

Координационная химия, 2020, T. 46, № 11, стр. 688-697
Гетерометаллические комплексы на основе хинон-катехолата трифенилсурьмы(V)
Л. С. Охлопкова 1, И. В. Смолянинов 2, А. И. Поддельский 1, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Южный научный центр РАН
Ростов-на-Дону, Россия
* E-mail: aip@iomc.ras.ru
Поступила в редакцию 04.05.2020
После доработки 10.06.2020
Принята к публикации 17.06.2020
Аннотация
На основе моноядерного хинон-катехолатного комплекса трифенилсурьмы(V) (Q-Cat)SbPh3, полученного из 4,4'-ди-(6-трет-бутил-3-метил-бензо-1,2-хинона) (Q-Q), синтезированы гетерометаллические комплексы хрома(III) CrIII[(SQ-Cat)SbPh3]3 (I), меди(II) CuII[(SQ-Cat)SbPh3]2 (II), цинка I2Zn(Q-Cat)SbPh3 (III) и IZn(SQ-Cat)SbPh3 (IV), где SQ – соответствующий анион-радикал о-семихинон, Cat – дианион катехолат – моно- и дивосстановленная формы хинонового фрагмента. Одноэлектронное окисление и одноэлектронное восстановление комплекса хрома I приводит к образованию парамагнитных производных [CrIII[(Q-Cat)SbPh3][(SQ-Cat)SbPh3]2]+ (I+) и [CrIII[(Cat-Cat)SbPh3][(SQ-Cat)SbPh3]2]– (I–) соответственно, легко фиксируемых с помощью спектроскопии ЭПР. Данные спектроскопии ЭПР и измерения температурной зависимости магнитной восприимчивости комплекса меди II свидетельствуют в пользу локализации неспаренного электрона на органическом лиганде, что можно объяснить антиферромагнитным обменом металл–лиганд в данном комплексе (J(SQ-Cu) = –157 см–1, J(SQ-SQ) = –10.0 см–1). Координация (Q-Cat)SbPh3 как нейтрального о-хинона на иодид цинка с образованием комплекса III значительно увеличивает окислительную способность хинонового фрагмента в данном комплексе (ERed1 = 0.43 В для III и –0.45 В (Q-Cat)SbPh3 относительно Ag/AgCl/KCl(нас.)), комплекс способен окислять иодид анион до иода с образованием парамагнитного производного IV, имеющего характерный для комплексов типа M(SQ-Cat) спектр ЭПР. Окислительно-восстановительные свойства комплексов I и III исследованы с помощью циклической вольтамперометрии.
Большой интерес ко многим химическим и биохимическим процессам, осуществляемым с помощью гетерометаллических соединений и ферментов, для использования в различных областях от каталитического синтеза топливных элементов до захвата и хранения солнечной энергии вызывает многочисленные попытки имитировать как структуру, так и функцию этих ферментов [1–7]. Значительное внимание уделяется изучению взаимодействия между различными металлами и металлсодержащими органическими фрагментами в синтетических гетерометаллических комплексах, а также влиянию этих взаимодействий на электронное строение, редокс-активные свойства, перенос электронов и химическую реакционную способность. Интересную область исследований представляют комплексные соединения на основе бис-о-хинонов, позволяющих связывать между собой два и более различных металлических центра и являющихся при этом редокс-активными лигандами, способными к обратимому присоединению одного или нескольких электронов. Это не только значительно расширяет диапазон окислительно-восстановительных свойств образующихся соединений, но и приводит к возможности получения комбинаций степеней окисления металла(ов) и восстановления редокс-активных лигандов [8–16].
Спектроскопия электронного парамагнитного резонанса (ЭПР) – важный и информативный метод исследования парамагнитных координационных соединений, содержащих парамагнитные ионы металлов и радикальные частицы, так как позволяет не только получать полезную информацию о строении координационного узла, о взаимном расположении лигандов в координационной сфере металла, но также исследовать динамические процессы, протекающие с изменением геометрии комплексов. Поскольку в редокс-ряду о-хинона одна из восстановленных форм – о-семихиноновая – парамагнитна, во многих случаях важную информацию о строении и химических свойствах о-семихиноновых и родственных комплексов можно получить с помощью спектроскопии ЭПР [17–21]. Не менее информативен метод ЭПР в случае производных бис-о-бензохинонов [12–26]. Ранее мы получили катехолатный комплекс трифенилсурьмы(V) (Q-Cat)SbPh3 на основе 4,4'-ди-(6-трет-бутил-3-метилбензо-1,2-хинона) (Q-Q) [27]. Было показано, что комплекс Q-Q может образовывать о-семихиноновые производные типа M(SQ-Cat)SbPh3 [24].
В настоящей работе исследованы гетерометаллические о-семихиноновые комплексы хрома(III) CrIII[(SQ-Cat)SbPh3]3 (I), меди(II) CuII[(SQ-Cat)SbPh3]2 (II), а также производные цинка типа I2Zn(Q-Cat)SbPh3 (III) с нейтральным координированным о-хиноновым фрагментом и его о-семихиноновое производное IZn(SQ-Cat)SbPh3 (IV).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез, выделение и исследование свойств комплексов проводили в вакуумированных ампулах в отсутствие кислорода. Органические растворители, используемые в работе, очищали по стандартным методикам [28]. Исходный (Q-Cat)SbPh3 синтезировали по описанному методу [27].
Синтез трис-[(4-(3-метил-6-трет-бутилбензосемихинолят-1,2)-3-метил-6-трет-бутилкатехолато)трифенилсурьма(V)]хрома(III) CrIII[(SQ-Cat)SbPh3]3 (I). Раствор (Q-Cat)SbPh3 (100 мг, 0.14 ммоль) в 25 мл ТГФ перемешивали в течение 20 мин с избытком амальгамы таллия. Цвет раствора изменился с вишневого на желто-зеленый. Раствор осторожно декантировали с амальгамы и добавляли при интенсивном перемешивании к раствору второй порции (Q-Cat)SbPh3 (100 мг, 0.14 ммоль) в 20 мл ТГФ. Через 10 мин полученный раствор семихинолята таллия(I) Tl(SQ-Cat)SbPh3 смешивали с раствором тетрагидрофураната трихлорида хрома(III) (35 мг, 0.093 ммоль) в 20 мл ТГФ. Реакционную смесь отфильтровывали от выпавшего осадка TlCl. Затем ТГФ удаляли, остаток растворяли в толуоле. Полученный раствор снова отфильтровывали и концентрировали. Выпавший мелкокристаллический продукт I фиолетового цвета отделяли фильтрованием, промывали холодным толуолом и высушивали в вакууме. Выход I 79%.
| Найдено, %: | C 66.25; | H 5.65; | Sb 16.73. |
| Для C120H123O12CrSb3 | |||
| вычислено, %: | C 66.27; | H 5.66; | Sb 16.84. |
ИК-спектр (ν, см–1): 1658 сл, 1642 сл, 1565 с, 1407 с, 1340 ср, 1316 ср, 1277 с, 1246 с, 1180 ср, 1102 сл, 1069 с, 997 ср, 960 с, 891 сл, 864 сл, 821 сл, 778 сл, 730 с, 694 с, 629 сл, 540 ср, 455 ср.
Синтез бис-[(4-(3-метил-6-трет-бутилбензосемихинолят-1,2)-3-метил-6-трет-бутил-катехолато)трифенилсурьма(V)]меди(II) CuII[(SQ-Cat)- SbPh3]2 (II) выполняли по методу, аналогичному для I, с использованием 19 мг (14 ммоль) СuCl2 и 200 мг (Q-Cat)SbPh3. Выход мелкокристаллического образца II 75%.
ИК-спектр (ν, см–1): 1403 ср, 1303 ср, 1244 сл, 1213 сл, 1169 сл, 1139 сл, 1062 ср, 1039 ср, 960 ср, 893 сл, 863 сл, 820 сл, 771 сл, 727 с, 694 сл, 676 ср, 626 сл, 560 сл, 512 сл.
Синтез O,O'-[4-(2-метил-5-трет-бутилциклогексадиен-1,5-дион-3,4-ил)-3-метил-6-трет-бутилкатехолато]трифенилсурьма(V)-дийодоцинка I2Zn(Q-Cat)SbPh3 (III). К толуольному раствору (Q-Cat)SbPh3 (100 мг, 0.14 ммоль) приливали свежеприготовленный раствор иодида цинка (44.6 мг, 0.14 ммоль) в толуоле. Цвет раствора сразу менялся с вишневого на фиолетовый. После концентрирования раствора и выдерживания его при комнатной температуре в течение 3 ч выделился фиолетовый мелкокристаллический продукт, который отфильтровывали и высушивали при пониженном давлении. Выход III 65%.
ИК-спектр (ν, см–1): 1363 c, 1291 с, 1274 с, 1246 ср, 1212 с, 1140 с, 1050 с, 963 с, 929 сл, 866 сл, 818 сл, 730 с, 671 с, 622 сл, 560 сл, 511 сл, 484 сл, 468 сл.
Синтез [(4-(3-метил-6-трет-бутилбензосемихинолят-1,2)-3-метил-6-трет-бутилкатехолато)трифенилсурьма(V)]иодоцинка IZn(SQ-Cat)SbPh3 (IV). Сине-фиолетовый мелкокристаллический продукт получали из маточного раствора (оставшегося после выделения комплекса III), который охлаждали и выдерживали в течение 2 сут при 0°С. Выход IV 15%.
ИК-спектр (ν, см–1): 1612 ср, 1591 ср, 1575 ср, 1500 ср, 1336 с, 1298 с, 1278 с, 1242 с, 1217 ср, 1184 ср, 1141 ср, 1063 ср, 996 ср, 963 ср, 862 сл, 819 сл, 735 с, 689 с, 624 сл, 562 сл, 521 сл, 504 сл, 454 с.
ИК-спектры регистрировали на Фурье-ИК спектрометре ФСМ 1201 в вазелиновом масле. Электронные спектры поглощения (ЭСП) комплексов регистрировали на спектрофотометрах Perkin Elmer UV/VIS Lambda 25 в диапазоне 280–1100 нм (кварцевые кюветы, толщина поглощающего слоя 10 мм) при 298 К. Элементный анализ проводили на C,H,N-анализаторе Euro EA 3000, а также методом пиролитического разложения в токе кислорода. Квантово-химические расчеты выполняли с использованием программного пакета Gaussian09 [29] с применением метода расчета: B3LYP/def2tzvp. Измерения статической магнитной восприимчивости образцов проводили в Международном Томографическом Центре СО РАН (г. Новосибирск) на MPMS-5S SQUID магнетометре (магнитное поле 0.5 Тл) в температурном интервале 2–330 K. Моделирование магнитных свойств комплексов и определение параметров обменного взаимодействия между парамагнитными центрами проводили с помощью программы Mjollnir [30]. Спектры ЭПР снимали на спектрометре ЭПР Bruker EMX (рабочая частота ~9.7 ГГц). В качестве стандарта при определении g-фактора использовали дифенилпикрилгидразил (ДФПГ, g = 2.0037). Симуляцию спектров ЭПР произвели при помощи WinEPR SimFonia (Bruker). Измерение потенциалов окисления проводили методом циклической вольтамперометрии (ЦВА) в трехэлектродной ячейке с помощью потенциостата IPC-pro в среде аргона. Рабочий электрод – стационарный стеклоуглеродный электрод с диаметром 2 мм, вспомогательный электрод – платиновая пластина (S = 18 мм2), электрод сравнения – Ag/AgCl/KCl(нас.) с водонепроницаемой диафрагмой. Скорость развертки потенциала 0.2 В/с. Масс-спектры электронной ионизации получили с использованием времяпролетного MALDI-Toff масс-спектрометра (матрица – DCTB).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Ранее в ИМХ РАН был синтезирован хиноно-катехолатный комплекс (Q-Cat)SbPh3 [27], представляющий собой удобный объект для получения на его основе гетерометаллических соединений [24]. Так, обменная реакция между о-семихиноновым производным M(SQ-Cat)SbPh3 (М = Na, K, Tl) и хлоридом хрома(III) должна привести к получению соответствующего трис-о-семихинонового производного хрома(III). Действительно, взаимодействие тетрагидрофуранового комплекса хлорида хрома(III) с полученным in situ о-семихинолятом таллия на основе (Q-Cat)SbPh3 в растворе ТГФ (схема 1 ) сопровождается мгновенным изменением окраски реакционной смеси.
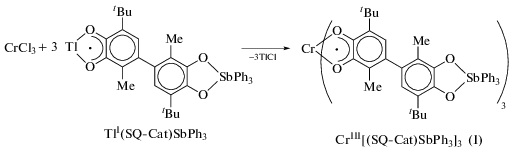
Схема 1 .
В качестве продукта обменной реакции был выделен комплекс, идентифицированный с помощью элементного анализа, ИК- и масс-спектроскопии как трис-лигандное производное хрома I.
Образование комплекса I, содержащего лиганд в катехолатной и семихиноновой формах, подтверждается характерным набором полос в ИК-спектре в области 700–1600 см–1. Так, в интервале 1350–1450 см–1 наблюдаются интенсивные полосы валентных колебаний полуторных связей С–О, а в области 1200–1300 см–1 – валентные колебания одинарной связи С–О, что подтверждает смешанную природу сурьмаорганического фрагмента. В масс-спектре комплекса I кроме пика, соответствующего молекулярному иону [I]+, также наблюдаются сигналы от продуктов фрагментации, основным направлением которой является разрыв С–С-связи, соединяющей два фенильных метил-трет-бутильных кольца. К сожалению, все попытки получить кристаллы, пригодные для РСА, оказались безуспешными.
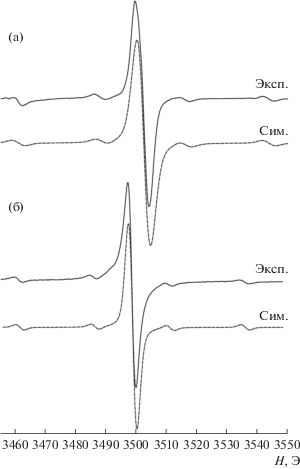
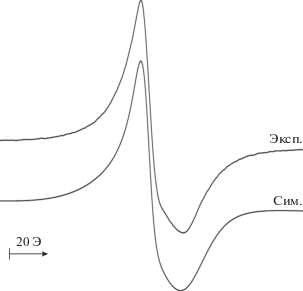
Окисление комплекса I трифлатом серебра в молярном соотношении 1 : 1 в растворе ТГФ приводит к одноэлектронному окислению комплекса с образованием катиона [CrIII[(Q-Cat)SbPh3]- [(SQ-Cat)SbPh3]2]+ (I+). Исходный комплекс имеет синглетное основное спиновое состояние вследствие антиферромагнитного обмена металл–лиганд между высокоспиновым хромом(III) (d3, S = 3/2) и тремя анион-радикальными о-семихиноновыми центрами (S = 1/2), подобное магнитное поведение было ранее неоднократно продемонстрировано на примере других трис-о-бензосемихиноновых комплексов хрома(III) [31–33]. Одноэлектронное окисление приводит к получению парамагнитного производного I+ с S = 1/2, изотропный спектр ЭПР которого в растворе ТГФ представляет собой сигнал с giso = 1.9700 с сателлитным расщеплением на магнитном изотопе атома хрома (53Cr, I = 3/2, 9.5%) с константой СТВ ai(53Cr) = 27.6 Э (рис. 1а). Данные параметры близки к параметрам спектра ЭПР комплекса хрома [Cr(Cl4BQ)(Cl4SQ)2]+[SbF6]– (giso = 1.969, ai(53Cr) = 27.5 Э) [34].
Рис. 1.
Спектр ЭПР [CrIII[(Q-Cat)SbPh3][(SQ-Cat)SbPh3]2]+[OTf]– (I+[OTf]–) (a) и Na+[CrIII[(Cat-Cat)SbPh3][(SQ-Cat)SbPh3]2]– (Na+I–) (б): эксп. – экспериментальный (ТГФ, 293 К); сим. – компьютерная симуляция (WINEPR SimFonia 1.26).
Близкие характеристики также имеют спектры ЭПР сходных комплексов переноса электрона на основе трис-тетрахлор-(бром)-о-семихинолятов хрома и кобальтоцена и тетратиафульвалена [35].
Одноэлектронное восстановление комплекса хрома I с помощью металлического натрия в растворе ТГФ приводит к восстановлению одного из о-бензосемихиноновых лигандов до катехолатного, в результате чего также образуется парамагнитное производное Na+[CrIII[(Cat-Cat)SbPh3][(SQ-Cat)- SbPh3]2]– (Na+I–), содержащее один неспаренный электрон, спектр ЭПР которого имеет несколько иные характеристики: giso = 1.9725, ai(53Cr) = 24.9 Э (рис. 1б). Подобный анионный комплекс хрома на основе 3,5-ди-трет-бутил-о-бензосемихиноновых лигандов K+[CrIII(3,5-DBCat)(3,5-DBSQ)2]– имеет спектр ЭПР с похожими характеристиками: giso = 1.972 и ai(53Cr) = 24.3 Э [36]. Дополнительной сверхтонкой структуры на магнитных ядрах лигандов не наблюдается. Параметры спектра ЭПР говорят о том, что неспаренный электрон находится на орбитали, имеющей значительно металлический характер (значительный вклад орбиталей металла).
Согласно данным ЦВА, для производного хрома I в катодной области наблюдаются три редокс-перехода: первые два являются квазиобратимыми и одноэлектронными, третий пик необратимый (табл. 1).
Таблица 1.
Значения редокс-потенциалов комплекса I по данным ЦВА (CH2Cl2, СУ-электрод, 0.15 М Bu4NClO4, с = 0.002–0.003 моль/л, V = 0.2 В/с, отноcительно Ag/AgCl/KCl(нас.))
| Комплекс | $E_{{{\text{рк}}}}^{1}$, В* | $E_{{{\text{рк}}}}^{2}$, В* | $E_{{{\text{рк}}}}^{3}$, В* | $E_{{{\text{ра}}}}^{1}$, В* | $E_{{{\text{ра}}}}^{2}$, В* |
|---|---|---|---|---|---|
| I | –0.09 | –0.74 | –1.15 | 1.00 | 1.29 |
| III | 0.43 | –0.49 | 0.85 | 0.96 | |
| (Q-Cat)SbPh3 | –0.45 | –0.62 | 1.10 | 1.40 |
* $Е_{{{\text{рк}}}}^{1}$ – потенциал пика первого катодного процесса; $Е_{{{\text{рк}}}}^{2}$ – потенциал пика второго катодного процесса; $Е_{{{\text{рк}}}}^{3}$ – потенциал пика третьего катодного процесса; $Е_{{{\text{рa}}}}^{1}$ – потенциал первого пика окисления; $Е_{{{\text{рa}}}}^{3}$ – потенциал пика второго анодного процесса.
Фиксируемые редокс-переходы в катодной области характеризуют электровосстановление о‑семихинонового фрагмента лиганда. В анодной области для I регистрируются два необратимых многоэлектронных пика окисления при 1.00 и 1.29 В. Наблюдаемую картину можно объяснить близостью значений потенциалов редокс-переходов Cat/SQ в катехолатном фрагменте лиганда [(SQ-Cat)SbPh3]– с редокс-переходом SQ/Q в о‑семихинолятном фрагменте лиганда. Для сравнения представлены результаты ЦВА исходного комплекса (Q-Cat)SbPh3, где в катодной области наблюдаются две необратимые стадии восстановления при –0.45 и –0.62 В. Потенциалы окисления (Q-Cat)SbPh3 и комплекса I хорошо согласуются.
По аналогичной схеме был получен комплекс меди II с подобным сурьмасодержащим лигандом (схема 2 ).
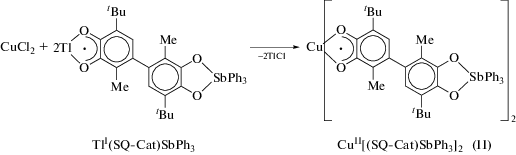
Схема 2 .
Комплекс II выделен в виде мелкокристаллического порошка, охарактеризованного данными ИК-, ЭПР-спектроскопии и элементного анализа. В ИК-спектре II присутствуют полосы поглощения валентных колебаний одинарной и полуторной связей С–О (1245, 1302 и 1450 см–1) и отсутствуют полосы, соответствующие колебаниям карбонильных групп С=О (1660 см–1).
Для полученного образца комплекса II были проведены измерения статической магнитной восприимчивости (μэфф). Температурная зависимость μэфф комплекса меди приведена на рис. 2.
Рис. 2.
Температурная зависимость эффективного магнитного момента комплекса II (сплошная линия – μрасч).
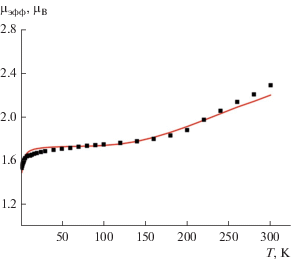
Эффективный магнитный момент при 300 К составляет 2.28 μв и плавно уменьшается при снижении температуры до значения 1.79 μв при 180 К. При дальнейшем снижении температуры до 25 К значение μэфф остается практически неизменным и составляет 1.68–1.78 μв. В пределе низких температур комплекс II демонстрирует резкое снижение μэфф, обусловленное слабым межмолекулярным обменом антиферромагнитного характера. Из анализа следует, что высокотемпературное значение существенно ниже ожидаемого для системы, включающей в себя три невзаимодействующих электрона (один на атоме меди и по одному на SQ-лигандах), а низкотемпературное незначительно отличается от величины μэфф для одного неспаренного электрона [37].
Параметры обменных взаимодействий в комплексе меди II были определены с применением модели для системы, содержащей три обменно-связанных спиновых центра (схема 3 ), с использованием Гамильтониана H = –2J(S1S2+ S2S3)× × 2J1S1S3, где S1= S3 = 1/2 спины семихиноновых лигандов, S2 – спин парамагнитного иона металла (S2 = 1/2 для иона Сu2+) с учетом межмолекулярного обменного взаимодействия (zJ) в приближении молекулярного поля.
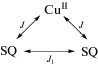
Схема 3 .
Удовлетворительное воспроизведение экспериментальных данных μэфф(Т) было получено при следующих параметрах: gSQ = 2.0 (фиксированное), gCu = 2.01, J(SQ-Cu) = –157 см–1, J(SQ-SQ) = = J1 = –10.0 см–1, zJ = –1.8 см–1, TIP = 0.0, R2 = 7.7 × × 10–4.
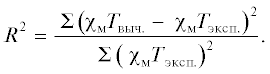
Соответствующая данным параметрам теоретическая кривая μэфф(Т) приведена на рис. 2 (сплошная линия).
Полученные результаты хорошо согласуются с данными ЭПР-спектроскопии. Спектр ЭПР комплекса II в твердом виде представлен на рис. 3. Таким образом, спектральные данные позволяют сделать вывод о том, что неспаренный электрон находится не на атоме меди(II), а на органическом лиганде.
Подобные примеры изменения магнитных и спектральных свойств комплексов меди(II) с редокс-активными радикальными лигандами в литературе известны. Например, в [38] описан бис-о-иминосемихинолят меди(II) на основе 4,6‑ди-трет-бутил-2-(2-метилселенофенил)аминофенола Cu(ISQ-SeMe)2. Спектральные и магнитные измерения показали, что данный комплекс имеет дублетное спиновое состояние (S = 1/2) с неспаренным электроном на органическом лиганде. Параметры спектра ЭПР в твердом состоянии при пониженной температуре для данного комплекса следующие: g1 = 2.012, g2 = 2.006, g3 = 1.998 (gav = 2.005). С повышением температуры для комплекса наблюдается заселение возбужденного состояния со спином S = 3/2 (квадруплетное состояние). Данные особенности магнитного и ЭПР-спектрального поведения комплекса меди(II) объясняются искажением координационной сферы атома меди за счет слабых взаимодействий между селенсодержащей боковой группой и центральным атомом меди. Вследствие этого между плоскостями о-иминосемихиноновых лигандов возникает определенный угол и изменяются характер и величина обменных магнитных взаимодействий металл–лиганд и лиганд–лиганд. Подобная ситуация наблюдается и для PhSe-содержащего аналога CuII(ISQ-SPh)2 [39]. Параметры магнитного обмена, рассчитанные для данного комплекса меди, составляют J = −64.0 см–1, J' = −23.0 см–1 (gCu(II) = 2.03, gR = 2.00). Спектр ЭПР комплекса имеет еще более слабовыраженную анизотропию и значения g-фактора ниже 2 (1.964, 1.992, 1.992, gav = 1.983). Авторы делают вывод о лиганд-центрированном характере неспаренного электрона в дублетном основном спиновом состоянии данного комплекса. В более поздних работах [40, 41] данные группы авторов исследуют серию подобных комплексов меди(II), для которых в зависимости от молекулярного строения, наличия или отсутствия дополнительных взаимодействий с центральным атомом меди наблюдаются различные по величине магнитные обмены. В некоторых случаях искажения координационного узла меди(II) в комплексах не связаны с наличием дополнительных взаимодействий функциональных групп в боковых звеньях лигандов с атомом меди, а определяются стерическими причинами, изогнутым строением лигандов и т.п. [42].
Как упоминалось выше, особенность о-хинонов заключается в их возможности реализовывать несколько устойчивых форм в комплексах с металлами. В последнее время в координационной химии наметилась тенденция исследования комплексов, где о-хиноны выступают в роли нейтральных лигандов [43]. Подобные комплексы позволяют получить новые свойства в реакциях активирующего комплексообразования [44]. Ранее был описан синтез устойчивого комплекса о‑хинона с иодидом цинка [43]. Для целенаправленного получения комплекса цинка с нейтральным сурьмасодержащим лигандом (Q-Cat)SbPh3 была проведена реакция последнего с иодидом цинка в среде толуола (схема 4 ).
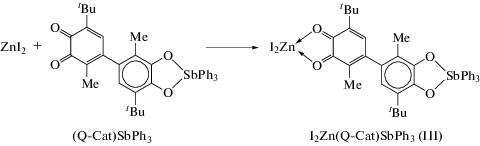
Схема 4 .
Данная реакция сопровождается резким изменением цвета с бордового на фиолетовый и выпадением мелкокристаллического продукта III. В ИК-спектре выделенного продукта в области 1200–1300 см–1 присутствуют полосы, характерные для катехолатных комплексов металлов, при 1605 см–1 наблюдается полоса, соответствующая колебаниям ν(С=О). Однако в интервале 1450–1480 см–1 наблюдаются интенсивные полосы, отвечающие колебаниям полуторных связей С–О. Спектр ЯМР 1Н содержит сигналы от протонов двух трет-бутильных групп различной ширины линии и от протонов фенильных заместителей при атоме сурьмы, однако сигналы от протонов в положении 5 и метильных групп значительно уширены. Полученный образец также имеет примесный анизотропный спектр ЭПР в твердом виде. Анализируя полученные результаты, предположено, что в результате образования молекулярного комплекса III происходит усиление акцепторных свойств о-хинона, что делает возможным дальнейшее протекание окислительно-восстановительных превращений (схема 5 ). Данная реакция лучше протекает в полярных растворителях.
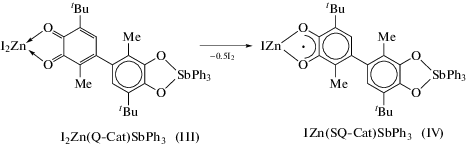
Схема 5 .
Комплекс IV выделен из маточного раствора после отделения комплекса III. Изотропный спектр ЭПР комплекса IV, зафиксированный в растворе ТГФ при комнатной температуре (рис. 4), представляет собой триплет мультиплетов с gi = = 2.0039.
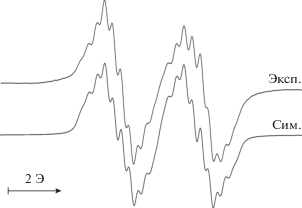
Спектр ЭПР был симулирован с помощью пакета программ Easyspin 5.2.25. Сверхтонкая структура спектра обусловлена сверхтонким взаимодействием (СТВ) неспаренного электрона с протоном в положении 5 ароматического кольца о-бензосемихинонового фрагмента с константой СТВ ai(11H) = 3.23 Э, а также с протонами двух неэквивалентных метильных групп (одна CH3-группа в о-семихиноновом фрагменте, вторая – в катехолатном) с константами СТВ ai(31H) = 0.64 Э и ai(31H) = 0.29 Э соответственно. Параметры данного спектра близки к параметрам спектров ЭПР подобных комплексов общего вида M(SQ-Cat)SbPh3, описанных ранее [24].
Было проведено дополнительное исследование электронной структуры комплекса III при помощи квантово-химических расчетов методом функционала плотности. Расчеты проводили на уровне B3LYP/DGDZVP. Согласно данным расчетов, верхняя занятая молекулярная орбиталь (ВЗМО –5.8 эВ), в основном локализована на иодид-анионах (рис. 5), а нижележащая ВЗМО-1 (–6.07 эВ) – на катехолатном лиганде (рис. 5).
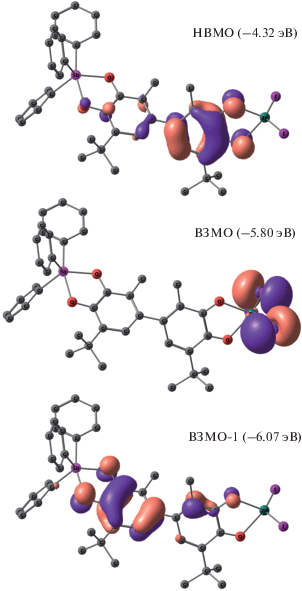
Низшая свободная молекулярная орбиталь (НСМО –4.32 эВ) преимущественно располагается на хиноновом фрагменте (рис. 5). Сравнение значения граничных орбиталей комплекса III и исходного хиноно-катехолата сурьмы (ВЗМО –5.49 эВ, НСМО –3.13 эВ) показало, что координация с иодидом цинка незначительно понижает ВЗМО, однако существенно увеличивает окислительную способность комплекса, сильно понижая НСМО. Полученные данные хорошо согласуются с данными УФ-спектроскопии, согласно которым в ближней ИК-области наблюдается интенсивная полоса поглощения с максимумом 910 нм и коэффициентом экстинкции 1 × 104 л/моль см (рис. 6).
Рис. 6.
ЭСП растворов комплекса III в растворителях: СH2Cl2 (1), CHCl3 (2), СH2Br2 (3), СCl4 (4). с = 1 × 10–4 моль/л, l = 1 см.
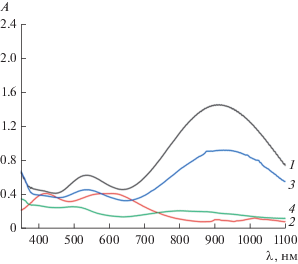
Энергия электронного перехода в максимуме полосы поглощения соответствует ~30 ккал/моль. Было проведено дополнительное исследование зависимости спектров от концентрации и температуры. Показано, что при комнатной температуре в диапазоне применяемых концентраций в хлористом метилене при последовательном разбавлении раствора в два и три раза для полосы поглощения выполняется закон Бугера–Ламберта–Бера, указывая на поглощение единой молекулярной системы. При понижении температуры образца от 20 до –30°С наблюдается увеличение интенсивности полосы с максимумом 910 нм на 30% (от 0.4 до 0.6 A) с одновременным смещением максимума до 930 нм, что характерно для полос переноса заряда. Следует отметить, что при регистрации спектров в бромистом метилене имеет место незначительное, а в четыреххлористом углероде – значительное уменьшение интенсивности этой полосы поглощения и отсутствие ее в таких растворителях, как ТГФ, ацетонитрил, толуол и хлороформ при тех же значениях концентрации.
Электрохимическое поведение комплекса III достаточно разнообразно (рис. 7) и протекает по схеме 6 .
Рис. 7.
ЦВА комплекса III при развертке потенциала от –0.8 до 1.2 В (CH2Cl2, СУ-анод, с = 0.002–0.003 M, 0.15 М NBu4ClO4, Ag/AgCl/KCl(нас.)).
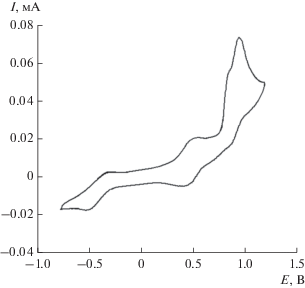
Схема 6 .
Электрохимическое поведение соединения III характеризуется наличием двух квазиобратимых пиков восстановления при +0.43 и –0.49 В (табл. 1), отвечающих превращению SQ/Q- и SQ/Cat-фрагментов лиганда. Координация кислоты Льюиса по о-хиноидной группе приводит к значительному смещению первого потенциала восстановления в анодную область, что обусловлено акцепторным влиянием ZnI2. Подобное поведение ранее наблюдалось для производных 3,6-ди-трет-бутил-о-бензохинона, содержащих координированный ZnBr2 [44], значения потенциалов редокс-переходов SQ/Q и SQ/Cat фиксировались при положительных значениях. В данном случае присутствие донорного катехолатного металлоцикла частично компенсирует акцепторное влияние ZnI2, поэтому первый редокс-переход наблюдается при 0.43 В. В анодной области наблюдаются два слабо разделенных пика окисления ($Е_{{\text{р}}}^{{{\text{ox1}}}}$ = 0.85 и $Е_{{\text{р}}}^{{{\text{ox2}}}}$ = = 0.96 В), которые можно отнести к превращению катехолатного фрагмента до соответствующего о‑хинона. Скорее всего, не происходит декоординация ZnI2, поскольку потенциалы окисления были бы идентичны полученным ранее для (Q-Cat)SbPh3 (1.10 и 1.40 В).
Таким образом, на основе хинон-катехолата трифенилсурьмы(V) получены гетерометаллические комплексы: трис-о-семихинолят хрома(III) CrIII[(SQ-Cat)SbPh3]3, бис-о-семихинолят меди(II) CuII[(SQ-Cat)SbPh3]2, и комплексы цинка с нейтральным координированным о-хиноновым лигандом I2Zn(Q-Cat)SbPh3 и моно-о-семихинолят иодоцинка IZn(SQ-Cat)SbPh3. С помощью спектроскопии ЭПР зафиксировано образование моновосстановленной и моноокисленной форм комплекса хрома(III): бис-о-семихинолят-катехолат хрома(III) [CrIII[(Cat-Cat)SbPh3][(SQ-Cat)SbPh3]2]– и о-хинон-бис-о-семихинолят хрома(III) [CrIII[(Q-Cat)SbPh3][(SQ-Cat)SbPh3]2]+ соответственно. Комплекс меди(II) CuII[(SQ-Cat)SbPh3]2 имеет основное дублетное спиновое состояние с неспаренным электроном на лиганде, обусловленное антиферромагнитным обменом металл–лиганд (J(SQ-Cu) = –157 см–1, J(SQ-SQ) = –10.0 см–1). Окислительные способности о-хинон-катехолата трифенилсурьмы(V) в нейтральном комплексе с иодидом цинка значительно увеличиваются (первый потенциал восстановления Q/SQ растет с ‒0.45 В для (Q-Cat)SbPh3) до 0.43 В (относительно Ag/AgCl/KCl(нас.)), данный комплекс способен окислять иодид анион в комплексе до элементарного иода с образованием парамагнитного о-семихинонового комплекса IZn(SQ-Cat)SbPh3.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Adams R.D., Cotton F.A. Catalysis by Di- and Polynuclear Metal Cluster Complexes. Wiley-VCH, 1998. P. 283.
Appel A.M., Bercaw J.E., Bocarsly A.B. et al. // Chem. Rev. 2013. V. 113. P. 6621.
Buchwalter P., Rosé J., Braunstein P. // Chem. Rev. 2015. V. 115. P. 28.
Karkas M.D., Verho O., Johnston E.V., Akermark B. // Chem. Rev. 2014. V. 114. P. 11863.
Lee S.C., Lo W., Holm R.H. // Chem. Rev. 2014. V. 114. P. 3579.
Tard C., Pickett C.J. // Chem. Rev. 2009. 109. P. 2245.
Nam W., Lee Y.M., Fukuzumi S. // Acc. Chem. Res. 2014. V. 47. P. 1146.
Starikov A.G., Starikova A.A., Minyaev R.M. et al. // Chem. Phys. Let. 2020. V. 740. 137073.
Albold U., Hoyer C., Neuman N.I. et al. // Inorg. Chem. 2019. V. 58. P. 3754.
Старикова А.А., Метелица Е.А., Минкин В.И. // Коорд. химия. 2019. Т. 45. С. 350 (Starikova A.A., Metelitsa E.A., Minkin V.I. // Russ. J. Coord. Chem. 2019. V. 45. P. 411).https://doi.org/10.1134/S1070328419060095
Starikova A.A., Minkin V.I. // Mendeleev Commun. 2016. V. 26. P. 423.
Klementieva S.V., Kuropatov V.A., Fukin G.K. et al. // Z. Anorg. Allgem. Chem. 2011. V. 637. P. 232.
Kuropatov V., Klementieva S., Fukin G. et al. // Tetrahedron. 2010. V. 66. P. 7605.
Poddel’sky A.I., Piskunov A.V., Druzhkov N.O. et al. // Z. Anorg. Allg. Chem. 2009. V. 635. P. 2563.
Shultz D.A., Fico R.M., Jr., Bodnar S.H. et al. // J. Am. Chem. Soc. 2003. V. 125. P. 11761.
Caneschi A., Dei A., Mussari C.P. et al. // Inorg. Chem. 2002. V. 41. P. 1086.
Poddel’sky A.I., Cherkasov V.K., Bubnov M.P. et al. // J. Organomet. Chem. 2005. V. 690. P. 145.
Abakumov G.A., Poddel’sky A.I., Bubnov M.P. et al. // Inorg. Chim. Acta. 2005. V. 358. P. 3829.
Smolyaninov I.V., Antonova N.A., Poddel’sky A.I. et al. // J. Organomet. Chem. 2011. V. 696. P. 2611.
Ilyakina E.V., Poddel’sky A.I., Piskunov A.V., Somov N.V. // Inorg. Chim. Acta. 2012. V. 380. P. 57.
Ilyakina E.V., Poddel’sky A.I., Cherkasov V.K., Abakumov G.A. // Mendeleev Commun. 2012. V. 22. P. 208–210.
Куропатов В.А., Клементьева С.В., Поддельский А.И. и др. // Изв. АН. Сер. хим. 2010. № 9. С. 1652 (Kuropatov V.A., Klementieva S.V., Poddel’sky A.I. et al. // Russ. Chem. Bull. 2010. V. 59. P. 1698).
Пискунов А.В., Черкасов В.К., Дружков Н.О. и др. // Изв. АН. Сер. хим. 2005. № 7. С. 1580 (Piskunov A.V., Cherkasov V.K., Druzhkov N.O. et al. // Russ. Chem. Bull. 2005. V. 54. P. 1627).
Черкасов В.К., Грунова Е.В., Абакумов Г.А. // Изв. АН. Сер. хим. 2005. № 9. С. 2004 (Cherkasov V.K., Grunova E.V., Abakumov G.A. // Russ. Chem. Bull. 2005. V. 54. P. 2067).
Bencini A., Daul C.A., Dei A. et al. // Inorg. Chem. 2001. V. 40. P. 1582.
Абакумов Г.А., Черкасов В.К., Неводчиков В.И. и др. // Изв. АН. Сер. хим. 1997. № 4. С. 804 (Abakumov G.A., Nevodchikov V.I., Druzhkov N.O. et al. // Russ. Chem. Bull. 1997. V. 46. P. 771).
Cherkasov V.K., Grunova E.V., Poddel’sky A.I. et al. // J. Organomet. Chem. 2005. V. 690. P. 1273.
Гордон А., Форд Р. Спутник химика. М.: Мир, 1976.
Frisch, M.J., Trucks, G.W., Schlegel, H.B. et al. Gaussian 09 (revision E.01). Wallingford: Gaussian Inc., 2013.
Litvinenko, A.S., Mikhaleva E.A., Kolotilov S.V., Pavlishchuk V.V. // Theor. Experiment. Chem. 2011. V. 46. P. 422.
Buchanan R. M., Kessel S.L., Downs H.H. et al. // J. Am. Chem. Soc. 1978. V. 100. P. 7894.
Buchanan R.M., Downs H.H., Shorthill W.B. et al. // J. Am. Chem. Soc. 1978. V. 100. P. 4318.
Rodriguez J.H., Wheeler D.E., McCusker J.K. // J. Am. Chem. Soc. 1998. V. 120. P. 12051.
Pierpont C.G., Downs H.H. // J. Am. Chem. Soc. 1976. V. 98. P. 4834.
Chang H.-Ch., Ishii T., Kondo M., Kitagawa S. // Dalton Trans. 1999. P. 2467.
Sofen S.R., Ware D.C., Cooper S.R., Raymond K.N. // Inorg. Chem. 1979. V. 18. P. 234.
Абакумов Г.А., Черкасов В.К., Бубнов М.П. и др. // Изв. АН. Сер. хим. 1992. Т. 10. С. 2315.
Bubrin M., Paretzki A., Hübner R. et al. // Z. Anorg. Allgem. Chem. 2017. V. 643. P. 1621.
Rakshit R., Ghorai S., Biswas S., Mukherjee C. // Inorg. Chem. 2014. V. 53. P. 3333.
Paretzki A., Hubner R., Ye Sh. et al. // J. Material. Chem. 2015. V. 3. P. 4801.
Mondal M.K., Mukherjee C. // Dalton Trans. 2016. V. 45. P. 13532.
Kaim W. // Dalton Trans. 2003. P. 761.
Абакумов Г.А., Черкасов В.К., Пискунов А.В. и др. // Докл. РАН. 2009. Т. 427. С. 330.
Абакумов Г.А., Черкасов В.К., Пискунов А.В. и др. // Докл. РАН. 2010. Т. 434. С. 344.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия