии кислорода и протекает с образованием соответствующех простых эфиров.
С использованием реакций окислительного присоединения была проведена функционализация трифенилсурьмой полимерных материалов, содержащих о-бензохиноновые фрагменты в боковых цепях, с получением сурьмасодержащих полимеров, обладающих активностью по отношению к молекулярному кислороду [275–277].
В некоторых случаях, когда соответствующий о-бензохинон неустойчив, синтез катехолатного комплекса производят по обменной реакции между пирокатехином и дигалогенидом триорганилсурьмы(V) в присутствии основания [231, 232, 269, 278, 279].
Катехолаты трифенилсурьмы способны образовывать комплексы с N-донорными лигандами, в которых КЧ центрального атома металла повышается до шести [280]. Так, из 3,6-ди-трет-бутилкатехолата трифенилсурьмы и 5-(2,6-диметилфенил)-3(4-пиридил)-1-фенилформазана (схема 98 ) получен комплекс шестикоординированной сурьмы (473).
Схема 98 .
Подобные шестикоординационные комплексы трифенилсурьмы (474−476) на основе различных замещенных о-бензохинонов и иминометилпиридина типа (Cat)SbPh3 · ImPy описаны в [281], молекулярное строение и особенности электрохимического поведения комплексов триарилсурьмы(V) (477−483) с п-N,N-диметиламинопиридином и п-цианопиридином рассмотрены в [282] (cхема 99). Обнаружено, что координация дополнительного лиганда может не только сдвигать потенциалы электрохимического окисления, но и менять механизм окисления редокс-активных катехолатных лигандов.
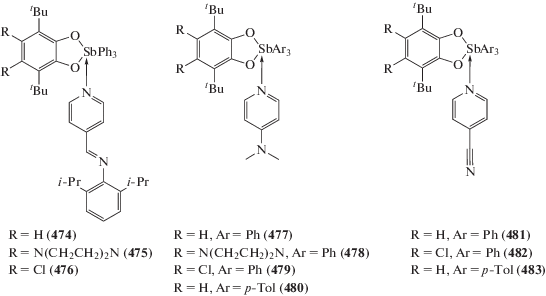
Схема 99 .
Авторы [283 ] показали, что обработка биядерного стибина орто-хлоранилом в дихлорметане при комнатной температуре приводит к образованию биядерного производного пятивалентной сурьмы (484) (cхема 100).
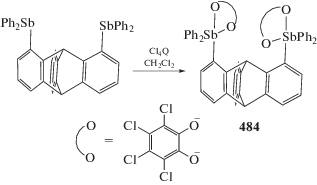
Схема 100 .
Показано, что органостибораны, содержащие 2,2'-бифениленфенильную группировку и остаток орто-хинона, при действии фторида трис(диметиламино)сульфония превращаются в соли (485 и 486), в анионах которых присутствуют шестикоординированные атомы сурьмы (cхема 101) [284 ] . Группа различных ионных комплексов типа [(Cat)SbPh3X]−[R4N]+ (487) получена при действии на катехолаты трифенилсурьмы тетраалкиламмониевых солей в ацетонитриле (cхема 101) [256].
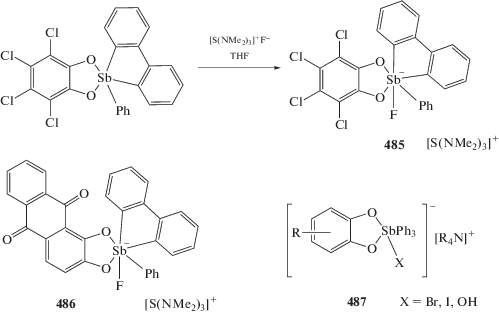
Схема 101 .
Продуктом реакции окислительного присоединения 3,4,5,6-тетрахлоро-1,2-бензохинона с 1,8-бис-(дифенилстибино)бифениленом является биядерное соединение сурьмы (488), в котором атомы металла имеют разные КЧ (4 и 6) (схема 102 ) [285].
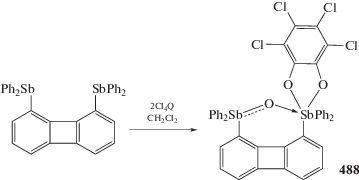
Схема 102 .
Соединение (489), полученное окислением 4,5-бис-(дифенилстибино)-9,9-диметилксантена двумя молями о-хлоранила Cl4Q, при действии [нBu4N]+[Ph3SiF2]− (схема 103 ) превращается в ионный комплекс (490) [286], анион которого содержит цепочку из атомов Sb−F−Sb (Sb(1)–F(1) 2.1684(17), Sb(2)–F(1) 2.1622(18) Å, что приближается по своему значению к сумме ковалентных радиусов указанных атомов − 2.12 Å [63].
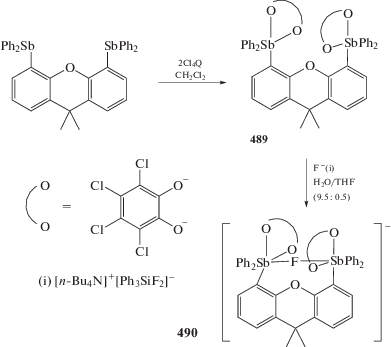
Схема 103 .
Окислением трис(пентафторфенил)сурьмы о-хлоранилом получен стиборан (491), который при действии трис(трет-бутил)фосфина с параформом (схема 104 ) превращается в цвиттер-ион (492), а реакция стиборана (491) с триэтилфосфиноксидом (L) приводит к образованию продукта присоединения (493), в котором лиганд L координируется на атом сурьмы через атом кислорода [287].
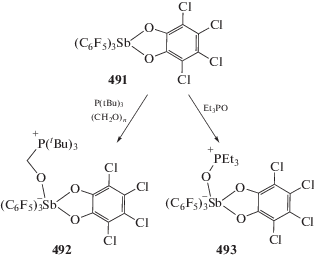
Схема 104 .
Присутствие в одном из арильных лигандов триарилсурьмы дифенилфосфиновой группировки не изменяет схемы окисления производного трехвалентной сурьмы, однако в целевых продуктах (494 и 495) имеет место координация атома фосфора на атом сурьмы, благодаря чему КЧ последнего увеличивается до шести (схема 105 ) [287].
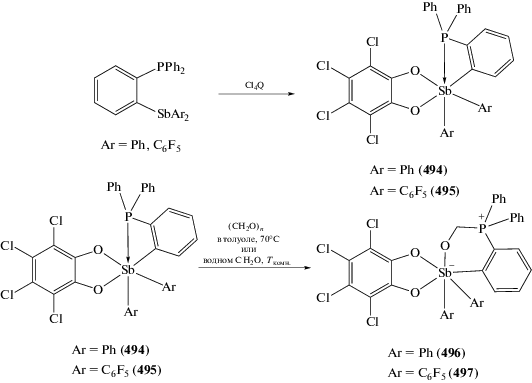
Схема 105 .
Взаимодействие полученных фосфиностиборанов (494 и 495) с параформом (толуол, 70°С) или формальдегидом (Н2О, Tкомн) приводит к образованию гетероциклических соединений сурьмы (496 и 497) (схема 105 ) [287].
Установлено, что окисление (o-(Ph2P)C6H4)3SbNi(PPh3) с помощью PhICl2 сопровождается конверсией стибина в стибораниловый лиганд [(o-(Ph2P)C6H4)3ClSb]NiCl (498). Кроме того, реакция (498) с катехолат-дианионом в присутствии циклогексилизоцианида (схема 106 ) приводит к образованию комплекса никеля [(o-(Ph2P)C6H4)3(Cl4Cat)Sb]Ni-(CNCy) (499), в котором атом никеля координируется с атомом сурьмы [288].
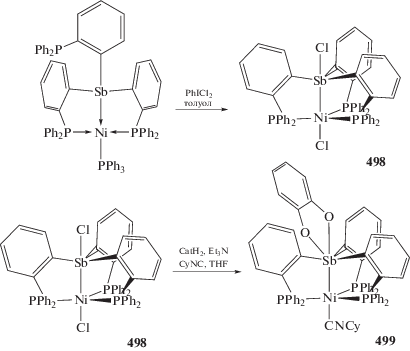
Схема 106 .
Катехолатный комплекс (500), полученный из [(o-(Ph2P)C6H4)2SbClPh]PtCl и о-хлоранила, в присутствии фторид-анионов (при действии избытка фторида калия; схема 107 ) превращается во фторостиборан [(o-(Ph2P)C6H4)2(Cl4Cat)SbF]PtClPh (501). Это преобразование сопровождается удалением хлоридного лиганда от атома платины и присоединением фторидного лиганда к атому сурьмы [289].
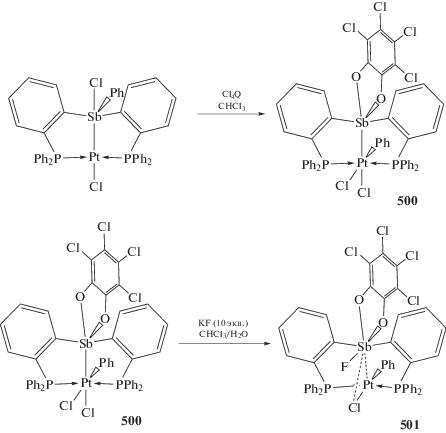
Схема 107 .
Следует отметить работу [290], в которой были выделены и структурно охарактеризованы продукты реакций (протекающих с участием диоксида углерода, кислоты и пентаарилсурьмы), имеющие необычное строение с трехкоординированным атомом углерода в катионе, например [(Ph4SbO)3C]+[OSO2C6H3(NO2)2-2,4]– (502) (схема 108 ).
Схема 108 .
ПРОИЗВОДНЫЕ СУРЬМЫ Ar2SbХ3, ArSbХ4 И ИХ РЕАКЦИИ
Вопросы синтеза и реакционной способности производных пятивалентной сурьмы с одной или двумя арильными группами при центральном атоме рассматриваются в значительно меньшей мере по сравнению с аналогичными соединениями с четырьмя и тремя органическими заместителями.
Производные сурьмы(V) с двумя или одной арильными группами при атоме сурьмы, имеющие молекулярную или ионную структуру, можно получить по реакциям перераспределения лигандов, окисления или замещения. Показано, что комплексы (503–508) с октаэдрическими анионами [R2SbCl4]− (схема 109 ) легко можно получить из солей фосфония, стибония или аммония и тригалогенидов диорганилсурьмы [291]. При этом иодид метилтриорганилсурьмы взаимодействует с выделяющимся иодом с образованием соединения (509) – ионного производного метилтриорганилстибониевого катиона.
Схема 109 .
Трихлорид дифенилсурьмы реагирует с хлоридом арилселения (схема 110 ) с образованием комплексного соединения (510), содержащего анионы дифенилтетрахлорсурьмы [292].
Схема 110 .
Окисление. Стибины LSbCl2 и LPhSbCl при действии хлористого сульфурила превращаются в нейтральные производные пятивалентной сурьмы LSbCl4 (327) и LPhSbCl3 (328) (схема 73 ) [152].
В роли окислителя могут быть использованы соединения платины. Так, окисление хлордиарилстибина производным платины (схема 111 ) с последующим действием хлористого фенилиодония приводит к последовательному хлорированию по атомам сурьмы и платины с образованием комплексов (511 и 512). При фотолизе конечного продукта окисления имеет место элиминирование хлора и образование соединения пятикоординированной сурьмы [293].
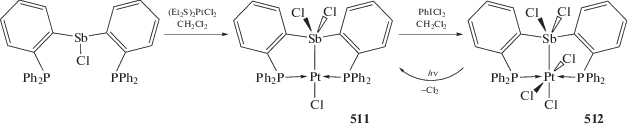
Схема 111 .
Действие хлористого фенилиодония на комплекс (513), полученный из хлорида палладия и хлордиарилстибина (схема 112 ), приводит к окислению атомов палладия и сурьмы с образованием производного (514). Образующийся комплекс (514) при фотолизе элиминирует хлор и восстанавливается до прежнего состояния [294].
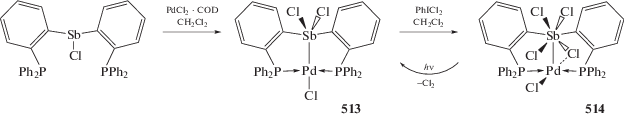
Схема 112 .
Производное пятикоординированной сурьмы (515) может быть получено из [(2-дифенилфосфанил)фенил]дихлорстибина и AuCl(Tht) в тетрагидрофуране (схема 113 ) [295].
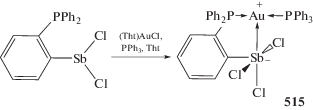
Схема 113 .
Замещение. Обработка комплекса (511) фторидом таллия (cхема 114) приводит к образованию трифторстиборанов (516 и 517) [296].
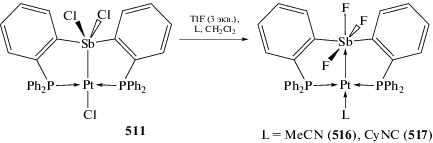
Схема 114 .
Реакции эквимолярных количеств пара-галогенофенилстибиновых кислот с дифенилсиланолом в кипящем толуоле (6 ч) приводит к образованию четырехъядерных комплексов сурьмы [(p-ClC6H4Sb)4(O)4(Ph-2SiO2)4] (518), [(p-BrC6H4Sb)4(O)4(Ph2SiO2)4] (519) (схема 115 ). Кластеры (518 и 519) изоструктурны и содержат центральный искаженный кубический фрагмент Sb4O4 [297].
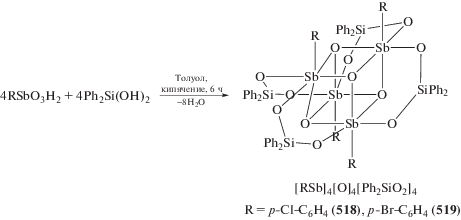
Схема 115 .
Взаимодействие трихлорида дифенилсурьмы с трифлатом серебра в присутствии N-оксида п-метилпиридина в различных соотношениях исходных реагентов (схема 116 ) приводит к образованию четырех типов соединений шестикоординированной сурьмы (520–523) [298].
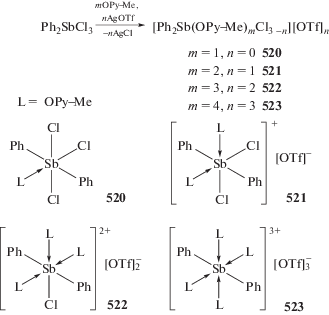
Схема 116 .
Взаимодействие замещенных бензойных кислот с трихлоридом дифенилсурьмы в бензоле в присутствии триэтиламина (схема 117 ) сопровождается образованием четырехядерных оксопроизводных дифенилсурьмы (524–526) с двумя симметричными остатками карбоновых кислот, расположенными напротив друг друга [299].
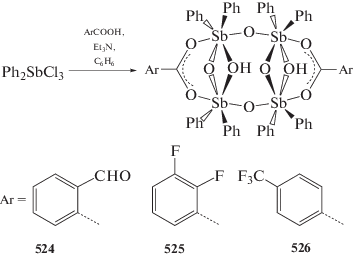
Схема 117 .
Трифлатный комплекс (527) получается из хлорсодержащего комплекса платины (511) и трифлата серебра в хлористом метилене (cхема 118) [300].
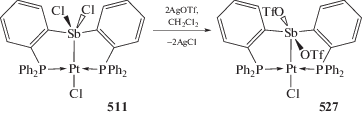
Схема 118 .
Серия солей вольфрамоарсенатов (528–530) (cхема 119), содержащих одну, две или три группировки PhSb, синтезированa из неорганических солей вольфрамоарсенатов и дихлорфенилстибина в растворе уксусной кислоты [301].
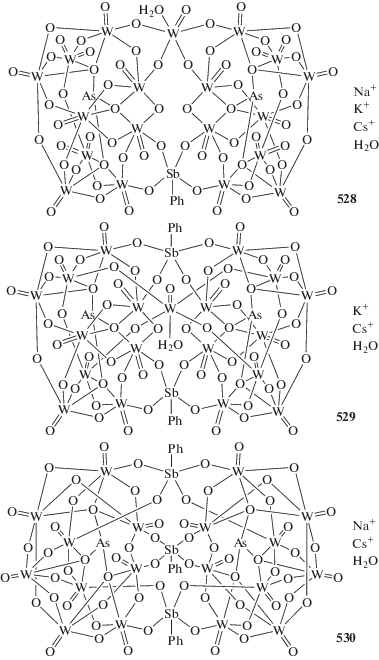
Схема 119 .
Полиядерные кластерные соединения [Co(p-TolSb)12O28{Co(H2O)3}4]Cl2 · 6H2O (531), [Co(p-Cl–C6H4–Sb)12O28{Co(H2O)3}4]Cl2 · 22H2O (532), (PhCH2NMe3)2[Zn(p-Cl–C6H4–Sb)12O28Zn4Cl2.54Br1.46] · · 8MeCN · H2O (533) и [BaCoH4(p-MeC6H4Sb)O28] · · 5H2O (534), содержащие в своем составе группы ArSb (Ar = 4-R–C6H4, R = Me, Cl) и атомы кобальта, цинка, бария, кислорода, синтезировали добавлением солей металлов к раствору арилстибиновой кислоты и аммиака в воде [302]. После перемешивания реакционной смеси до полного растворения исходных реагентов, удаления растворителя и перекристаллизации остатка из водного ацетонитрила получали целевые продукты.
В результате реакции хлорирования дихлорида арилсурьмы сульфурилхлоридом получен тетрахлорид арилсурьмы ArSbCl4535 (cхема 120), гидролиз которого приводил к образованию кислородсодержащего соединения сурьмы (ArSb)4O6(OH)4 (536) адамантанобразной структуры [303].
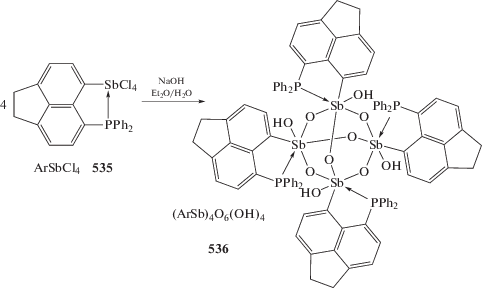
Схема 120 .
При взаимодействии комплекса (ArSb)4(O)2- (O2P(O)Ph)4(OP(O)(OH)Ph)4 (537) (продукта конденсации 1 экв. хлорфенилстибиновой кислоты ArSbO(OH)2 (Ar = p-Cl–C6H4) и 2 экв. фенилфосфиновой кислоты PhPO(OH)2) с ацетатом кобальта(II) впервые получено полиядерное производное (538) [304] (cхема 121).
Схема 121 .
О синтезе полиядерного соединения (Ar-Sb)2O(OP(O)(OH)t-Bu)6 (539) из пара-хлорфенилстибиновой и трет-бутилфофиновой кислот сообщалось в [305]. Данный комплекс содержит два атома сурьмы, связанных мостиковым атомом кислорода, два мостиковых и четыре терминальных остатка трет-бутилфосфиновой кислоты (схема 122 ).
Схема 122 .
Взаимодействие комплекса (539 с ацетатом меди приводит в зависимости от условий проведения к образованию 4-, 3-, 5- или 8-ядерных комплексов меди, например комплекса (540).
Продуктами реакций полиядерных кислородсодержащих сурьмаорганических соединений (ArSb)4(O)2(O2P(O)Ph)4(OP(O)(OH)Ph)4 (537) и (ArSb)2O(OP(O)(OH)tBu)6 (539) с ацетатом кобальта(II) в присутствии метоксида лития и пиридина в метаноле являются, соответственно, комплексы кобальта (538 и 541) (схема 122 ) [306]. Молекулы пиридина в комплексах могут быть заменены на иные амины, которые вводятся в реакционную смесь взамен пиридина. Например, замена пиридина в (538) на пиразин приводит к формированию координационного полимера (542) (схема 122 ).
Синтез новых полиоксометаллатов Sb12 (543) и Sb14 (544), полученных из пара-толилстибиновой кислоты и солей металлов в присутствии основания, описали авторы [307].
Реакции Ph2SbCl3 с RSi(OH)3, где R – трет-Bu, цикло-C6H11, и Ph2Si(OH)2 в толуоле в присутствии триэтиламина привели к образованию соединений сурьмы(V) и оксогидроксокластеров сурьмы смешанной валентности (III/V). Интересно, что во всех реакциях наблюдается, по крайней мере, один разрыв связи Sb–C, что приводит к формированию новых кластерных производных [(Ph2Sb)4(PhSb)2–(C4H9SiO3)2(O)6(OH)2] (545), [(Ph2Sb)4(PhSb)2(C6H11–SiO3)2(O)6(OH)2] (546), [(Ph2Sb)(PhSb)2(Ph2SiO2)2(O)3–(OH)2]–Et3NH+ (547) и [(Ph2Sb)4(Sb)2(Ph2SiO2)2(O)6–(OH)2] (548) соответственно (схема 123 ) [308].
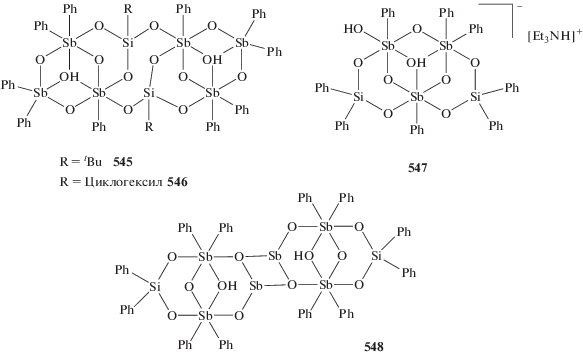
Схема 123 .
Трехъядерные производные сурьмы типа {[C5H5NH] · [(ArSb)3{(tBu)4Si4O9}{(tBu)2Si2O5}(µ3-O)(µ-OH)]}, где Ar – p-Cl–C6H4 (549), p-Br–C6H4 (550), 3,5-Cl2–C6H3 (551), и типа [(ArSb)3{(tBuSiO2)4 (tBuSiO2OH)2}(µ3-O)(µ-OH)2] (Ar = p-I–PrC6H4, 552) образуются из арилстибониевой кислоты и трет-бутилсилантриола (толуол, 110°С, 8 ч) [309].
Особенности синтеза и строения арилстибониевых кислот и их калиевых и натриевых солей обсуждены в [310]. Показано, что после растворения арилстибониевых кислот в водном растворе щелочи, удаления воды и перекристаллизации остатка из водного ацетонитрила образуются кристаллы комплексов, содержащие 12-ядерные анионные соединения сурьмы, например анион [K2H8(p-Cl–C6H4Sb)12O30]2– (553).
Фосфонаты и фосфоселенаты моноарилсурьмы (554–558) (схема 124 ) получены с 54–86% выходом из арилстибониевых кислот и органилфосфиновых и фенилселеновой кислот в ацетонитриле при перемешивании в течение 24 ч при комнатной температуре [311].
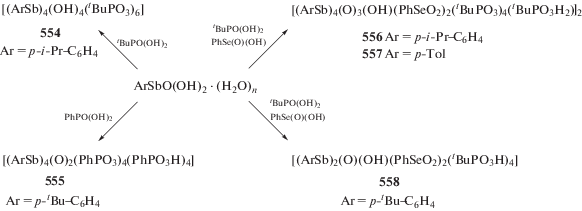
Схема 124 .
Биядерный оксид (2,6-димезитилфенил)сурьмы при обработке серной кислотой превращается в биядерный комплекс (559) с мостиковым сульфатным лигандом; действие на оксид водного раствора гидроксида натрия приводит к образованию тетраядерного сурьмаорганического производного (560) (cхема 125), в состав которого входит гидратированные катионы натрия [312].
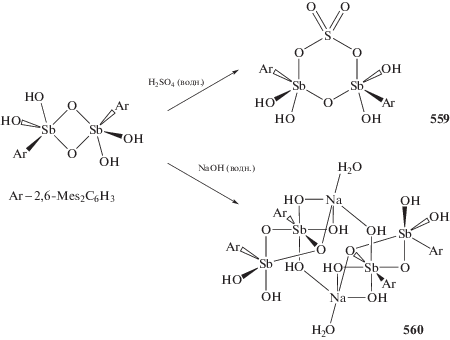
Схема 125 .
Три полиядерных арилстибоната тетрабутиламмония [(p-Cl–C6H4Sb)16(µ3-O)8(µ2-OH)5(µ2-O)23]3–$[{\text{E}}{{{\text{t}}}_{{\text{4}}}}{\text{N}}]_{{\text{3}}}^{ + }$ (561), [(p-Cl–C6H4Sb)16(µ3-O)8(µ2-OH)6 (µ2-O)22]4–$[{\text{E}}{{{\text{t}}}_{{\text{4}}}}{\text{N}}]_{{\text{4}}}^{ + }$ (562, cхема 126) и [Na2(p-Cl–C6H4Sb)12(µ4-O)3(µ3-O)12(µ2-O)9(OH)6(H2O)3]4–$[{\text{E}}{{{\text{t}}}_{{\text{4}}}}{\text{N}}]_{{\text{3}}}^{ + }$ (563), содержащих сольватные молекулы воды и ацетонитрила, синтезированы из пара-хлорфенилстибониевой кислоты и гидроксида тетрабутиламмония [313].
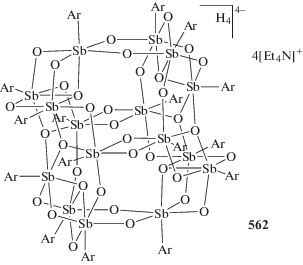
Схема 126 .
При взаимодействии оксида дифенилтеллурия с органостибониевой кислотой или полимерным оксидом трифенилсурьмы образуются 14- (564) и восьмиядерные (565) сурьмаорганические кластерные соединения (cхема 127) [314].
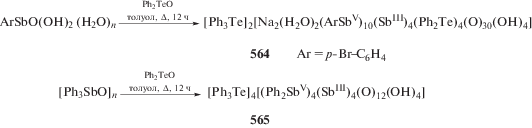
Схема 127 .
Из пиразолильных лигандов H2LR, HL и арилстибониевых кислот синтезированы 4- и 16-ядерные кластеры сурьмы (566–571) (cхема 128), в которых наряду со связями Sb−O может иметь место координация атомов азота с атомом металла, определяемая, по мнению авторов, объемом пиразолильного лиганда [315].
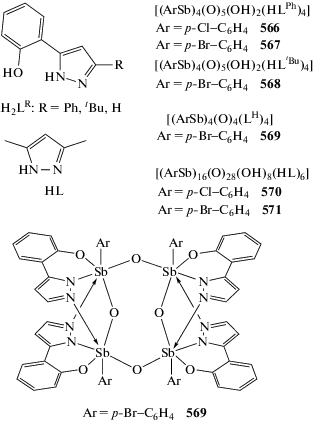
Схема 128 .
Взаимодействие эквимолярных количеств арилстибониевых кислот с 8-гидроксихинолином (HL1) или 2-(пиразол-5-ил)-α-нафтолом (HL2) в толуоле при кипячении позволяет получить адамантаноподобные кластеры типа L4(RSb)4O6 (cхема 129): [(p-X–C6H4Sb)4(µ2-O)6(L1)4], где X = Cl (572), Br (573), [(p-Cl–C6H4Sb)4(µ2-O)6(L2)4] · HL2 (574) и [(p-Br–C6H4Sb)4(µ2-O)6(L2)4]2 · HL2 (575) [316]. Интересно, что (572–575) по структуре напоминают димерную форму оксида сурьмы Sb2O3. Тетрамер [(p-Cl–C6H4Sb)4(µ2-O)4(L2-H)4] (576), имеющий строение, подобное структуре тетраядерного комплекса сурьмы (569), также был выделен в качестве побочного продукта. В данном комплексе нафтольный лиганд депротонирован не только по гидроксильной группе, но и по атому азота пиразолильной группы, и наблюдается дополнительное ковалентное взаимодействие этого атома азота с соседним атомом сурьмы в четырехъядерном кластере (576) (cхема 129).
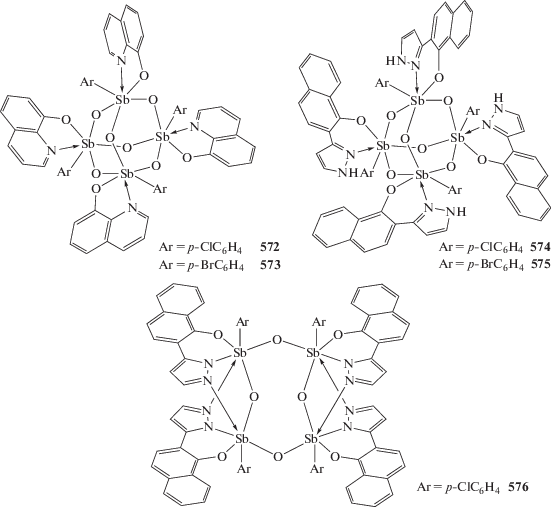
Схема 129 .
ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ АРИЛЬНЫХ СОЕДИНЕНИЙ СУРЬМЫ(V)
Несмотря на то, что соединения сурьмы достаточно токсичны, в течение почти столетия они широко используются в терапии в качестве противопаразитарных средств, особенно при лечении лейшманиоза [317]. В целом же можно сказать, что работ, посвященных практическому использованию и исследованию потенциальных возможностей использования арильных производных пятивалентной сурьмы, известно относительно мало. Показано, что некоторые дикарбоксилаты триарилсурьмы (259–266) (схема 56 ), (284–291) (схема 58 ) являются биологически активными веществами [129, 134], сурьмаорганическое производное лапахола (215) ингибирует рост клеток хронической лейкемии [108]. Сурьмаорганические производные ацетилсалициловой, 3-ацетоксибензойной кислот (соединения 223 и 224) [113] и диакрилаты трифенил- и три-пара-толилсурьмы (227–236) (схема 49 ) [116]; (279, 280) [131]) обладают антибактериальной активностью и эффективны в лечении лейшманиоза и стафилоккока. Противоопухолевой активностью обладают некоторые арильные соединения пятивалентной сурьмы: (213, 124) [107], (218, 219) [111], (237–241) (схема 51 ) [117], (255–258) (схема 55 ) [128], (281) и др. бис-(N-фенилглицинаты) триарилсурьмы(V) [132], (403) [195]. Отдельно следует выделить комплексы бис-(N-оксифтальимид) и бис-(N-оксисукцинимид) трифенилсурьмы(V) (213 и 214) (схема 47 ) [107], которые являются более активными противораковыми агентами, чем цисплатин.
Высокая каталитическая активность арильных соединений сурьмы описывалась на примерах трифлата (321), катализирующего реакцию альдольной конденсации (схема 69 ) [148]; комплексов (336 и 337), последний из которых показан эффективным катализатором в реакции гидросилилировании бензальдегида с использованием триэтилсилана (схема 130 ) [156].
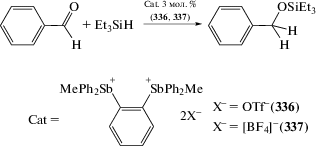
Схема 130 .
Каталитические свойства этих соединений стибония были исследованы в гидросилилировании бензальдегида с использованием триэтилсилана в CDCl3 (схема 130 ). Хотя [Ph3MeSb]+ [OTf]– и [Ph3MeSb]+[BF4]– (3 мол. %) не способствуют реакции при комнатной температуре, некоторая каталитическая активность наблюдалась в случае (336) (1.5 мол. %) с конверсией 11% через 8 ч. Удивительно контрастное поведение наблюдалось в случае (337) (1.5 мол. %), который оказывается гораздо более активным, когда гидросилилирование бензальдегида протекает до полной конверсии в течение 8 ч.
Платинасодержащий комплекс дихлорида триарилсурьмы (346) (схема 81 ) катализирует реакцию присоединения мезитилена к этиловому эфиру ацетиленкарбоновой кислоты (схема 131 ) [159].
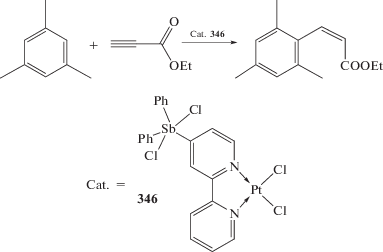
Схема 131 .
Другой платинасодержащий трифлатный комплекс (527) (схема 118 ) является эффективным катализатором реакций циклоприсоединения (схема 132 ) [300].
Схема 132 .
Примером активного катализатора в реакции димеризации 1,1-дифенилэтилена (схема 133 ) является комплекс (182), при этом выход продукта конденсации достигает 99% [90].
Схема 133 .
Показано, что трифторметансульфонат хлоротрифенилсурьмы Ph3SbCl(OTf) (577) катализирует реакции циклоприсоединения нитрилов с циклотрифосфинами (схема 134 ), которые протекают с образованием 1-аза-2,3,4-трифосфоленов с выходом до 90% [318]. Подобной активностью в данной реакции обладает и бис-трифлат трифенилсурьмы (308) (схема 135 ).
Схема 134 .
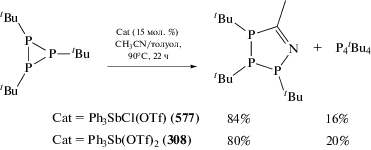
Схема 135 .
ЗАКЛЮЧЕНИЕ
Химия металлоорганических и координационных соединений сурьмы(V) в настоящее время развивается достаточно интенсивно, и в последние 10–15 лет получено большое количество разнообразных моно-, би- и полиядерных соединений; показано, что данные соединения сурьмы демонстрируют большое разнообразие структурных типов, проявляют химическую активность в самых разнообразных процессах. При изучении данных соединений обнаружена высокая каталитическая активность в ряде интересных и перспективных каталитических процессов (альдольная конденсация, гидросилилирование, образование новых связей углерод–углерод, циклоприсоединение и др.), показана возможность избирательной фиксации фторид-анионов; установлена возможность стабилизации соединения трехкоординированного углерода; впервые продемонстрирована возможность обратимого связывания молекулярного кислорода комплексами металлов главных групп. Ряд сурьмаорганических производных является биологически активными веществами, обладают антибактериальной, противогрибковой и противоопухолевой активностью. Сурьмаорганические и координационные соединения к настоящему времени остаются значительно менее изученными по сравнению с фосфор- и мышьяксодержащими соединениями, однако анализ публикаций по данной теме позволяет сделать вывод о том, что интерес к данным соединениям постоянно растет, так как производные сурьмы(V) имеют большие перспективы применения в качестве катализаторов самых разнообразных реакций тонкого органического синтеза, в качестве биохимически активных веществ и компонентов лекарственных препаратов, а также в качестве переносчиков малых молекул, сенсоров на различные анионы и молекулярные группы и многое другое.
Список литературы
Schröder G., Okinaka T., Mimura Y. et al. // Chem. Eur. J. 2007. V. 13. P. 2517.
García-Monforte M.A., Alonso P.J., Ara I. et al. // Angew. Chem. Int. Ed. 2012. V. 51. P. 2754.
Pan B., Gabbai F.P. // J. Am. Chem. Soc. 2014. V. 136. P. 9564.
Chalmers B.A., Bìhl M., Arachchige K.S.A. et al. // Chem. Eur. J. 2015. V. 21. P. 7520.
Шарутин В.В., Жидков В.В., Муслин Д.В. и др. // Изв. АН. Сер. хим. 1995. № 5. С. 958.
Шарутин В.В., Шарутина О.К., Сенчурин В.С., Щелоков А.О. // Журн. общ. химии. 2016. Т. 86. № 1. С. 92.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2017. Т. 62. № 3. С. 290 (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Inorg. Chem. 2017. V. 62. № 3. P. 295). https://doi.org/10.1134/S0036023617030147
Шарутин В.В., Шарутина О.К., Ефремов А.Н., Андреев П.В. // Журн. неорган. химии. 2017. Т. 62. № 10. С. 1330 (Sharutin V.V., Sharutina O.K., Efremov A.N., Andreev P.V. // Russ. J. Inorg. Chem. 2017. V. 62. № 10. P. 1320). https://doi.org/10.1134/S0036023617100163
Sharutin V.V., Sharutina O.K., Senchurin S.V. // Bull. South Ural State Univ. Ser. Chem. 2015. V. 7. № 4. P. 86.
Шарутин В.В., Шарутина О.К., Андреев П.В. // Коорд. химия. 2016. Т. 42. № 7. С. 412. (Sharutin V.V., Sharutina O.K., Andreev P.V. // Russ. J. Coord. Chem. 2016. Т. 42. № 7. P. 449.) https://doi.org/10.1134/S1070328416060075
Шарутин В.В., Сенчурин В.С., Шарутина О.К. // Журн. общ. химии. 2009. Т. 79. № 10. С. 1636.
Шарутин В.В., Шарутина О.К., Хныкина К.А. // Журн. неорган. химии. 2016. Т. 61. № 2. С. 192. (Sharutin V.V., Sharutina O.K., Khnykina K.A. // Russ. J. Inorg. Chem. 2016. V. 61. № 2. P. 180.) https://doi.org/10.1134/S0036023616020194
Шарутин В.В., Шарутина О.К., Котляров А.Р. // Журн. неорган. химии. 2015. Т. 60. № 4. С. 525. (Sharutin V.V., Sharutina O.K., Kotlyarov A.R. // Russ. J. Inorg. Chem. 2015. V. 60. № 4. P. 465). https://doi.org/10.1134/S0036023615040221
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2014. Т. 59. № 9. С. 1182. (Sharutin V.V., Sharutina O.K., Senchurin S.V. // Russ. J. Inorg. Chem. 2014. V. 59. P. 951). https://doi.org/10.1134/S0036023614090174
Шарутин В.В., Шарутина О.К., Мельникова И.Г. и др. // Изв. АН. Сер. хим. 1996. № 8. С. 2082.
Шарутин В.В., Сенчурин В.С., Шарутина О.К. // Журн. неорган. химии. 2014. Т. 59. № 2. С. 247. (Sharutin V.V., Senchurin V.S., Sharutina, O.K. // Russ. J. Inorg. Chem. 2014. V. 59. P. 115). https://doi.org/10.1134/S003602361402017X
Шарутин В.В., Шарутина О.К. // Коорд. химия. 2014. Т. 40. № 9. С. 559. (Sharutin V.V., Sharutina O.K. // Russ. J. Coord. Chem. 2014. V. 40. № 9. P. 643). https://doi.org/10.1134/S1070328414090073
Sharutin V.V., Sharutina O.K, Gubanova Yu.O. // Bull. South Ural State Univ. Ser. Chem. 2015. V. 7. P. 17.
Шарутин В.В., Шарутина О.К., Губанова Ю.О. и др. // Коорд. химия. 2017. Т. 43. № 7. С. 444. (Sharutin V.V., Sharutina O.K., Gubanova Y.O. et al. // Russ. J. Coord. Chem. 2017. V. 43. № 7. P. 453). https://doi.org/10.1134/S1070328417060070
Шарутин В.В., Шарутина О.К. // Журн. неорган. химии. 2015. Т. 60. № 3. С. 340. (Sharutin V.V., Sharutina O.K. // Russ. J. Inorg. Chem. 2015. V. 60. № 3. P. 292). https://doi.org/10.1134/S0036023615030171
Sharutin V.V., Sharutina O.K., Gubanova Y.O. et al. // J. Organomet. Chem. 2015. V. 798. P. 41.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Коорд. химия. 2014. Т. 40. № 2. С. 108. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Coord. Chem. 2014. V. 40. № 2. P. 109). https://doi.org/10.1134/S1070328414020109
Sharutin V.V., Sharutina O.K., Gubanova Y.O. et al. // Mendeleev Commun. 2018. V. 28. P. 621.
Шарутин В.В., Шарутина О.К., Губанова Ю.О. // Вест. Южно-Урал. гос. ун-та. Сер. хим. 2017. Т. 9. № 4. С. 56.
Sharutin V.V., Sharutina O.K., Gubanova Y.O., Eltsov O.S. // Inorg. Chim. Acta. 2019. V. 494. P. 211.
Шарутин В.В., Шарутина О.К., Молокова О.В. // Журн. неорган. химии. 2012. Т. 57. № 6. С. 902. (Sharutin V.V., Sharutina O.K., Molokova O.V. // Russ. J. Inorg. Chem. 2012. V. 57. № 6. P. 832). https://doi.org/10.1134/S0036023612010226
Шарутин В.В., Шарутина О.К. // Коорд. химия. 2017. Т. 43. № 4. С. 244. (Sharutin V.V., Sharutina O.K. // Russ. J. Coord. Chem. 2017. V. 43. № 4. P. 232). https://doi.org/10.1134/S1070328417040054
Шарутин В.В., Шарутина О.К. // Журн. неорган. химии. 2017. Т. 62. № 7. С. 925. (Sharutin V.V., Sharutina O.K. // Russ. J. Inorg. Chem. 2017. V. 62. № 7. P. 905). https://doi.org/10.1134/S003602361707021X
Шарутин В.В., Молокова О.В., Шарутина О.К. // Журн. неорган. химии. 2013. Т. 58. № 4. С. 460. (Sharutin V.V., Molokova O.V., Sharutina O.K. // Russ. J. Inorg. Chem. 2013. V. 58. № 4. С. 400). https://doi.org/10.1134/S0036023613040177
Шарутин В.В., Молокова О.В., Шарутина О.К. и др. // Коорд. химия. 2005. Т. 31. № 3. С. 172. (Sharutin V.V., Molokova O.V., Sharutina O.K. et al. // Russ. J. Coord. Chem. 2005. V. 31. № 3. P. 159). https://doi.org/10.1007/s11173-005-0068-4
Шарутин В.В., Молокова О.В., Шарутина О.К. и др. // Журн. общ. химии. 2004. Т. 74. № 10. С. 1600. (Sharutin V.V., Molokova O.V., Sharutina O.K. et al. // Russ. J. Gen. Chem. 2010. V. 74. № 10. P. 1485).
Шарутин В.В., Шарутина О.К. // Журн. общ. химии 2014. Т. 84. № 3. С. 457. (Sharutin V.V., Sharutina O.K. // Russ. J. Gen. Chem. 2014. V. 84. № 3. P. 515).
Шарутин В.В., Сенчурин В.С., Шарутина О.К., Панова Л.П. // Журн. неорган. химии. 2008. Т. 53. № 7. С. 1194. (Sharutin V.V., Senchurin V.S., Sharutina O.K., Panova L.P. // Russ. J. Inorg. Chem. 2008. V. 53. № 7. С. 1110). https://doi.org/10.1134/S0036023608070206
Шарутин В.В., Сенчурин В.С., Фастовец О.А. и др. // Коорд. химия. 2008. Т. 34. № 5. С. 373 (Sharutin V.V., Senchurin V.S., Fastovets O.A. et al. // Russ. J. Coord. Chem. 2008. V. 34. № 5. P. 367). https://doi.org/10.1134/S1070328408050096
Sharutin V.V., Sharutina O.K., Senchurin V.S // Bull. South Ural State Univ. Ser. Chem. 2015. V. 7. P. 98.
Шарутин В.В., Сенчурин В.С., Шарутина О.К. // Бутлеровские сообщения. 2011. Т. 28. С. 35.
Шарутин В.В., Шарутина О.К., Задачина О.П. и др. // Коорд. химия. 2003. Т. 29. № 1. С. 8. (Sharutin V.V., Sharutina O.K., Zadachina O.P. et al. // Russ. J. Coord. Chem. 2003. V. 29. № 1. P. 6). https://doi.org/10.1023/A:1021878530695
Шарутин В.В., Пакусина А.П., Егорова И.В. и др. // Коорд. химия. 2008. Т. 34. № 4. С. 267. (Russ. J. Coord. Chem. 2008. V. 34. № 4. P. 259). https://doi.org/10.1134/S1070328408040040
Шарутин В.В., Шарутина О.К., Сенчурин В.С., Муковоз П.П. // Журн. неорган. химии. 2014. Т. 59. № 5. С. 678. (Sharutin V.V., Sharutina O.K., Senchurin V.S., Mukovoz P.P. // Russ. J. Inorg. Chem. 2014. V. 59. № 5. С. 508). https://doi.org/10.1134/S0036023614050155
Шарутин В.В., Сенчурин В.С., Фастовец О.А. и др. // Журн. неорган. химии 2009. Т. 54. № 3. С. 436. (Sharutin V.V., Senchurin V.S., Fastovets O.A. et al. // Russ. J. Inorg. Chem. 2009. V. 54. № 3. С. 389). https://doi.org/10.1134/S0036023609030103
Yang M., Pati N., Belanger-Chabot G., Gabbai F.P. // Dalton Trans. 2013. V. 47. P. 11843.
Quan L., Yin H., Cui J. et al. // J. Organomet. Chem. 2009. V. 694. P. 3683.
Quan L., Yin H., Cui J. et al. // J. Organomet. Chem. 2009. V. 694. P. 3708.
Geng H., Hong M., Yang Y. et al. // J. Coord. Chem. 2015. V. 68. P. 2938.
Иванов М.А., Шарутин В.В., Иванов А.В. и др. // Коорд. химия. 2008. Т. 34. № 7. С. 533. (Ivanov M.A., Sharutin V.V., Ivanov A.V. et al. // Russ. J. Coord. Chem. 2008. V. 34. P. 527). https://doi.org/10.1134/S1070328408070099
Иванов М.А., Иванов А.В., Шарутин В.В. и др. // Журн. неорган. химии. 2009. Т. 54. № 5. С. 766. (Ivanov M.A., Ivanov A.V., Sharutin V.V. et al. // Russ. J. Inorg. Chem. 2009. V. 54. № 5. P. 708). https://doi.org/10.1134/S0036023609050088
Иванов М.А., Герасименко А.В., Иванов А.В. и др. // Журн. неорган. химии. 2013. Т. 58. № 2. С. 234. (Ivanov M.A., Gerasimenko A.V., Ivanov A.V. et al. // Russ. J. Inorg. Chem. 2013. V. 58. № 2. P. 197). https://doi.org/10.1134/S0036023613020071
Ivanov M.A., Ivanov A.V., Antzutkin O.N. et al. // Inorg. Chim. Acta. 2007. V. 360. P. 2897.
Артемьева Е.В., Шарутина О.К., Шарутин В.В. // Журн. общ. химии. 2017. Т. 87. № 12. С. 2094.
Yin H.-D., Wen L.-Y., Cui J.-C., Li W.-K. // Polyhedron. 2009. V. 28. P. 2919.
Robertson A.P.M., Chitnis S.S., Jenkins H.A. et al. // Chem. Eur. J. 2015. V. 21. P. 7902.
Matsukawa S., Yamamichi H., Yamamoto Y., Ando K. // J. Am. Chem. Soc. 2009. V. 131. P. 3418.
Lin T., Wade C.R., Perez L.M., Gabbai F.P. // Angew. Chem. Int. Ed. 2010. V. 49. P. 6357.
Hirai M., Myahkostupov M., Castellano F.N., Gabbai F.P. // Organometallics. 2016. V. 35. P. 1854.
Yamamichi H., Matsukawa S., Kojima S. et al. // Heteroatom Chem. 2011. V. 22. P. 553.
Ke I.-S., Myahkostupov M., Castellano F.N., Gabbai F.P. // J. Am. Chem. Soc. 2012. V. 134. P. 15309.
Wade C.R., Ke I.-S., Gabbai F.P. // Angew. Chem. 2012. V. 124. P. 493.
Wang G.-C., Xiao J., Yu L. et al. // J. Organomet. Chem. 2004. V. 689. P. 1631.
Wang G.-C., Lu Y.-N., Xiao J. et al. // J. Organomet. Chem. 2005. V. 690. P. 151.
Li J.-S., Liu R.-C., Chi X.-B. et al. // Inorg. Chim. Acta. 2004. V. 357. P. 2176.
Егорова И.В., Жидков В.В., Гринишак И.П., Раханский А.А. // Журн. общ. химии. 2014. Т. 84. С. 1176.
Шарутин В.В., Попкова М.А., Тарасова Н.М. // Вест. Южно-Урал. гос. ун-та. Сер. Хим. 2018. Т. 10. № 1. С. 55.
Бацанов С.С. // Журн. неорган. химии. 1991. Т. 36. № 12. С. 3015.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Коорд. химия. 2016. Т. 42. № 3. С. 178. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Coord. Chem. 2016. V. 42. № 3. P. 201). https://doi.org/10.1134/S1070328416030088
Шарутин В.В., Шарутина О.К., Сенчурин В.С., Пельков П.А. // Журн. неорган. химии. 2016. Т. 61. С. 195. (Sharutin V.V., Sharutina O.K., Senchurin V.S., Pel’kov P.A. // Russ. J. Inorg. Chem. 2016. V. 61. P. 183).
Sharutin V.V., Sharutina O.K., Senchurin V.S. // Bul. South Ural State Univ. Ser. Chem. 2017. V. 9. P. 58.
Sharutin V.V., Sharutina O.K., Senchurin V.S. // Bull. South Ural State Univ. Ser. Chem. 2015. V. 7. P. 80.
Шарутин В.В., Шарутина О.К., Сенчурин В.С., Сомов Н.В. // Журн. неорган. химии. 2016. Т. 61. № 8. С. 1017. (Sharutin V.V., Sharutina O.K., Senchurin V.S., Somov N.V. // Russ. J. Inorg. Chem. 2016. V. 61. № 8. P. 969). https://doi.org/10.1134/S0036023616080143
Шарутин В.В., Шарутина О.К., Сенчурин В.С., Сомов Н.В. // Коорд. химия. 2016. Т. 42. № 12. С. 758. (Sharutin V.V., Sharutina O.K., Senchurin V.S., Somov N.V. // Russ. J. Coord. Chem. 2016. V. 42. № 12. P. 779). https://doi.org/10.1134/S1070328416120058
Cotton F.A., Francis R. // J. Am. Chem. Soc. 1960. V. 82. № 12. P. 2986.
Шарутин В.В., Сенчурин В.С., Шарутина О.К. // Журн. неорган. химии. 2013. Т. 58. № 5. С. 616. (Sharutin V.V., Senchurin V.S., Sharutina O.K. // Russ. J. Inorg. Chem. 2013. V. 58. № 5. P. 543). https://doi.org/10.1134/S0036023613050203
Шарутин В.В., Сенчурин В.С., Шарутина О.К., Гущин А.В. // Бутлеровские сообщения. 2012. Т. 29. № 2. С. 26.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Бутлеровские сообщения. 2012. Т. 30. № 6. С. 50.
Кочешков К.А., Сколдинов А.П., Землянский Н.Н. Методы элементоорганической химии. Сурьма, висмут. М.: Наука, 1976. 483 с.
Шарутин В.В., Сенчурин В.С., Пакусина А.П. и др. // Журн. неорган. химии. 2010. Т. 55. № 1. С. 64. (Sharutin V.V., Senchurin V.S., Pakusina A.P. et al. // Russ. J. Inorg. Chem. 2010. V. 55. № 1. P. 61). https://doi.org/10.1134/S0036023610010122
Ткачева А.Р., Шарутин В.В. // Вест. Южно-Урал. гос. ун-та. Сер. хим. 2018. Т. 10. № 3. С. 59.
Шарутин В.В., Сенчурин В.С., Шарутина О.К., Гущин А.В. // Бутлеровские сообщения. 2012. Т. 30. № 4. С. 55.
Шарутин В.В., Шарутина О.К., Тарасова Н.М., Ельцов О.С. // Изв. АН. Сер. хим. 2019. № 1. С. 24. (Sharutin V.V., Sharutina O.K., Tarasova N.M., El-tsov O.S. // Russ. Chem. Bull. 2019. V. 68. № 1. P. 24.)
Ferguson G., Hawley D.M. // Acta Crystrallog. B. 1974. V. 30. P. 103.
Millington P.L., Sowerby D.B. // Dalton Trans. 1992. P. 1199.
Шарутин В.В., Сенчурин В.С., Шарутина О.К. и др. // Журн. общ. химии. 1996. Т. 66. С. 1755.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2013. Т. 58. № 11. С. 1454 (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Inorg. Chem. 2013. V. 58. № 11. P. 1302). https://doi.org/10.1134/S0036023613110181
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Изв. АН. Сер. хим. 2016. № 3. С. 751. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. Chem. Bull. 2016. V. 65. № 3. P. 751).
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2015. Т. 60. № 9. С. 1200. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Inorg. Chem. 2015. V. 60. № 9. P. 1093). https://doi.org/10.1134/S0036023615060145
Шарутин В.В., Шарутина О.К., Молокова О.В. // Бутлеровские сообщения. 2011. Т. 28. № 19. С. 45.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2015. Т. 60. № 2. С. 203. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Inorg. Chem. 2015. V. 60. № 2. P. 166). https://doi.org/10.1134/S0036023615020126
Сенчурин В.С., Шарутин В.В., Шарутина О.К., Щелоков А.О. // Журн. неорган. химии. 2015. Т. 60. № 10. С. 1320. (Senchurin V.S., Sharutin V.V., Sharutina O.K., Shchelokov A.O. // Russ. J. Inorg. Chem. 2015. V. 60. № 10. P. 1204). https://doi.org/10.1134/S0036023615100174
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Коорд. химия. 2016. Т. 42. № 1. С. 34. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Coord. Chem. 2016. V. 42. № 1. P. 32). https://doi.org/10.1134/S1070328415120076
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2016. Т. 61. № 6. С. 744. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Inorg. Chem. 2016. V. 61. № 6. P. 708). https://doi.org/10.1134/S0036023616060164
Yang M., Gabbai F.P. // Inorg.Chem. 2017. V. 56. P. 8644.
Sharutin V.V., Sharutina O.K., Efremov A.N. et al. // J. Fluor. Chem. 2018. V. 216. P. 7.
Sharutin V.V., Sharutina O.K. // Bull. South Ural State Univ. Ser. Chem. 2017. V. 9. P. 57.
Шарутин В.В., Пакусина А.П., Шарутина О.К. и др. // Коорд. химия. 2004. Т. 30. № 8. С. 578. (Sharutin V.V., Pakusina A.P., Sharutina O.K. et al. // Russ. J. Coord. Chem. 2004. V. 30. № 8. P. 541). https://doi.org/10.1023/B:RUCO.0000037432.61330.07
Шарутин В.В., Егорова И.В., Шарутина О.К. и др. // Коорд. химия. 2004. Т. 30. № 12. С. 925. (Sharutin V.V., Egorova I.V., Sharutina O.K. et al. // Russ. J. Coord. Chem. 2004. V. 30. № 12. P. 874). https://doi.org/10.1007/s11173-005-0042-1
Шарутин В.В., Егорова И.В., Дорофеева О.А. и др. // Журн. неорган. химии. 2004. Т. 49. № 11. С. 1821.
Sharutin V.V., Sharutina O.K., Senchurin V.S. et al. // Bull. South Ural State Univ. Ser. Chem. 2015. V. 7. № 4. P. 44.
Шарутин В.В., Шарутина О.К., Хисамов Р.М., Сенчурин В.С. // Журн. неорган. химии. 2017. Т. 62. № 6. С. 782. (Sharutin V.V., Sharutina O.K., Khisamov R.M., Senchurin V.S. // Russ. J. Inorg. Chem. 2017. V. 62. № 6. P. 766). https://doi.org/10.1134/S0036023617060201
Шарутин В.В., Пакусина А.П., Шарутина О.К. и др. // Коорд. химия. 2007. Т. 33. № 2. С. 101. (Sharutin V.V., Pakusina A.P., Sharutina O.K. et al. // Russ. J. Coord. Chem. 2007. V. 33. № 2. P. 96). https://doi.org/10.1134/S1070328407020042
Шарутин В.В., Сенчурин В.С., Шарутина О.К. и др. // Журн. неорган. химии. 2011. Т. 56. № 4. С. 599. (Sharutin V.V., Senchurin V.S., Sharutina O.K. et al. // Russ. J. Inorg. Chem. 2011. V. 56. № 4. P. 558). https://doi.org/10.1134/S0036023610071010
Lin T.-P., Nelson R.C., Wu T. et al. // Chem. Sci. 2012. V. 3. P. 1128.
Шарутин В.В., Сенчурин В.С., Шарутина О.К., Акулова Е.В. // Журн. общ. химии. 2008. Т. 78. № 12. С. 1999. (Sharutin V.V., Senchurin V.S., Sharutina O.K. et al. // Russ. J. Gen. Chem. 2008. V. 78. № 12. P. 2344).
Егорова И.В., Жидков В.В., Гринишак И.П. и др. // Журн. неорган. химии. 2018. Т. 63. № 6. С. 745. (Egorova I.V., Zhidkov V.V., Grinishak I.P. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 6. P. 781). https://doi.org/10.1134/S0036023618060086
Yin H., Quan L., Li L. // Inorg. Chem. Commun. 2008. V. 11. P. 1121.
Fan J. // Acta Crystallogr. E. 2009. V. 65. m12.
Asghar F., Badshah A., Shah A. et al. // J. Organomet. Chem. 2012. V. 717. P. 1.
Zhang X.-Y., Cui L.-S., Zhang X. et al. // J. Mol. Struct. 2017. V. 1134. P. 742.
Wang F., Yin H., Yue C. et al. // J. Organomet. Chem. 2013. V. 738. P. 35.
Oliveira L.G., Silva M.M., Paula F.C.S. et al. // Molecules. 2011. V. 16. P. 10314.
Dong L., Yin H., Wen L., Wang D. // Acta Crystallogr. E. 2009. V. 65. m1438.
Wen L., Yin H., Wang C. // Acta Crystallogr. E. 2009. V. 65. m1442.
Islam A., Rodrigues B.L., Marzano I.M. et al. // Eur. J. Med. Chem. 2016. V. 109. P. 254.
Wen L., Yin H., Li W., Wang D. // Inorg. Chim. Acta. 2010. V. 363. P. 676.
Islam A., Da Silva J.G., Berbet F.M. et al. // Molecules. 2014. V. 19. P. 6009.
Wen L., Yin H., Wang C. // Acta Crystallogr. E. 2009. V. 65. m1443.
Quan L., Yin H., Cui L. et al. // Acta Crystallogr. E. 2009. V. 65. m656.
Mushtaq R., Rauf M.K., Bolte M. et al. // Appl. Organomet. Chem. 2017. V. 31. e3606.
Iftikhar T., Rauf M.K., Sarwar S. et al. // J. Organomet. Chem. 2017. V. 851. P. 89.
Wu Q., Yin H., Yue C. et al. // J. Coord. Chem. 2012. V. 65. P. 2098.
Wen L., Yin H., Quan L., Wang D. // Acta Crystallogr. E. 2008. V. 64. m1303.
Wen L., Yin H., Wang D. et al. // Acta Crystallogr. E. 2008. V. 64. m1426.
Yin H.-D., Wen L.-Y., Cui J.-C., Li W.-K. // Polyhedron. 2009. V. 28. P. 2919.
Hong M., Yin H.-D., Li W.-K., You X.-Y. // Inorg. Chem. Commun. 2011. V. 14. P. 1616.
Quan L., Yin H., Fu W. // Acta Crystallogr. E. 2011. V. 67. m713.
Quan L., Yin H., Wang D. // Acta Crystallogr. E. 2008. V. 64. m1302.
Beniwal S., Kumar A., Chhimpa S. et al. // Appl. Organomet. Chem. 2019. P. 4712.
Quan L., Yin H., Wang D. // Acta Crystallogr. E. 2008. V. 64. m349.
Quan L., Yin H., Wang D. // Acta Crystallogr. E. 2009. V. 65. m99.
Jiang J.,Yin H., Wang D. et al. // Dalton Trans. 2013. V. 42. P. 8563.
Mushtaq R., Rauf M.K., Bond M. et al. // Appl. Organomet. Chem. 2016. V. 30. P. 465.
Sarwar S., Iftikhar T.,Rauf M.K. et al. // Inorg. Chim. Acta. 2018. V. 476. P. 12.
Yu L., Ma Y.-Q., Liu R.-C. et al. // Polyhedron. 2004. V. 23. P. 823.
Yu L., Ma Y.-Q., Wang G.-C., Li J.-S. // Heteroatom Chem. 2004. V. 15. P. 32.
Kishore P., Ali J., Narasimhulu G., Baskar V. // J. Chem. Sci. 2018. V. 130. P. 100.
Saleem L., Altaf A.A., Badshah A. et al. // Inorg. Chim. Acta. 2018. V. 474. P. 148.
Kather R., Svoboda T., Wehrhahn M. et al. // Chem. Commun. 2015. V. 51. P. 5932.
Betz R., Junggeburth S., Klufers P., Mayer P. // Acta Crystallogr. E. 2010. V. 66. m28.
Srungavruksham N.K., Baskar V. // Eur. J. Inorg. Chem. 2013. P. 4345.
Шарутин В.В., Шарутина O.К., Пакусина А.П., Смирнова С.А. // Журн. неорган. химии. 2009. Т. 54. № 10. С. 1705. (Sharutin V.V., Sharutina O.K., Pakusina A.P., Smirnova S.A. // Russ. J. Inorg. Chem. 2009. V. 54. № 10. P. 1630). https://doi.org/10.1134/S0036023609100209
Шарутин В.В., Шарутина O.К., // Журн. общ. химии 2016. Т. 86. № 8. С. 1360.
Шарутин В.В., Шарутина O.К., Сенчурин В.С., Андреев П.А. // Журн. общ. химии. 2018. Т. 88. № 5. С. 866.
Шарутин В.В., Пакусина А.П., Шарутина O.К. и др. // Коорд. химия. 2007. Т. 33. № 2. С. 109. (Sharutin V.V., Pakusina, A.P., Sharutina O.K., et al. // Russ. J. Coord. Chem. 2007. V. 33. № 2. P. 104). https://doi.org/10.1134/S1070328407020054
Li N., Qiu R., Zhang X. et al. // Tetrahedron. 2015. V. 71. P. 4275.
Robertson A.P.M., Burford N., McDonald R., Ferguson M.J. // Angew. Chem. Int. Ed. 2014. V. 53. P. 3480.
Slawin A.M.Z., Waddell P.G., Woollins J.D. // Acta Crystallogr. E. 2010. V. 66. m418.
Wade C.R., Lin T.-P., Nelson R.C. et al. // J. Am. Chem. Soc. 2011. V. 133. P. 8948.
Wade C.R., Gabbai F.P. // Angew. Chem. Int. Ed. 2011. V. 50. P. 7369.
Copsey M.C., Gallon S.B., Grocott S.K. et al. // Inorg. Chem. 2005. V. 44. P. 5495.
Ugarte R.A., Devarajan D., Mushinski R.M., Hudnall T.W. // Dalton Trans. 2016. V. 45. P. 11150.
Dinsdale N., Jeffery J.C., Kilby R.J. et al. // Inorg. Chim. Acta. 2007. V. 360. P. 418.
Chandrasekhar V., Thirumoorthi R. // Organometallics. 2009. V. 28. P. 2637.
Mills M.B., Hollingshead A.G., Maahs A.C. et al. // CrystEngComm. 2015. V. 17. P. 7816.
Urbanova I., Jambor R., Ruzicka A. et al. // Dalton Trans. 2014. V. 43. P. 505.
Benjamin S.L., Levason W., Reid G. // Organometallics. 2013. V. 32. P. 2760.
Wade C.R., Gabbai F.P. // Organometallics. 2011. V. 30. P. 4479.
Christianson A.M., Gabbai F.P. // Chem.Commun. 2017. V.53. P. 2471.
Hirai M., Cho J., Gabbai F.P. // Chem. Eur. J. 2016. V. 22. P. 6537.
Uchiyama Y., Sugimoto J., Shibata M. et al. // Bull. Chem. Soc. Jpn. 2009. V. 82. P. 819.
Qiao Y., Jiang J., Cui J. // Acta Crystallogr. E. 2012. V. 68. m1552.
Lo Y.-H., Gabbai F.P. // Organometallics. 2018. V. 37. P. 2500.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Бутлеровские сообщения. 2013. Т. 36. № 11. С. 98.
Jones J.S., Wade C.R., Gabbai F.P. // Organometallics. 2015. V. 34. P. 2647.
Sen S., Ke I.-S., Gabbai F.P. // Inorg. Chem. 2016. V. 55. P. 9162.
Wade C.R., Gabbai F.P. // Angew. Chem. 2011. V. 123. P. 7369.
Sen S., Ke I.-S., Gabbai F.P. // Organometallics. 2017. V. 36. P. 4224.
Yang H., Gabbai F.P. // J. Am. Chem. Soc. 2015. V. 137. P. 13425.
Ke I.-S., Gabbai F.P. // Inorg. Chem. 2013. V. 52. P. 7145.
Christianson A.M., Rivard E., Gabbai F.P. // Organometallics. 2017. V. 36. P. 2670.
Neumüller B., Dehnicke K. // Z. Anorg. Allg. Chem. 2004. V. 630. P. 1360.
Шарутин В.В., Сенчурин В.С., Шарутина О.К., Казаков М.В. // Журн. общ. химии. 2012. Т. 82. № 1. С. 99.
Gkaniatsou E.I., Banti C.N., Kourkoumelis N. et al. // J. Inorg. Biochem. 2015. V. 150. P. 108.
Thepe T.C., Garascia R.J., Selvoski M.A., Patel A.N. // Ohio J. Sci. 1977. V. 77. P. 134. http://hdl.handle.net/1811/22448.
Гущин А.В., Калистратова О.С., Верховых Р.А. и др. // Вест. Нижегород. ун-та им. Н.И. Лобачевского. 2013. № 1. С. 86.
Шарутин В.В., Шарутина О.К., Пакусина А.П. и др. // Журн. неорган. химии. 2008. Т. 53. № 8. С. 1335. (Sharutin V.V., Sharutina O.K., Pakusina A.P. et al. // Russ. J. Inorg. Chem. 2008. V. 53. № 9. P. 1242). https://doi.org/10.1134/S0036023608080160
Артемьева Е.В., Шарутина О.К., Шарутин В.В., Буланова А.В. // Журн. неорган. химии. 2020. Т. 65. № 1. С. 25. (Artem’eva E.V., Sharutin V.V., Sharutina O.K., Bulanova A.V. // Russ. J. Inorg. Chem. 2020. V. 65. № 1. P. 22). https://doi.org/10.1134/S0036023620010039
Шарутин В.В., Сенчурин В.С., Шарутина О.К. // Бутлеровские сообщения. 2010. Т. 22. № 12. С. 7.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. и др. // Журн. неорган. химии. 2011. Т. 56. № 7. С. 1125. (Sharutin V.V., Sharutina O.K., Senchurin V.S. et al. // Russ. J. Inorg. Chem. 2011. V. 56. № 7. P. 1060). https://doi.org/10.1134/S0036023611070242
Шарутин В.В., Сенчурин В.С., Шарутина О.К.и др. // Бутлеровские сообщения. 2011. Т. 28. № 19. С. 54.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. и др.// Бутлеровские сообщения. 2012. Т. 29. № 3. С. 51.
Шарутин В.В., Сенчурин В.С., Шарутина О.К. // Бутлеровские сообщения. 2010. Т. 22. № 11. С. 46.
Шарутин В.В., Шарутина О.К., Сенчурин В.С., Чагарова О.В. // Журн. общ. химии. 2012. Т. 82. № 10. С. 1646.
Fukin G.K., Samsonov M.A., Kalistratova O.S., Gushchin A.V. // Struct. Chem. 2016. V. 27. № 1. P. 357.
Шарутин В.В., Сенчурин В.С., Шарутина О.К., Чагарова О.В. // Журн. неорган. химии. 2011. Т. 56. № 7. С. 1129. (Sharutin V.V., Senchurin V.S., Sharutina O.K., Chagarova O.V. // Russ. J. Inorg. Chem. 2011. V. 56. № 7. P. 1064). https://doi.org/10.1134/S0036023611070254
Шарутин В.В., Сенчурин В.С., Шарутина О.К., Чагарова О.В. // Журн. общ. химии 2011. Т. 81. № 11. С. 1789.
Шарутин В.В., Сенчурин В.С., Шарутина О.К. и др. // Коорд. химия. 2011. Т. 37. № 10. С. 782. (Sharutin V.V., Senchurin V.S., Sharutina O.K. et al. // Russ. J. Coord. Chem. 2011. V. 37. № 10. P. 781). https://doi.org/10.1134/S1070328411090089
Шарутин В.В., Шарутина О.К., Толстогузов Д.С. // Журн. общ. химии. 2014. Т. 84. № 9. С. 1516.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2014. Т. 59. № 4. С. 481. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Inorg. Chem. 2014. V. 59. № 4. P. 326). https://doi.org/10.1134/S0036023614040202
Шарутин В.В., Егорова И.В., Пакусина А.П. и др. // Коорд. химия. 2007. Т. 33. № 3. С. 176. (Sharutin V.V., Egorova I.V., Pakusina A.P. et al. // Russ. J. Coord. Chem. 2007. V. 33. № 3. P. 168). https://doi.org/10.1134/S1070328407030037
Andreev P.V., Somov N.V., Kalistratova O.S. et al. // Acta Crystallogr. E. 2013. V. 69. m167.
Гущин А.В., Шашкин Д.В., Прыткова Л.К. и др. // Журн. общ. химии. 2011. Т. 81. № 3. С. 397.
Duffin R.N., Blair V.L., Kedzierski L., Andrews P.C. // Dalton Trans. 2018. V. 47. P. 971.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2014. Т. 59. № 9. С. 1178. (Sharutin V.V., Sharutina O.K., Senchurin V.S. // Russ. J. Inorg. Chem. 2014. V. 59. № 9. P. 947). https://doi.org/10.1134/S0036023614090162
Шарутин В.В., Молокова О.В., Шарутина О.К., Акимова Т.И. // Журн. общ. химии. 2009. Т. 79. № 8. С. 1297.
Шарутин В.В., Шарутина О.К., Пакусина А.П. и др. // Коорд. химия. 2004. Т. 30. № 1. С. 15. (Sharutin V.V., Sharutina O.K., Pakusina A.P. et al. // Russ. J. Coord. Chem. 2004. V. 30. № 1. P. 13). https://doi.org/10.1023/B:RUCO.0000011636.28262.d3
Шарутин В.В., Молокова О.В., Шарутина О.К., Смирнова С.А. // Журн. неорган. химии. 2012. Т. 57. № 9. С. 1334. (Sharutin V.V., Molokova O.V., Sharutina O.K., Smirnova S.A. // Russ. J. Inorg. Chem. 2012. V. 57. № 9. P. 1252). https://doi.org/10.1134/S0036023612090185
Polychronis N.M., Banti C.N., Raptopoulou C.P. et al. // Inorg. Chim. Acta. 2019. V. 489. P. 39.
Шарутин В.В., Пакусина А.П., Шарутина О.К., Почекутова Т.С. // Журн. неорган. химии. 2008. Т. 53. № 11. С. 1857. (Sharutin V.V., Pakusina A.P., Sharutina O.K., Pochekutova T.S. // Russ. J. Inorg. Chem. 2008. V. 53. № 11. P. 1737). https://doi.org/10.1134/S0036023608110119
Шарутин В.В., Шарутина О.К., Ефремов А.Н., Артемьева Е.В. // Журн. неорган. химии. 2019. Т. 64. № 5. С. 482. (Sharutin V.V., Sharutina O.K., Efremov A.N., Artem’eva E.V. // Russ. J. Inorg. Chem. 2019. V. 64. № 5. P. 597). https://doi.org/10.1134/S0036023619050188
Шарутин В.В., Шарутина О.К., Артемьева Е.В., Макерова М.С. // Журн. неорган. химии. 2015. Т. 60. № 2. С. 207. (Sharutin V.V., Sharutina O.K., Artem’eva E.V., Makerova M.S. // Russ. J. Inorg. Chem. 2015. V. 60. № 2. P. 170). https://doi.org/10.1134/S0036023615020138
Шарутин В.В., Шарутина О.К., Габитова Д.М., Шайхвалеева С.Я. // Журн. неорган. химии. 2017. Т. 62. № 1. С. 61. (Sharutin V.V., Sharutina O.K., Gabitova D.M., Shaikhvaleeva S.Y. // Russ. J. Inorg. Chem. 2017. V. 62. № 1. P. 55). https://doi.org/10.1134/S003602361701017X
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Коорд. химия. 2017. Т. 43. № 8. С. 496. (Sharutin V.V., Sharutina O.K., Efremov A.N. // Russ. J. Coord. Chem. 2017. V. 43. № 8. P. 526). https://doi.org/10.1134/S1070328417080073
Шарутин В.В., Шарутина О.К., Артемьева Е.В., Макерова М.С. // Журн. общ. химии. 2016. Т. 86. № 12. С. 2039.
Sharutin V.V., Sharutina O.K., Artem’eva E.V., Makerova M.S. // Bull. South Ural State Univ. Ser. Chem. 2015. V. 7. № 2. P. 17.
Sharutin V.V., Sharutina O.K., Artem’eva E.V., Makerova M.S. // Bull. South Ural State Univ. Ser. Chem. 2016. V. 8. № 2. P. 61.
Artem'eva E.V., Makerova M.S., Sharutin V.V., Sharutina O.K. // Bull. South Ural State Univ. Ser. Chem. 2017. V. 9. № 2. P. 50.
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Журн. общ. химии. 2016. Т. 86. № 5. С. 876.
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Коорд. химия. 2017. Т. 43. № 9. С. 521. (Sharutin V.V., Sharutina O.K., Efremov A.N. // Russ. J. Coord. Chem. 2017. V. 43. № 9. P. 565). https://doi.org/10.1134/S1070328417090081
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Коорд. химия. 2016. Т. 42. № 11. С. 712. (Sharutin V.V., Sharutina O.K., Efremov A.N. // Russ. J. Coord. Chem. 2016. V. 42. № 11. P. 737). https://doi.org/10.1134/S1070328416110087
Sharutin V.V., Sharutina O.K. // Bull. South Ural State Univ. Ser. Chem. 2016. V. 8. № 1. P. 57.
Шарутин В.В., Шарутина О.К., Ефремов А.Н., Андреев П.В. // Журн. неорган. химии. 2018. Т. 63. № 2. С. 164. (Sharutin V.V., Sharutina O.K., Efremov A.N., Andreev P.V. // Russ. J. Inorg. Chem. 2018. V. 63. № 2. P. 174). https://doi.org/10.1134/S0036023618020195
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Коорд. химия. 2020. Т. 46. № 1. С. 45. (Sharutin V.V., Sharutina O.K., EfremovA.N. // Russ. J. Coord. Chem. 2020. V. 46. № 1. P. 42). https://doi.org/10.1134/S1070328419120066
Шарутин В.В., Шарутина О.К. // Изв. АН. Сер. хим. 2017. № 4. С. 707. (Sharutin V.V., Sharutina O.K. // Russ. Chem. Bull. 2017. V. 66. № 4. P. 707).
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Журн. неорган. химии. 2016. Т. 61. № 1. С. 46. (Sharutin V.V., Sharutina O.K., Efremov A.N. // Russ. J. Inorg. Chem. 2016. V. 61. № 1. P. 43). https://doi.org/10.1134/S003602361601023X
Шарутин В.В., Шарутина О.К. // Журн. общ. химии. 2016. Т. 86. № 8. С. 1366.
Шарутин В.В., Шарутина О.К., Ефремов А.Н., Андреев П.В. // Коорд. химия. 2018. Т. 44. № 5. С. 333. (Sharutin V.V., Sharutina O.K., Efremov A.N., Andreev P.V. // Russ. J. Coord. Chem. 2018. V. 44. № 10. P. 635). https://doi.org/10.1134/S107032841810010X
Шарутин В.В., Шарутина О.К., Решетникова Р.В. и др. // Журн. неорган. химии. 2017. Т. 62. № 11. С. 1457. (Sharutin V.V., Sharutina O.K., Reshetnikova R.V. et al. // Russ. J. Inorg. Chem. 2017. V. 62. № 11. P. 1450). https://doi.org/10.1134/S003602361711016X
Гущин А.В., Прыткова Л.К., Шашкин Д.В. и др. // Вест. Нижегород. ун-та им. Н.И. Лобачевского. 2010. № 1. С. 95.
Fukin G.K., Samsonov M.A., Arapova A.V. et al. // J. Solid State Chem. 2017. V. 254. P. 32.
Sharutin V.V., Sharutina O.K., Senchutin V.S. // Bull. South Ural State Univ. Ser. Chem. 2015. V. 7. № 4. P. 93.
Шарутин В.В., Шарутина О.К., Казаков М.В. // Журн. неорган. химии. 2014. Т. 59. № 10. С. 1352. (Sharutin V.V., Sharutina O.K., Kazakov M.V. // Russ. J. Inorg. Chem. 2014. V. 59. № 10. P. 1115). https://doi.org/10.1134/S0036023614100167
Шарутин В.В., Шарутина О.К., Сенчурин В.С. и др. // Журн. неорган. химии. 2018. Т. 63. № 7. С. 823. (Sharutin V.V., Sharutina O.K., Senchurin V.S. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 7. P. 867. https://doi.org/10.1134/S0036023618070185
Шарутин В.В., Шарутина О.К., Шалабанова Н.А. // Коорд. химия. 2018. Т. 44. № 6. С. 402. (Sharutin V.V., Sharutina O.K., Shalabanova N.A. // Russ. J. Coord. Chem. 2018. V. 44. P. 765). https://doi.org/10.1134/S1070328418120138
Sharutin V.V., Sharutina O.K. // Bull. South Ural State Univ. Ser. Chem. 2016. V. 8. № 4. P. 61.
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Журн. неорган. химии. 2018. Т. 63. № 3. С. 327. (Sharutin V.V., Sharutina O.K., Efremov A.N. // Russ. J. Inorg. Chem. 2018. V. 63. № 3. P. 343). https://doi.org/10.1134/S0036023618030208
Шарутин В.В., Шарутина О.К. // Журн. неорган. химии. 2015. Т. 60. № 12. С. 1631. (Sharutin V.V., Sharutina O.K. // Russ. J. Inorg. Chem. 2015. V. 60. № 12. P. 1491). https://doi.org/10.1134/S0036023615120219
Шарутин В.В., Шарутина О.К. // Журн. неорган. химии. 2014. Т. 59. № 11. С. 1507. (Sharutin V.V., Sharutina O.K. // Russ. J. Inorg. Chem. 2014. V. 59. № 11. P. 1263). https://doi.org/10.1134/S0036023614110229
Шарутин В.В., Шарутина О.К., Чагарова О.В., Молокова О.В. // Журн. общ. химии. 2011. Т. 81. № 11. С. 1793.
Betz R., Lindner C., Klufers P., Mayer P. // Acta Crystallogr. E. 2009. V. 65. m253.
Holmes R.R., Day R.O., Chandrasekhar V., Holmes J.M. // Inorg. Chem. 1987. V. 26. P. 157.
Piskunov A.V., Poddel’sky A.I. // Glob. J. Inorg. Chem. 2011. V. 2. № 2. P. 110.
Poddel’sky A.I. // Antimony: Characteristics, Compounds and Applications / Eds M. Razeghi. N.Y.: Nova Science Publishers, 2012. P. 267.
Cherkasov V.K., Grunova E.V., Poddel’sky A.I. et al. // J. Organomet. Chem. 2005. V. 690. № 5. P. 1273.
Poddel’sky A.I., Vavilina N.N., Somov N.V. et al. // J. Organomet. Chem. 2009. V. 694. № 21. P. 3462.
Poddel’sky A.I., Piskunov A.V., Druzhkov N.O. et al. // Z. Anorg. Allg. Chem. 2009. V. 635. № 15. P. 2563.
Poddel’sky A.I., Smolyaninov I.V., Somov N.V. et al. // J. Organomet. Chem. 2010. V. 695. № 4. P. 530.
Куропатов В.А., Клементьева С.В., Поддельский А.И. и др. // Изв. АН. Сер. хим. 2010. № 9. С. 1652. (Kuropatov V.A., Klementieva S.V., Poddel’sky A.I. et al. // Russ. Chem. Bull., Int. Ed., 2010. V. 59. № 9. P. 1698).
Poddel’sky A.I., Somov N.V., Druzhkov N.O. et al. // J. Organomet. Chem. 2011. V. 696. № 2. P. 517.
Поддельский А.И., Смолянинов И.В., Вавилина Н.Н. и др. // Коорд. химия. 2012. Т. 38. № 4. С. 296. (Poddel’sky A.I., Smolyaninov I.V., Vavilina N.N. et al. // Russ. J. Coord. Chem. 2012. V. 38. № 4. P. 284). https://doi.org/10.1134/S1070328412040094
Poddel’sky A.I., Baranov E.V., Fukin G.K. et al. // J. Organomet. Chem. 2013. V. 733. P. 44.
Poddel’sky A.I., Smolyaninov I.V., Fukin G.K. et al. // J. Organomet. Chem. 2016. V. 824. P. 1.
Poddel’sky A.I., Druzhkov N.O., Fukin G.K. et al. // Polyhedron. 2017. V. 124. P. 41.
Poddel’sky A.I., Arsenyev M.V., Astaf’eva T.V. et al. // J. Organomet. Chem. 2017. V. 835. P. 17.
Арсеньев М.В., Охлопкова Л.С., Поддельский А.И., Фукин Г.К. // Коорд. химия. 2018. Т. 44. № 1. С. 71. (Arsen’ev M.V., Okhlopkova L.S., Poddel’skii A.I., Fukin G.K. // Russ. J. Coord. Chem. 2018. V. 44. № 2. P. 162). https://doi.org/10.1134/S1070328418020021
Arsenyev M.V., Astaf’eva T.V., Baranov E.V. et al. // Mendeleev Commun. 2018. V. 28. P. 76.
Abakumov G.A., Poddel’sky A.I., Grunova E.V. et al. // Angew. Chem. Int. Ed. 2005. V. 44. P. 2767.
Абакумов Г.А., Черкасов В.К., Грунова Е.В. и др. // Докл. РАН. 2005. Т. 405. № 2. С. 199. (Abakumov G.A., Cherkasov V.K., Grunova E.V. et al. // Dokl. Chem. 2005. V. 405. № 1–3. P. 222).
Cherkasov V.K., Abakumov G.A., Grunova E.V. et al. // Chem. Eur. J. 2006. V. 12. № 14. P. 3916.
Poddel’sky A.I., Kurskii Yu.A., Piskunov A.V. et al. // Appl. Organomet. Chem. 2011. V. 25. № 3. P. 180.
Fukin G.K., Baranov E.V., Jelsch C. et al. // J. Phys. Chem. A. 2011. V. 115. № 29. P. 8271.
Fukin G.K., Baranov E.V., Poddel’sky A.I. et al. // Chem. Phys. Chem. 2012. V. 13. № 17. P. 3773.
Фукин Г.К., Самсонов М.А., Баранов Е.В. и др. // Изв. АН. Сер. хим. 2016. № 1. С. 54. (Fukin G.K., Samsonov M.A., Baranov E.V. et al. // Russ. Chem. Bull. 2016. V. 65. № 1. P. 54).
Фукин Г.К., Самсонов М.А., Поддельский А.И., Черкасов В.К. // Изв. АН. Сер. хим. 2016. № 1. С. 61. (Fukin G.K., Samsonov M.A., Poddel’sky A.I., Cherkasov V.K. // Russ. Chem. Bull. 2016. V. 65. № 1. P. 61).
Фукин Г.К., Самсонов М.А., Баранов Е.В. и др. // Коорд. химия. 2017. Т. 43. № 12. С. 759. (Fukin G.K., Samsonov M.A., Baranov E.V. et al. // Russ. J. Coord. Chem. 2017. V. 43. № 12. P. 858). https://doi.org/10.1134/S1070328417120028
Поддельский А.И., Смолянинов И.В., Курский Ю.А. и др. // Изв. АН. Сер. хим. 2009. № 3. С. 520. (Poddel’sky A.I., Smolyaninov I.V., Kurskii Yu.A. et al. // Russ. Chem. Bull. 2009. V. 58. № 3. P. 532).
Poddel’sky A.I., Smolyaninov I.V., Kurskii Yu.A. et al. // J. Organomet. Chem. 2010. V. 695. № 8. P. 1215.
Смолянинов И.В., Поддельский А.И., Берберова Н.Т. и др. // Коорд. химия. 2010. Т. 36. № 9. С. 650. (Smolyaninov I.V., Poddel’skiy A.I., Berberova N.T. et al. // Russ. J. Coord. Chem. 2010. V. 36. № 9. P. 644). https://doi.org/10.1134/S1070328410090022
Поддельский А.И., Илякина Е.В., Смолянинов И.В. и др. // Изв. АН. Сер. хим. 2014. № 4. С. 923. (Poddel’skii A.I., Ilyakina E.V., Smolyaninov I.V. et al. // Russ. Chem. Bull. 2014. V. 63. № 4. P. 923).
Поддельский А.И., Охлопкова Л.С., Мещерякова И.Н. и др. // Коорд. химия. 2019. Т. 45. № 2. С. 120. (Poddel’skii A.I., Okhlopkova L.S., Meshcheryakova I.N. et al. // Russ. J. Coord. Chem. 2019. V. 45. № 2. P. 133). https://doi.org/10.1134/S1070328419010093
Поддельский А.И., Смолянинов И.В. // Журн. общ. химии. 2010. Т. 80. № 3. С. 538.
Смолянинова С.А., Поддельский А.И., Смолянинов И.В., Берберова Н.Т. // Коорд. химия. 2014. V. 40. № 5. С. 274. (Smolyaninova S.A., Poddel’sky A.I., Smolyaninov I.V., Berberova N.T. // Russ. J. Coord. Chem. 2014. V. 40. № 5. P. 273). https://doi.org/10.1134/S107032841405011X
Смолянинов И.В., Поддельский А.И., Смолянинова С.А., Мовчан Н.О. // Журн. общ. химии. 2014. V. 84. № 9. С. 1523.
Poddel’sky A.I., Smolyaninov I.V., Berberova N.T. et al. // J. Organomet. Chem. 2015. V. 789–790. P. 8.
Poddel’sky A.I., Smolyaninov I.V., Fukin G.K. et al. // J. Organomet. Chem. 2018. V. 867. P. 238.
Poddel’sky A.I., Astaf’eva T.V., Smolyaninov I.V. et al. // J. Organomet. Chem. 2018. V. 873. P. 57.
Smolyaninov I.V., Antonova N.A., Poddel’sky A.I. et al. // J. Organomet. Chem. 2011. V. 696. № 13. P. 2611.
Смолянинов И.В., Антонова Н.А., Поддельский А.И. и др. // Докл. АН. 2012. Т. 443. № 1. С. 64 (Smolyaninov I.V., Antonova N.A., Poddel’sky A.I. et al. // Dokl. Chem. 2012. V. 443. № 1. P. 72).
Smolyaninov I.V., Antonova N.A., Poddel’sky A.I. et al. // Appl. Organomet. Chem. 2012. V. 26. № 6. P. 277.
Смолянинов И.В., Поддельский А.И., Антонова Н.А. и др. // Коорд. химия. 2013. Т. 39. № 2. С. 75. (Smolyaninov I.V., Poddel’skii A.I., Antonova N.A. et al. // Russ. J. Coord. Chem. 2013. V. 39. № 2. P. 165). https://doi.org/10.1134/S1070328413020073
Smolyaninov I.V., Antonova N.A., Poddel’sky A.I. et al. // Appl. Organomet. Chem. 2014. V. 28. P. 274.
Смолянинов И.В., Поддельский А.И., Корчагина Е.О. и др. // Докл. РАН. 2015. Т. 460. № 5. С. 561. (Smolyaninov I.V., Poddel’sky A.I., Korchagina E.O. et al. // Dokl. Phys. Chem. 2015. V. 460. № 2. P. 45).
Смолянинов И.В., Поддельский А.И., Смолянинова С.А. и др. // Изв. АН. Сер. хим. 2015. № 9. С. 2223. (Smolyaninov I.V., Poddel’sky A.I., Smolyaninova S.A. et al. // Russ. Chem. Bull. 2015. V. 64. № 9. P. 2223).
Смолянинов И.В., Поддельский А.И., Смолянинова С.А., Берберова Н.Т. // Электрохимия. 2015. Т. 51. № 11. С. 1155. (Smolyaninov I.V., Pod-del’skii A.I., Smolyaninova S.A., Berberova N.T. // Russ. J. Electrochem. 2015. V. 51. № 11. P. 1021).
Poddel’sky A.I., Somov N.V., Kurskii Yu.A. et al. // J. Organomet. Chem. 2008. V. 693. № 21–22. P. 3451.
Смолянинов И.В., Поддельский А.И., Берберова Н.Т. // Электрохимия. 2011. Т. 47. № 11. С. 1295. (Smolyaninov I.V., Poddel’skii A.I., Berberova N.T.// Russ. J. Electrochem. 2011. V. 47. № 11. P. 1211).
Смолянинов И.В., Поддельский А.И., Смолянинова С.А., Берберова Н.Т. // Коорд. химия. 2014. Т. 40. № 10. С. 608. (Smolyaninov I.V., Poddel’sky A.I., Smolyaninova S.A., Berberova N.T. // Russ. J. Coord. Chem. 2014. V. 40. № 10. P. 726). https://doi.org/10.1134/S1070328414090097
Arsenyev M.V., Shurygina M.P., Poddel’sky A.I. et al. // J. Polym. Res. 2013. V. 20. № 3. P. 98.
Lenshina N.A., Shurygina M.P., Arsenyev M.V. et al. // J. Coord. Chem. 2015. V. 68. № 23. P. 4159.
Chesnokov S.A., Lenshina N.A., Arsenyev M.V. et al. // Appl. Organomet. Chem. 2017. V. 31. P. e3553.
Baryshnikova S.V., Bellan E.V., Poddel’sky A.I. et al. // Eur. J. Inorg. Chem. 2016. V. 2016. № 33. P. 5230.
Поддельский А.И., Арсеньев М.В., Охлопкова Л.С. и др. // Коорд. химия. 2017. Т. 43. № 12. С. 744. (Poddel’sky A.I., Arsen’ev M.V., Okhlopkova L.S. et al. // Russ. J. Coord. Chem. 2017. V. 43. № 12. P. 843). https://doi.org/10.1134/S1070328417120089
Протасенко Н.А., Поддельский А.И., Смолянинов И.В. и др. // Изв. АН. Сер. хим. 2014. № 4. С. 930. (Protasenko N.A., Poddel’skii A.I., Smolyaninov I.V. et al. // Russ. Chem. Bull. 2014. V. 63. № 4. P. 930).
Okhlopkova L.S., Poddel’sky A.I., Smolyaninov I.V. et al. // J. Organomet. Chem. 2019. V. 897. P. 32.
Охлопкова Л.С., Смолянинов И.В., Баранов E.В., Поддельский А.И. // Коорд. химия. 2020. Т. 46. № 7. С. 410. (Okhlopkova L.S., Smolyaninov I.V., Baranov E.V., Poddel’skii A.I. // Russ. J. Coord. Chem. 2020. V. 46. № 7. P. 466). https://doi.org/10.1134/S1070328420060081
Chen C.-H., Gabbai F.P. // Angew. Chem. Int. Ed. 2017. V. 56. P. 1799.
Hirai M., Gabbai F.P. // Chem. Sci. 2014. V. 5. P. 1886.
Chen C.-H., Gabbai F.P. // Dalton Trans. 2018. V. 47. P. 12075.
Hirai M., Gabbai F.P. // Angew. Chem. Int. Ed. 2015. V. 54. P. 1205.
Tofan D., Gabbai F.P. // Chem. Sci. 2016. V. 7. P. 6768.
Jones J.S., Wade C.R., Gabbai F.P. // Angew. Chem. Int. Ed. 2014. V.53. P. 8876.
Ke I.-S., Jones J.S., Gabbai F.P. // Angew. Chem. Int. Ed. 2014. V. 53. P. 2633.
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Журн. неорган. химии. 2020. Т. 65. № 1. С. 49. (Sharutin V.V., Sharutina O.K., Efremov A.N. // Russ. J. Inorg. Chem. 2020. V. 65. № 1. P. 45). https://doi.org/10.1134/S0036023620010155
Breunig H.J., Koehne T., Moldovan O. et al. // J. Organomet. Chem. 2010. V. 695. P. 1307.
Pop A., Silvestru A., Juarez-Perez E.J. et al. // Dalton Trans. 2014. V. 43. P. 2221.
Yang H., Gabbai F.P. // J. Am. Chem. Soc. 2014. V. 136. P. 10866.
Sahu S., Gabbai F.P. // J. Am. Chem. Soc. 2017. V. 139. P. 5035.
Jones J.S., Gabbai F.P. // Chem. Eur. J. 2017. V. 23. P. 1136.
You D., Yang H., Sen S., Gabbai F.P. // J. Am. Chem. Soc. 2018. V. 140. P. 9644.
Prabhu M.S.R., Jami A.K., Baskar V. // Organometallics. 2009. V.28. P. 3953.
Frazee C., Burford N., McDonald R. et al. // Chem. Eur. J. 2018. V. 24. P. 4011.
Yin H., Wu Q., Hong M., Li W. // Z. Anorg. Allg. Chem. 2012. V. 638. P. 725.
You D., Gabbai F.P. // J. Am. Chem. Soc. 2017. V. 139. P. 6843.
Yang P., Bassil B.S., Lin Z. et al. // Chem. Eur. J. 2015. V. 21. P. 15600.
Nicholson B.K., Clark C.J., Telfer S.G., Groutso T. // Dalton Trans. 2012. V. 41. P. 9964.
Brunig J., Hupf E., Lork E. et al. // Dalton Trans. 2015. V. 44. P. 7105.
Ali S., Baskar V., Muryn C.A., Winpenny R.E.P. // Chem. Commun. 2008. P. 6375.
Ali S., Muryn C.A., Tuna F., Winpenny R.E.P. // Dalton Trans. 2010. V. 39. P. 124.
Ali S., Muryn C.A., Tuna F., Winpenny R.E.P. // Dalton Trans. 2010. V. 39. P. 9588.
Nicholson B.K., Clark C.J., Wright C.E. et al. // Organometallics. 2011. V. 30. P. 6612.
Kishore P.V.V.N., Baskar V. // Inorg. Chem. 2014. V. 53. P. 6737.
Prabhu M.S.R., Ugandhar U., Baskar V. // Dalton Trans. 2016. V. 45. P. 6963.
Nicholson B.K., Clark C.J., Wright C.E., Groutso T. // Organometallics. 2010. V. 29. P. 6518.
Ugandhar U., Baskar V. // Dalton Trans. 2016. V. 45. P. 6269.
Beckmann J., Hesse M. // Organometallics. 2009. V. 28. P. 2345.
Liu Z.-Q., Ozawa Y., Yagasaki A. // Bull. Chem. Soc. Jpn. 2014. V. 87. P. 1245.
Srungavruksham N.K., Baskar V. // Dalton Trans. 2015. V. 44. P. 6358.
Jami A.K., Prabhu M.S.R., Baskar V. // Organometallics. 2010. V. 29. P. 1137.
Jami A.K., Baskar V. // Dalton Trans. 2012. V. 41. P. 12524.
Mishra J., Saxena A., Singh S. // Curr. Med. Chem. 2007. V. 14. P. 1153.
Chitnis S.S., Sparkes H.A., Annibale V.T. et al. // Angew. Chem. Int. Ed. 2017. V. 56. P. 9536.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия