

Координационная химия, 2020, T. 46, № 4, стр. 204-220
Гомолигандные трис-о-диоксоленовые комплексы. Особенности молекулярной структуры и магнитные свойства
М. П. Бубнов 1, *, А. В. Пискунов 1, А. А. Золотухин 1, И. Н. Мещерякова 1, Н. А. Скородумова 1, А. С. Богомяков 2, Е. В. Баранов 1, Г. К. Фукин 1, В. К. Черкасов 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Международный томографический центр СО РАН
Новосибирск, Россия
* E-mail: bmp@iomc.ras.ru
Поступила в редакцию 04.10.2019
После доработки 22.10.2019
Принята к публикации 31.10.2019
Аннотация
Трис-о-диоксоленовые (о-хиноновые, о-семихиноновые и катехолатные) комплексы металлов представляют собой интересную и наглядную модельную систему для демонстрации взаимосвязи структуры и магнитных свойств при рассмотрении рядов однотипных объектов. После опубликования обзоров [1, 2] появился ряд работ, описывающих трис-о-диоксоленовые комплексы металлов, имеющие в своем составе производные симметричного 3,6-ди-трет-бутил-о-бензохинона с заместителями в положениях 4,5. Кроме того, авторы обзора предлагают вниманию несколько ранее неопубликованных структур комплексов такого типа. Появление заместителей в боковых цепях приводит к увеличению стерической загруженности металлоцентра и, как следствие, к нехарактерным для трис-диоксоленовых комплексов координационным полиэдрам, таким как тригональная призма. Искажение структуры, в свою очередь, отражается на магнитных свойствах комплексов. Следует отметить, что в настоящем обзоре рассмотрены только комплексы с производными стерически затрудненных о-хинонов: 3,5- и 3,6-ди-трет-бутил-о-бензохинонов. Объемные трет-бутильные группы препятствуют образованию тесных прямых межмолекулярных контактов, с одной стороны, и предотвращают протекание органических реакций по бензохиноновому циклу, с другой.
Сочетание переходного металла с лигандами переменной валентности, к числу которых относится о-диоксолен, порождает неоднозначность в интерпретации распределения зарядов между ними. Среди факторов, определяющих валентное и спиновое состояние обоих, на первом месте стоит соотношение их редокс-потенциалов. Однако следует учитывать и стерическую загруженность о-диоксолена, которая, с одной стороны, наряду с ионным радиусом металла определяет геометрию координационного узла, с другой – может непосредственно влиять на расстояние металл–лиганд. Редокс превращения замещенного о-хинона представлены на схеме 1 :
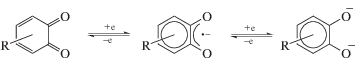
Схема 1 .
Примером влияния пространственного экранирования о-диоксоленов на свойства гомолигандных комплексов, образуемых этими диоксоленами, может служить ряд соединений кобальта с ди-трет-бутил-о-бензохинонами. Несимметричный 3,5-ди-трет-бутил-о-бензохинон (3,5-DBQ), благодаря наличию неэкранированного трет-бутильной группой атома кислорода, образует тетрамерный комплекс [Co(3,5-DBSQ)2]4 (3,5-DBSQ = = анион-радикал (семихинон) 3,5-DBQ), в котором кобальт находится в двухвалентном высоко-спиновом состоянии [3]. Симметричный 3,6-ди-трет-бутил-о-бензохинон (3,6-DBQ) формирует псевдооктаэдрический полиэдр с трехвалентным низкоспиновым кобальтом [4]. Наличие в 3,6-DBQ в положениях 4,5 пиперазинового бицикла, аннелированного через атомы азота N,N' (3,6-DBQ-pipe), приводит к структуре идеальной тригональной призмы. Магнитные свойства такого комплекса однозначно интерпретировать не представляется возможным [5]. Стерически затрудненные о-хиноны, описываемые в настоящем обзоре, представлены на схеме 2 (3,6-DBQ-gly – 3,6-ди-трет-бутил-о-бензосемихинон с O,O'-этилендиолатным циклическим заместителем в – положениях 4,5; здесь и далее Q, SQ, Cat это нейтральный о-хинон, его анион-радикал и дианион соответственно):
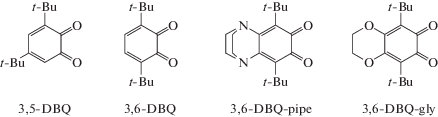
Схема 2 .
Различные металлы, находясь в однотипном окружении из трех диоксоленовых лигандов, существуют в разных валентных и спиновых состояниях. В сочетании с разнообразием структурных полиэдров это приводит к широкому спектру магнитных свойств трис-диоксоленовых комплексов. Обменные взаимодействия варьируют от сильно антиферромагнитного для трис-семихинолятов хрома [6] до слабого ферромагнитного для комплексов алюминия и галлия [4].
Комплексы общего вида M(диоксолен)3, полученные в прошлом веке, подробно описаны в обзоре [1]. Автором этого обзора (проф. Пьепонтом) на основании анализа длин связей во многих о-хиноновых комплексах переходных металлов сделаны выводы о том, как по длинам связей в координационном пятичленном цикле –O–C–C–O–M– можно определить редокс-состояние диоксоленового лиганда без учета ионного радиуса металла [7]. Позднее справедливость этих наблюдений подтверждена в расчетах С. Брауна [8]. Задача настоящей работы – описание новых комплексов в рамках общих закономерностей, обнаруженных в [1], прежде всего их структурных особенностей и связанных с ними магнитных свойств. Кроме того, в обзоре рассмотрен ограниченный круг комплексов: соединения со стерически-затрудненными о‑хинонами. Оставшаяся часть комплексов – это соединения с о-хлоранилом и 9,10-фенантренхиноном, для них структурные особенности во многом определяются упаковкой, а магнитные свойства – межмолекулярным магнитным обменом.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Наиболее логичным представляется описание свойств комплексов на основании распределения по группам в периодической системе (ПС), к которым принадлежат металлы-комплексообразователи. Кадмий – единственный из металлов II группы ПС, для которого структурно охарактеризован комплекс состава M(диоксолен)3. Аналогичное ему соединение магния [9] структурно охарактеризовано не было. Соединение (3,6-DBQ)Cd(3,6-DBSQ)2 (1) получено по реакции амальгамы кадмия с соответствующим о-хиноном [10] и исследовано РСА. В каждой из двух кристаллографически независимых молекул комплекса наблюдаются два лиганда с распределением длин связей, характерным для семихиноновой формы координации, и один лиганд – с выраженными двойными С=О и С=С связями, принадлежащими координированному о-хинону. Геометрия внутренней координационной сферы обеих молекул искажена настолько, что ее трудно отнести к какому-либо типу координационного окружения. Здесь для дальнейшего обсуждения координационных полиэдров следует ввести понятие “угол скручивания” (twist angle) – величину, количественно характеризующую промежуточные формы шестикоординационных трис-хелатных комплексов между октаэдром и тригональной призмой [11]. В октаэдре и тригональной призме есть ось симметрии третьего порядка. Проекция, сделанная вдоль этой оси в октаэдре, будет иметь вид двух равнобедренных треугольников, повернутых на 60° и наложенных друг на друга (рис. 1). Такая же проекция в тригональной призме будет выглядеть как треугольник. Таким образом, угол скручивания α для октаэдра составляет 60°, а для тригональной призмы – 0°. В случае трис-диоксоленовых комплексов угол скручивания определялся для каждого лиганда в отдельности следующим образом: в проекции вдоль оси третьего порядка строится центроид на основе трех атомов кислорода, формирующий нижний треугольник, и центроид, формирующий верхний треугольник. По четырем точкам: центроид 1–O(1)–O(2)–центроид 2 определяется торсионный угол для данного лиганда.
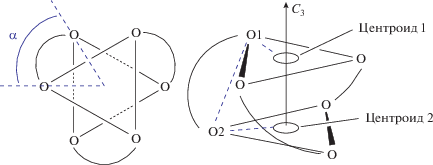
Угол скручивания в комплексе (1) составляет 34.09°–38.03°. Углы OMO для атомов кислорода, расположенных в транс-положении по отношению друг к другу, составляют 102.47°, 140.42°, 161.13° для одной молекулы и 145.05°, 152.70°, 170.96° – для другой. Для октаэдра этот угол составляет 180°, для тригональной призмы 133°. Отличительная особенность геометрии нейтрального о-хинона – излом по линии О…О, угол которого составляет 28.64° для одной молекулы и 25.55° для другой. Это заметно больше, чем для семихинонов: 9.12°, 18.38° для одной молекулы и 8.25°, 15.57° для другой.
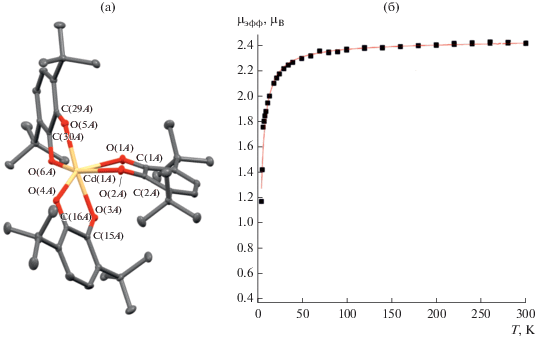
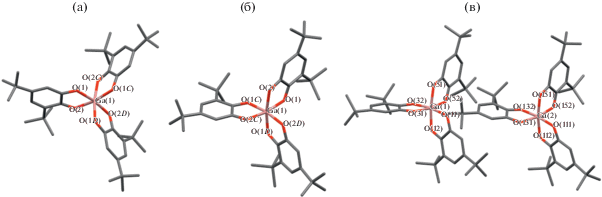
Магнитное поведение комплекса отражает его состав: два неспаренных электрона на двух семихинонах практически не взаимодействуют между собой (рис. 2). Магнитный момент при комнатной температуре 2.41 µB близок к расчетной величине 2.45 µB, вычисленной для двух невзаимодействующих спинов.
Рис. 2.
Общий вид одной из двух кристаллографически независимых молекул комплекса (1) (а) и температурная зависимость его магнитного момента (б).
Следует также рассмотреть аналогичный по зарядовому распределению комплекс никеля (3,6-DBQ)Ni(3,6-DBSQ)2 (2) [12]. Комплекс получали по реакции карбонила никеля с избытком о-хинона. Судя по распределению длин связей в хелатных циклах, так же как и в случае кадмия, один из лигандов – координированный о-хинон. Геометрия внутренней координационной сферы близка к октаэдру. Угол скручивания составляет 41.32°–41.37°. Молекула (2) имеет ось симметрии С2, проходящую через атом никеля и совпадающую с локальной осью симметрии лиганда. В отличие от (1) нейтральный о-хиноновый лиганд плоский, а анион-радикальные – имеют излом по линии O…O 13.22°.
Магнитный момент комплекса (2) практически не изменяется с температурой и составляет 4.57 µB (20–300 К). Ниже 20 К он незначительно падает до 4.38 µB. Авторы [12] пишут о слабом антиферромагнитном взаимодействии никеля (Ni(II), d8, S = 1) с двумя семихинонами (S = 1/2) J = –0.2 см–1, g = 2.43.
Третья группа металлов ПС представлена в ряду трис-лигандных о-диоксоленовых комплексов семью соединениями: два комплекса алюминия, три – галлия и два – индия. Комплексы галлия с тремя 3,5-ди-трет-бутил-о-бензосемихинонами (Ga(3,5-DBSQ)3 (3) описаны тремя независимыми авторскими группами [13–15]. Во всех трех работах обнаружено, что о-диоксоленовые лиганды находятся в анион-радикальной форме, геометрия комплексов – искаженный октаэдр (рис. 3). Поскольку 3,5-ди-трет-бутил-о-бензохинон не имеет оси симметрии, то образуемые им трис-лигандные комплексы могут быть структурными и оптическими изомерами. Так в [14] описан оптический изомер комплекса, исследованного в [13].
Несмотря на то, что образцы комплекса синтезированы по похожим методикам (растворение галлия в толуольном растворе о-хинона с последующим упариванием растворителя), магнитные свойства, описанные авторами, отличаются. В [13] показано, что магнитный момент комплекса монотонно растет от 2.95 µB (320 К) с понижением температуры до 3.58 µB (9 К), а затем падает до 3.26 µB при 2 К. Значение энергии ферромагнитного взаимодействия между тремя неспаренными электронами на трех семихиноновых лигандах составляет 7.8 см–1 (g = 1.92). Таким образом, основное состояние комплекса – квадруплет (S = 3/2) – ниже по энергии двух вырожденных дублетных состояний (S = 1/2) на 23.4 см–1. Ферромагнитный характер обменного взаимодействия подтверждается полевой зависимостью намагниченности при 2–4 К. Авторы считают, что преобладающее ферромагнитное взаимодействие реализуется через вакантные 4p-орбитали галлия. В [14] авторы описывают несколько иное магнитное поведение. Значение χТ (~0.7 см3K/моль) мало зависит от температуры и уменьшается только при температурах ниже 30 К. Высокотемпературное значение заметно выше такового для дублетного состояния и намного ниже значения χТ для квадруплетного. Авторы пишут о трирадикальном основном состоянии, в котором квартетное состояние выше по энергии двух вырожденных дублетных на 8–20 см–1. В [15] описана полиморфная модификация, в которой ячейка содержит две симметрически независимые молекулы того же оптического изомера, что и в [14]. Магнитные свойства, представленные в [15], аналогичны описанным в [13]: величина χТ монотонно растет с понижением температуры, достигая 1.8 см3 K/моль при 5 К, что близко к теоретическому значению для квадруплетного спинового состояния. Энергия ферромагнитного обмена между лигандами составляет J = 17.63 см–1 (g = 2.0). Рассматривая противоречия в выводах авторов работ [13, 15] и [14], особенно с учетом магнитных взаимодействий в комплексах галлия и алюминия с симметричным 3,6-ди-трет-бутил-о-бензосемихиноном, описанных ниже, можно предположить, что в [14] или есть ошибка в результатах измерений, или авторы имели дело с какой-то другой полиморфной модификацией комплекса (в массе).
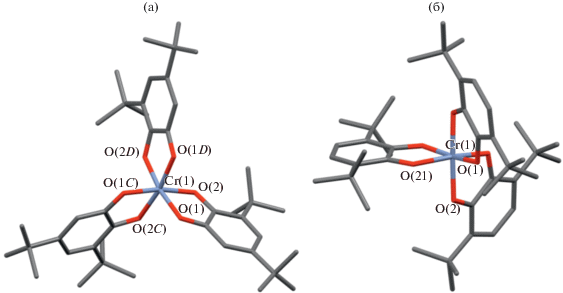
Известны комплексы металлов III группы с симметричным 3,6-ди-трет-бутил-о-бензосемихиноном и его производным – 3,6-ди-трет-бутил-о-бензосемихиноном, имеющим в положениях 4,5 конденсированный N,N'-пиперазиновый бицикл. Трис-лигандные комплексы алюминия и галлия (M(3,6-DBSQ)3, M = Al (4) [16], Ga (5)) получали по обменной реакции соответствующих галогенидов с о-семихинолятом таллия [4]. Комплексы изоструктурны, поэтому в [4] подробно описана молекулярная структура только соединения галлия. Структура комплекса алюминия подробно исследована в настоящей работе. Судя по длинам связей, все три лиганда находятся в анион-радикальной форме (C–O 1.289–1.294 Å). Структура внутренней координационной сферы – слабо искаженный октаэдр (рис. 4). Угол скручивания составляет 44.13°–45.33°. Молекула имеет ось симметрии второго порядка, проходящую через атом алюминия и середину одного из лигандов. Как это часто наблюдается в подобных структурах, плоскости о-семихиноновых лигандов слегка скручены вдоль локальной оси симметрии лиганда. Кроме того, имеет место небольшой излом по линии O…O каждого лиганда. Однако суммарно (скручивание + излом) искажения не превышают 20° (это значение максимального торсионного угла AlO(1)C(1)C(6)).
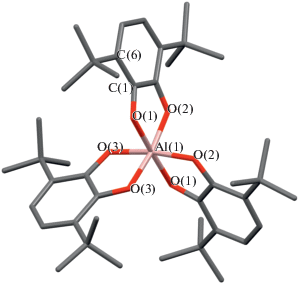
Также как и в описанных выше комплексах галлия с 3,5-DBSQ, в соединениях (4) и (5) преобладают ферромагнитные обменные взаимодействия. Значение магнитного момента комплексов (4 и 5) при комнатной температуре близко к 3.0 µB, что соответствует трем невзаимодействующим спинам. Понижение температуры приводит к монотонному увеличению эффективного магнитного момента (µэфф) до 3.62 µB (5 К) для галлиевого комплекса и 3.86 µB (5 К) для алюминиевого. При расчете энергии обменного взаимодействия получены следующие значения: 6.23 см–1 (g = 1.91) для комплекса галлия и 8.55 см–1 (g = 1.93) для алюминия. Эти значения вполне согласуются с результатами, полученными в [13] для Ga(3,5-DBSQ)3 (7.8 см–1, g = 1.92).
В отличие от всех вышеописанных соединений, комплекс индия In(3,6-DBSQ)3 (6) имеет тригонально-призматическую геометрию (рис. 5) [17]. Комплекс получали растворением металлического индия в толуольном растворе о-хинона. По длинам связей в хелатных циклах лиганды находятся в анион-радикальной форме (C–O 1.270–1.289 Å).
Рис. 5.
Молекулярная структура комплекса In(3,6-DBSQ)3. Вид вдоль оси С3 (а), вид сбоку (б), температурная зависимость магнитного момента (в).
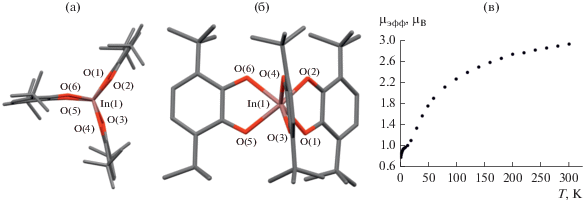
Тригональная призма слегка искажена. Угол скручивания составляет 4.25°–10.21°. Семихиноновые лиганды неплоские. Углы изгиба по линии O…O составляют 4.13°, 7.04° и 12.99°. Комплекс (6) – единственный представитель трис-о-диоксоленовых соединений 3,6-DBSQ с тригонально-призматическим координационным окружением. Эта особенность, вероятно, связана с большим ионным радиусом индия. Ионные радиусы металлов III группы по Н.В. Белову и Г.Б. Бокию [18]: Al 0.57, Ga 0.62, In 0.92 Å. Отметим, что тригонально-призматическая геометрия комплекса индия сохраняется и в анионной форме [19]. Восстановление проходит по одному из лигандов.
В отличие от вышеописанных комплексов III группы обменные взаимодействия в комп-лексе (6) имеют антиферромагнитный характер. Значение µэфф при комнатной температуре близкo к расчетному для трех невзаимодействующих спинов (3.0 µB). Охлаждение приводит к уменьшению µэфф до 0.75 µB (4 К) (рис. 5). Такой результат связан с симметрией комплекса. Если в октаэдре вакантные р-орбитали металлов (Ga, Al) и π*-орбитали семихинонов относятся к разным неприводимым представлениям и, следовательно, имеют нулевой интеграл перекрывания, то в точечной группе D3h две из трех р-орбиталей металла и две из трех π*-орбиталей семихинонов относятся к неприводимому представлению Е '. Таким образом, возникает ненулевой интеграл перекрывания магнитных орбиталей лигандов и, как следствие, канал для антиферромагнитного взаимодействия.
Получено четыре гомолигандных комплекса с производным 3,6-ди-трет-бутил-о-бензосемихинона, имеющим в положениях 4,5 конденсированный N,N'-пиперазиновый бицикл (3,6-DBSQ-pipe) [20]. Все комплексы M(3,6-DBSQ-pipe)3 (M = Al (7), Ga (8), In (9)) получены по обменной реакции галогенидов металлов с о-семихинолятом натрия. Кроме того, комплекс (8) можно получить растворением металлического галлия (в виде амальгамы) в растворе о-хинона. Комплекс галлия (8) выделен с двумя гостевыми молекулами ТГФ, алюминия (7) – с молекулой н-гексана. Получен комплекс галлия аналогичный (8), но не имеющий гостевых молекул растворителя (8а). Поскольку молекулярные структуры комплексов (8 и 8а) различаются мало, в настоящей работе обсуждается структура только (8). Геометрия соединений алюминия и галлия – сильно искаженный октаэдр (рис. 6). Однако назвать это искажение призматическим нельзя. Углы скручивания у комплексов: Al (7) 38.54°–48.87°, Ga (8) 37.98°–42.18°. Углы OAlO и OGaO по атомам кислорода, расположенным в транс-положении по отношению друг к другу составляют 166.42°, 169.35°, 170.02° для алюминия и 164.29°, 168.98°, 170.51° для галлия соответственно. Длины связей в хелатных циклах свидетельствуют об анион-радикальной форме лигандов (C–O 1.287–1.295 Å). Следует отметить, что 3,6-DBQ-pipe – один из наиболее стерически затрудненных о-хинонов. Об этом свидетельствует самый большой торсионный угол O=C–C=O в нейтральном о-хиноне 43.7°. При появлении заместителей в положениях 4,5 торсионный угол значительно увеличивается до величин 25°–33° [21–23]. В 3,6-DBQ этот угол составляет 1.12°. Отличие 3,6-DBQ-pipe от других о‑хинонов состоит в том, что жесткий пиперазиновый бицикл конкурирует с карбонильной группой за пространство, занимаемое метильными группами трет-бутильных заместителей. Стерическая загруженность о-хинона отражается на геометрии его анион-радикала в комплексах. В комплексах с 3,6-DBQ метильные группы трет-бутильных заместителей практически всегда находятся в заторможенной конформации по отношению к ближайшему атому кислорода карбонильной группы о-семихинона. В комплексах с 3,6-DBSQ-pipe можно встретить как заторможенную, так и заслоненную конформации. В комплексе алюминия (7) два семихинона имеют излом по линии O…O16.68° и 22.49°, третий из лигандов не имеет излома, но скручен по С2-локальной оси симметрии о-семихинона на угол 10.98°. Аналогичные искажения, только с другими углами, можно обнаружить и в комплексе галлия (8).
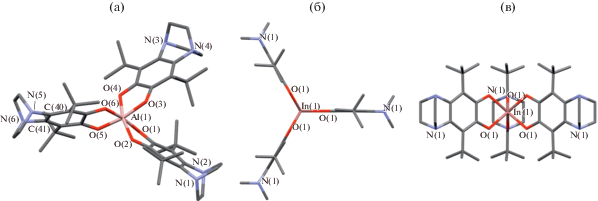
Как и в случае (6) комплекс индия In(3,6-DBSQ-pipe)3 (9) [20] отличается от комплексов алюминия и галлия. Соединение (9) имеет идеальную тригонально-призматическую геометрию (рис. 6). Молекула комплекса высокосимметрична и относится к точечной группе D3h. Длины связей в хелатных циклах свидетельствуют о том, что лиганды находятся в анион-радикальной форме (C–O 1.28 Å). Интересна упаковка (9). Оси третьего порядка всех молекул коллинеарны, а сами молекулы образуют слои (рис. 7).
Рис. 7.
Фрагмент упаковки комплекса In(3,6-DBSQ-pipe)3 (9): вид вдоль молекулярных осей С3 (a) и С2 (б). Температурные зависимости магнитного момента комплексов M(3,6-DBSQ-pipe)3 (M= Al (7), Ga (8), In (9)) (в).
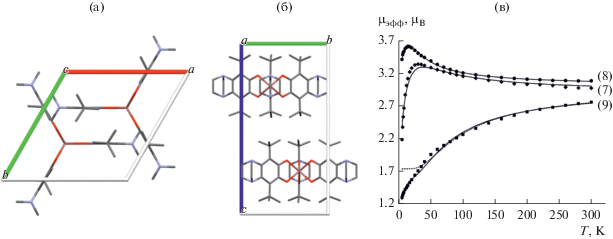
Магнитные свойства комплексов M(3,6-DBSQ-pipe)3, M = Al, Ga, In аналогичны описанным выше комплексам с 3,6-DBSQ (рис. 7). В соединениях алюминия и галлия преобладают ферромагнитные обменные взаимодействия, в комплексе индия – антиферромагнитные (J = 18.6 см–1, zJ' = = –2.2 см–1; J = 10.3 см–1, zJ' = –0.41см–1, g = 2.0 для M = Al, Ga соответственно, J = –28.3 см–1, zJ' = –6.1 см–1, g = 2.0 для M = In).
Сведения о структурно охарактеризованных трис-о-диоксоленовых комплексах металлов IV группы в структурной базе данных отсутствуют.
Среди металлов V группы охарактеризован комплекс ванадия V(3,6-DBCat)2(3,6-DBSQ) (3,6-DBCat = дианион (катехолат) 3,6-DBQ) [24] (10). Геометрия внутренней координационной сферы комплекса близка к октаэдрической. Угол скручивания 40.42°–41.25°. Углы OVO для атомов кислорода, расположенных в транс-положениях по отношению друг к другу составляют 164.25° и 177.81°. По длинам связей в хелатном цикле один из лигандов можно охарактеризовать как анион-радикал (C–O 1.298 Å). Длины связей в двух других имеют значения, характерные для дианионно-катехолатной формы координации: C–O 1.331, 1.326 Å. Это вполне согласуется со степенью окисления ванадия +5, d0. Магнитные свойства в работе не описаны. Спектр ЭПР раствора комплекса, в котором наблюдается СТВ с двумя протонами в положениях 4,5 (aизо(H) = 4.53 G (2H)) и СТС на ядре 51V (I = 7/2), aизо(V) = 3.47 G подтверждает дублетное лиганд-центрированное состояние. Таким образом, единственный неспаренный электрон локализован на одном семихиноне.
Нейтральные трис-о-диоксоленовые комплексы металлов VI группы представлены многочисленными структурами соединений хрома. Первый трис-о-семихинолят хрома Cr(3,5-DBSQ)3 (11) описан в 1979 г. [25], аналогичный комплекс Cr(3,6-DBSQ)3 (12) – в 2007 г. [26]. Комплекс (11) получен из карбонила хрома и о-хинона, (12) – по реакции тетрагидрофуранататрихлорида металла с пирокатехином в присутствии основания и кислорода воздуха. Для обоих соединений геометрия координационного полиэдра близка к октаэдрической (рис. 8). В (11) углы OCrO для атомов кислорода, расположенных в транс-положениях по отношению друг к другу составляют 170.44°; для (12) 169.25° и 177.75° соответственно. Интересно, что для (11) выделен и описан только один структурный и оптический изомер, молекула которого имеет идеальную симметрию С3. Угол скручивания 41.99°. Длины связей С–О находятся в диапазоне, характерном для семихиноновой формы координации (С–О 1.279 и 1.299 Å). Симметрия молекулы комплекса (12) слабо искаженная C3 (идеальная С2). Угол скручивания 42.80°–43.23°. Расстояния С–О чуть больше, чем в (11) (1.303–1.310 Å). Таким образом, зарядовое распределение в обоих комплексах соответствует формулировке Cr(III)(SQ– ·)3.
Однако, несмотря на наличие формально трех анион-радикалов, оба соединения диамагнитны. Трехвалентный хром (Cr3+, d3, S = 3/2) в октаэдрическом окружении имеет три неспаренных электрона в t2g-блоке атомных орбиталей. Орбитали семихинонов, на которых локализованы неспаренные электроны (в C2v локальной симметрии лиганда) в октаэдре имеют ту же симметрию, что и t2g-орбитали металла. Это приводит к большому интегралу перекрывания магнитных орбиталей лигандов и металла и, как результат, к сильному антиферромагнитному взаимодействию, иногда к диамагнетизму. Одноэлектронное восстановление обоих комплексов протекает по лиганду. Спектры ЭПР для анионов (11–) и (12–) указывают на то, что неспаренный электрон локализован на металле.
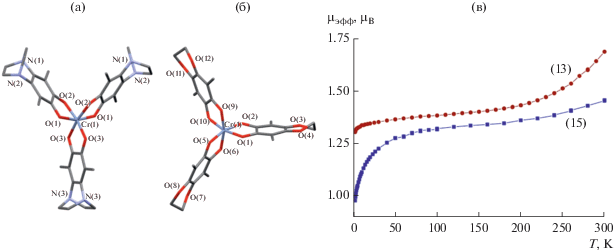
По реакции (THF)3CrCl3 с о-семихинолятом натрия мы получили и охарактеризовали структурно три новых комплекса хрома: Cr(3,6-DBSQ-pipe)3 · · C6H14 (13); Cr(3,6-DBSQ-pipe)3 · (изо-C5H11)2O (14); Cr(3,6-DBSQ-gly)3 (15), где 3,6-DBSQ-gly – 3,6-ди-трет-бутил-о-бензосемихинон с O,O'-этилендиолатным циклическим заместителем в положениях 4,5 (настоящая работа). В (13) геометрия внутренней координационной сферы близка к октаэдру (угол скручивания 37.13°–39.35°), все три лиганда различаются. Молекулярная геометрия (14) мало отличается от (13) (угол скручивания 37.22°–40.84°). Молекула (14) имеет ось С2, проходящую через атом хрома и середину одного из лигандов. Молекула (15) так же, как и (13), не имеет элементов симметрии – все лиганды разные (угол скручивания 36.24°–38.54°). Длины С–О связей в комплексах (13)–(15) лежат в диапазоне 1.295–1.307 Å, что соответствует зарядовому распределению Cr3+(SQ–•)3. Температурная зависимость магнитной восприимчивости исследована для (13) и (15).
В отличие от двух комплексов хрома (11) и (12), описанных выше, соединения (13) и (15) парамагнитны (рис. 9). Значение µэфф при 2.0 К 1.30 µB для (13), 0.97 µB для (15); высокотемпературные значения (300 К): 1.69 µB для (13), 1.46 µB для (15). Система Cr(SQ)3 четно-спиновая при любом зарядовом распределении, т.е. ее основным состоянием могут быть синглет S = 0, µэфф = 0, триплет S = 1, µэфф = 2.83 и т.д. Опираясь на рассуждения, приведенные выше, высокотемпературный момент комплексов (13) и (15), вероятнее всего, свидетельствует об остаточном парамагнетизме.
Рис. 9.
Молекулярная геометрия комплексов хрома 14 (а), (15) (б); температурные зависимости магнитных моментов для (13) и (15) (в).
Трис-диоксоленовые комплексы VII группы ПС представлены четырьмя комплексами: двумя соединениями марганца Mn(3,6-DBSQ)2(3,6-DBCat) (16) [27]; Mn(3,6-DBSQ-pipe)2(3,6-D-BCat-pipe) · 2THF (17) [28] и комплексами рения и технеция: Re(3,5-DBCat)3 (18) [29, 30]; Tc(3,5-DBCat)3 (19) [31]. Комплексы (16) и (18) получены по реакции соответствующих карбонилов с о-хиноном. Авторы работы [27] отмечают, что получили комплекс (16) случайно, проведя реакцию в присутствии 2,2'-дитиофена. Соединение (17) получено по обменной реакции дихлорида марганца с двумя эквивалентами о-семихинолята таллия в присутствии свободного о-хинона.
Геометрия внутренней координационной сферы комплексов марганца (16) и (17) близка к октаэдрической (углы скручивания: 43.93°–45.20° для (16); 40.64°–44.71°для (17)). Длины связей С–О в (16) составляют: 1.278/1.279; 1.305/1.321; 1.311/1.311 Å. Если первый лиганд однозначно интерпретируется как семихиноновый анион-радикал, то длины связей С–О в двух оставшихся имеют промежуточные значения между катехолатом и семихиноном. Учитывая наличие интенсивной полосы переноса заряда Cat–SQ, характерной для смешанолигандных комплексов, авторы [27] трактуют зарядовое распределение как Mn(IV)(3,6-DBCat)(3,6-DBSQ)2 с делокализованым зарядом между двумя лигандами: Mn(IV)(SQ)(dioxolene–1.5)2. Небольшое падение интенсивности этой полосы при нагревании комплекса свидетельствует о начинающемся редокс-изомерном переходе:
Кристаллическая решетка (17) содержит две гостевые молекулы ТГФ. Распределение зарядов в комплексе марганца (17) аналогично таковому в (16). Магнитный момент комплекса (16) монотонно и равномерно растет с повышением температуры от 1.67 µB при 5 К до 3.71 µB при 305 К, в то время как µэфф комплекса (17) остается практически неизменным (1.70–1.82 µB) в интервале 2–300 К. Основное дублетное состояние для системы спинов: Mn(IV), d3; 2(SQ–•) (2 × S = 1/2) легко объясняется сильным антиферромагнитным взаимодействием спинов благодаря одинаковой симметрии пограничных орбиталей, как и в случае комплексов хрома, т.е. неспаренные электроны двух о-семихинонов на π*-орбиталях сильно антиферромагнитно взаимодействуют с неспаренными электронами в t2g-блоке атомных орбиталей марганца. Магнетизм комплексов определяется спином на марганце. Авторы [27] не приводят энергий магнитного обмена для (16). Очевидно, что магнетизм комплекса определяется существованием термически достижимых состояний с более высокой мультиплетностью. В то же время для комплекса (17) эти состояния лежат выше по энергии и термически недостижимы.
Для комплекса рения (18) описана оригинальная методика получения [32].
Геометрия внутренней координационной сферы комплекса промежуточная между октаэдром и тригональной призмой (угол скручивания 33.08°–37.01°). Длины связей С–О в хелатных узлах лигандов отличаются от всех вышеописанных: 1.361/1.363; 1.356/1.363; 1.361/1.372 Å. Такие значения соответствуют катехолатной форме координации лигандов, и, следовательно, зарядовому распределению Re(3,5-DBCat)3. Магнетизм комплекса определяется единственным неспаренным электроном на металле [29]. Заниженную величину магнитного момента (1.18 µB при комнатной температуре) авторы [29] объясняют заметным вкладом спин-орбитального взаимодействия. Дублетное металл-центрированное состояние подтверждается и спектром ЭПР в растворе (〈g〉 = 2.010 и а = 0.002 см–1, СТВ на ядрах 185Re и 187Re, I = 5/2).
Аналогичный по составу и зарядовому распределению комплекс технеция (19) получен кипячением смеси соли NH4TcO4 с пирокатехином H2(3,5-DBCat) в метаноле [31]. Его структура похожа на структуру (18). Как и в случае с рением, магнетизм комплекса (19) определяется единственным неспаренным электроном на металле S = 1/2, µэфф = 1.28 µB (комнатная температура). ЭПР спектр раствора комплекса содержит СТС на ядре 99Tc (I = 9/2) (〈g〉 = 2.0134).
Гомолигандные о-диоксоленовые комплексы металлов VIII группы представлены четырьмя комплексами кобальта: Co(3,6-DBSQ)3 (20) [4]; Co(3,6-DBSQ-gly)3 (21) [5]; Co(3,6-DBSQ-pipe)3 (22) [5]; Co(3,6-DBSQ-pipe)3 · C6H14 (23) (настоящая работа); двумя комплексами железа Fe(3,5-DBSQ)3 (24) [33] и Fe(3,6-DBSQ)3 (25) [34], а также соединениями рутения Ru(3,5-DBSQ)3 (26), осмия Os(3,5-DBSQ)3 (27) [35, 36] и родия Rh(3,5-DBSQ)3 (28) [37]. Комплекс никеля (3,6-DBQ)Ni(3,6-DBSQ)2 (2) обсуждали выше.
Комплекс Co(3,6-DBSQ)3 (20) получен по реакции карбонила кобальта с о-хиноном. Геометрия внутренней координационной сферы – слабо искаженный октаэдр (угол скручивания 47.06°–47.60°). Молекула симметрична: ось С2 проходит через атом металла и делит один из лигандов пополам. Плоскости лигандов искажены мало. Длины связей С–О укладываются в интервал, характерный для координированного анион-радикала семихинона (1.290, 1.295 Å). Таким образом, зарядовое распределение в комплексе (20): Co(III), d6, низкоспиновый в окружении трех анион-радикалов SQ–•. Согласно данным магнетохимических измерений, основное спиновое состояние комплекса (20) – дублет µэфф = 1.82 µB при 5.0 К. Нагревание приводит к монотонному росту магнитного момента до 3.04 µB при 350 К. Высокотемпературное значение 3.04 µB очень близко к расчетной для трех спинов, локализованных на трех лигандах (3.0 µB). Иными словами, три неспаренных электрона лигандов антиферромагнитно взаимодействуют посредством заполненных d-орбиталей диамагнитного металла. Энергия взаимодействия авторами [4] оценивается в J = –39.1 см–1 (g = 2.17). Кроме того, авторы отмечают тот факт, что в трех изоструктурных комплексах: Al(3,6-DBSQ)3 (4), Ga(3,6-DBSQ)3 (5) и Co(3,6-DBSQ)3 (20), содержащих диамагнитные атомы металла, энергии магнитного обмена лиганд–лиганд имеют противоположные знаки для переходных и непереходных металлов: 8.55, 6.23, –39.1 см–1 для (4), (5), (20) соответственно. Авторы делают вывод о том, что участие заполненных d-орбиталей (t2g в Oh (октаэдр)) в групповых молекулярных орбиталях, содержащих неспаренные электроны в (20), обеспечивает канал антиферромагнитного взаимодействия лиганд–лиганд.
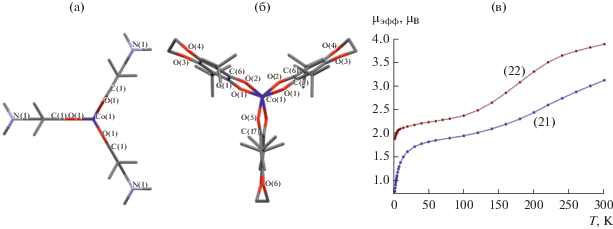
Позднее из карбонила кобальта получены три трис-лигандных комплекса кобальта, содержащие циклические заместители в о-хиноновых лигандах: (21) [5]; (22) [5]; (23) (настоящая работа). Структура комплекса (22) – тригональная призма – идентична структуре индиевого соединения (9) аналогичного состава. Молекула имеет группу симметрии D3h. Молекулярная геометрия (21) близка к тригональной призме, угол скручивания 20.07°–21.21° (рис. 10). Молекула имеет ось симметрии С2, проходящую через атом металла и совпадающую с локальной осью симметрии одного из лигандов. Согласно выводам авторов, тригонально-призматическая геометрия обеспечивается C···H и O···H межмолекулярными взаимодействиями. Длина связей C–O в (22) (1.256 Å) имеет пограничное значение длин связей, характерных для анион-радикала семихинона. Длина связей Co–O (1.995 Å) имеет промежуточное значение между Co(II) и Co(III) [38]. В комплексе (21), напротив, связи Co–O ближе к Co(II) высокоспиновому (2.020–2.030 Å), а связи C–O типично семихиноновые (1.270–1.273 Å). Но если магнетизм (9) трактуется однозначно, то магнитные свойства (21) и (22) однозначно интерпретировать не удается. Кривые температурной зависимости магнитного момента (21)и (22) похожи, с той разницей, что значения µэфф для (22) немного выше, чем для (21). Высокотемпературные значения µэфф (3.11 µB для (21) и 3.88 µB для (22) при 300 К) превышают чисто спиновое значение (3.0 µB) для трех невзаимодействующих спинов на трех лигандах, связанных с диамагнитным ионом (Co(III), d6, низкоспиновый). Таким образом, объяснить высокотемпературное значение магнитного момента наличием только радикальных лигандов невозможно.
Рис. 10.
Молекулярные структуры комплексов (22) (а), (21) (вид вдоль оси С3 (б)) и их температурные зависимости магнитного момента (в).
Авторами [5] рассматривались две модели для объяснения магнитных свойств комплексов (21) и (22). В рамках первой модели предполагалось, что кобальт(III) имеет промежуточный спин S = 1. В совокупности с тремя неспаренными электронами на семихинонах расчетное значение µэфф комплекса составляет 4.12 µB, что близко к экспериментальному при 300 К. Падение магнитного момента с охлаждением объясняется антиферромагнитным обменом. В рамках второй модели предполагалось, что в комплексе происходит обратимый перенос электрона (редокс-изомерия) с образованием формы высокоспинового Co(II) в окружении двух семихинонов и одного координированного нейтрального хинона с делокализацией двух неспаренных электронов по трем лигандам. Обе модели имеют свои аргументы “за” и “против”.
Используя другой растворитель для кристаллизации, мы получили комплекс, аналогичный по составу (22), но имеющий другую геометрию координационной сферы – искаженный октаэдр – соединение (23). Наличия гостевой молекулы растворителя – гексана – оказалось достаточно, чтобы разорвать взаимодействия C···H и O···H, которые удерживали (22) в геометрии тригональной призмы. Магнетизм комплекса изучен не был, но судя по длинам связей это типичный комплекс Co(III) с тремя семихинонами: Co–O 1.862–1.899 Å; С–O 1.283–1.313 Å.
Комплексы железа (24) [33] и (25) [34] получали по реакции карбонила железа с соответствующим о-хиноном. Геометрия внутренней координационной сферы промежуточная между октаэдром и тригональной призмой: углы скручивания для (24) 33.27°–36.95°; для (25) 37.71°–38.59°. Молекулярных осей симметрии в комплексах нет. Длины связей C–O свидетельствуют о том, что оба комплекса – соединения Fe(III) с тремя семихинонами: (24) C–O 1.275–1.287 Å; (25) C–O 1.261–1.288 Å. По данным магнетохимии, основное спиновое состояние комплекса (24) триплет S = 1. Магнитный момент практически не зависит от температуры и составляет 2.95 µB (285 К). Авторы [33] интерпретируют его как результат взаимодействия трех неспаренных электронов высокоспинового железа Fe(III), d5, с тремя неспаренными электронами семихинонов, поскольку орбитали железа (t2g в искаженном октаэдре) имеют одинаковую симметрию с π*-орбиталями семихинонов. Оставшиеся два электрона на атоме железа определяют магнетизм комплекса. В комплексе (25) ситуация несколько сложнее. Магнитный момент растет от 2.18 µB при 5 К до 4.6 µB при 300 К. Авторы [34] полагают, что основное состояние комплекса триплетное S = 1. Заниженное значение µэфф при низкой температуре объясняют межмолекулярным обменным взаимодействием, рост магнитного момента – заселением термически достижимых состояний с большей мультиплетностью. Однако расчетного значения 6.63 µB для системы спинов Fe(III), d5, S = 5/2; 3 × SQ, 3 × S = 1/2 магнитный момент не достигает. Аппроксимация дает энергии антиферромагнитного обменного взаимодействия: JFe–SQ = –139 cм–1; JSQ–SQ = –235 cм–1, g = 2.12.
Комплекс рутения (26) получали по обменной реакции калийной соли 3,5-ди-трет-бутил-о-семихинона с трихлоридом рутения. Структурно охарактеризованы два изомера: цис- и транс- по классификации [35]. Цис-изомер имеет ось симметрии С3, и все трет-бутильные группы, находящиеся в орто-положении к атому кислорода, имеют одно направление. В другом направлении – один из лигандов развернут. Геометрия внутренней координационной сферы обоих изомеров – октаэдр, искаженный в направлении тригональной призмы. Угол скручивания 49° в транс- и 48° – в цис-изомерах. Авторы не дают однозначной трактовки валентного состояния металла и лигандов. Отмечается, что длины связей С–О имеют промежуточное значение между семихиноном и катехолатом: 1.32 Å для цис- и 1.30 Å для транс-изомеров. Анализ связей Ru–O позволяет утверждать, что валентность рутения больше трех.
Комплекс осмия (27), описанный в той же работе, изоструктурен транс-изомеру (26). Угол скручивания 49°. Усредненная длина связей С–О 1.33 Å. Все три комплекса (26) (цис-), (26) (транс-) и (27) диамагнитны. Авторы [35] отмечают, что по данным ЯМР 1H комплексы рутения проявляют структурную жесткость, в то время как соединение осмия в растворе изомеризуется и образует рацемическую смесь оптических изомеров.
По реакции трихлорида родия с пирокатехином получен трис-лигандный комплекс (28) [37]. В элементарной ячейке две симметрически независимые молекулы представляют собой оптические изомеры. Координационный полиэдр – искаженный октаэдр. Углы скручивания 43.65°–46.87° для одной молекулы и 42.22°–48.12° – для другой. Следует отметить, что несмотря на очень большой разброс в длинах связей С–О (1.26–1.37 Å в одной молекуле и 1.28–1.34 Å – в другой), усредненное значение 1.31 Å характерно для координированного семихинона. Основное состояние комплекса – дублет, о чем свидетельствует спектр ЭПР с gi = 2.00. Соединение (28) и аналогичный ему по составу комплекс кобальта (20) демонстрируют схожее магнитное поведение, но в соединении родия состояния с более высокой мультиплетностью термически недостижимы. Таким образом, диамагнитный Rh(III) (d6) координирован тремя семихинонами, два из которых связаны сильным антиферромагнитным взаимодействием.
Проанализировав вышеперечисленные комплексы (табл. 1), можно сделать несколько выводов.
Таблица 1.
Группа симметрии, параметры ячейки, литературный источник, температура эксперимента для комплексов (1)–(26)
| Металл | Комплекс | Группа симметрии | Параметры ячейки (Å, град) | Литература | Условия* |
|---|---|---|---|---|---|
| Cd | Cd(3,6-DBSQ)2(3,6-DBQ) (1) | $P1\_$ | a = 14.5473(16), b = 17.5614(19), c = 17.6309(19) Å, α = 93.821(2)°, β = 101.263(2)°, γ = 109.323(2)°, Z = 4 | 10 | 100 K |
| Al | Al(3,6-DBSQ)3 (4) | C2/c | a = 21.8759(13), b = 19.1976(9),
c = 10.1066(5) Å, α = 90°, β = 95.189(4)°, γ = 90°, Z = 4 |
4 | RT |
| Al(3,6-DBSQ-pipe)3 · C6H14 (7) | Cc | a = 11.0539(17), b = 26.734(4),
c = 19.629(3) Å, α = 90°, β = 95.180(4)°, γ = 90°, Z = 4 |
20 | RT | |
| Ga | Ga(3,5-DBSQ)3 (3) | R3 | a = 16.419(2), b = 16.419(2),
c = 13.899(3) Å, α = 90°, β = 90°, γ = 120°, Z = 3 |
13 | RT |
| Ga(3,5-DBSQ)3 (3) | R3 | a = 16.408(6), b = 16.408(6),
c = 13.898(4) Å, α = 90°, β = 90°, γ = 120°, Z = 3 |
14 | RT | |
| Ga(3,5-DBSQ)3 (3) | $P1\_$ | a = 12.7500(16), b = 13.1264(16),
c = 13.4477(16) Å, α = 76.352(5)°, β = 77.706(5)°, γ = 74.690(5)°, Z = 2 |
15 | 100 K | |
| Ga(3,6-DBSQ)3 (5) | C2/c | a = 22.069(6), b = 19.365(5),
c = 10.163(3) Å, α = 90°, β = 94.90(2)°, γ = 90°, Z = 4 |
4 | RT | |
| Ga(3,6-DBSQ-pipe)3 · 2THF (8а) | Pca21 | a = 26.7864(11), b = 11.1805(5),
c = 19.6718(8) Å, α = 90°, β = 90°, γ = 90°, Z = 4 |
20 | 100 K | |
| Ga(3,6-DBSQ-pipe)3 (8) | P21/n | a = 13.9391(7), b = 24.4087(12),
c = 15.0333(7) Å, α= 90°, β = 92.844(1)°, γ = 90°, Z = 4 |
** | 100 K | |
| In | In(3,6-DBSQ)3 (6) | P21/n | a = 11.3435(4), b = 10.0725(4),
c = 36.4349(14) Å, α = 90°, β = 90.265(1)°, γ = 90°, Z = 4 |
4 | 100 K |
| In(3,6-DBSQ-pipe)3 (9) | P63/m | a = 12.0295(4), b = 12.0295(4),
c = 20.5222(11) Å, α = 90°, β = 90°, γ = 120°, Z = 2 |
20 | 100 K | |
| V | V (3,6-DBCat)2(3,6-DBSQ) (10) | Ccca | a = 19.9969(17), b = 23.740(2),
c = 18.1741(16) Å, α = 90°, β = 90°, γ = 90°, Z = 8 |
22 | RT |
| Cr | Cr(3,5-DBSQ)3 (11) | R3 | a = 16.385(3), b = 16.385(3),
c = 13.874(4) Å, α = 90°, β = 90°, γ = 120°, Z = 3 |
23 | RT |
| Cr(3,6-DBSQ)3 (12) | C2/c | a = 22.1182(9), b = 19.1114(9),
c = 10.0921(3) Å, α = 90°, β = 95.184(5)°, γ = 90°, Z = 4 |
24 | 100 K | |
| Cr(3,6-DBSQ-pipe)3 · C6H14 (13) | P21/c | a = 15.0706(11), b = 26.400(2),
c = 14.2269(11) Å, α = 90°, β = 91.525(2)°, γ = 90°, Z = 3 |
** | 100 K | |
| Cr(3,6-DBSQ-pipe)3 · iso-(C5H11)2O (14) | C2/c | a = 15.0692(13), b = 23.844(2),
c = 19.0140(16) Å, α = 90°, β = 112.880(2)°, γ = 90°, Z = 4 |
** | 100 K | |
| Cr(3,6-DBSQ-gly)3 (15) | P21/c | a = 21.0214(8), b = 19.7460(7),
c = 11.8423(4) Å, α = 90°, β = 103.577(1), γ = 90°, Z = 3 |
** | 100 K | |
| Mn | Mn(3,6-DBCat) (3,6-DBSQ)2 (16) | $P1\_$ | a = 10.3000(10), b = 10.9510(10),
c = 19.298(3) Å, α = 88.970(10)°, β = 79.600(10)°, γ = 82.270(10)°, Z = 2 |
25 | RT |
| Mn(3,6-DBCat-pipe)(3,6-DBSQ-pipe)2 · · 2THF (17) | Pca21 | a = 26.6369(18), b = 11.0435(7),
c = 19.5293(13) Å, α = 90°, β = 90°, γ = 90°, Z = 4 |
** | 100 K | |
| Re | Re(3,5-DBCat)3 (18) | P21/n | a = 15.8455(3), b = 15.8343(3),
c = 16.3301(3) Å, α = 90°, β = 93.3339(9)°, γ = 90°, Z = 4 |
27 | 200 K |
| Tc | Tc(3,5-DBCat)3 (19) | P21/n | a = 15.892(3), b = 15.878(4),
c = 16.367(3) Å, α = 90°, β = 93.13(1)°, γ = 90°, Z = 4 |
28 | RT |
| Co | Co(3,6-DBSQ)3(20) | C2/c | a = 21.799(9), b = 19.376(8),
c = 10.167(3) Å, α = 90°, β = 94.99(3)°, γ = 90°, Z = 4 |
4 | RT |
| Co(3,6-DBSQ-gly)3 (21) | Pbcn | a = 20.0108(12), b = 11.4870(8),
c = 20.8327(13) Å, α = 90°, β = 90°, γ = 90°, Z = 4 |
5 | 150 K | |
| Co(3,6-DBSQ-pipe)3 (22) | P63/m | a = 11.9045(3), b = 11.9045(3),
c = 20.6413(9) Å, α = 90°, β = 90°, γ = 120°, Z = 2 |
5 | 100 K | |
| Co(3,6-DBSQ-pipe)3 · C6H14 (23) | Pbca | a = 19.5724(18), b = 21.9542(19), c = 26.825(3) Å, α = 90°, β = 90°, γ = 90°, Z = 8 |
** | 100 K | |
| Fe | Fe(3,5-DBSQ)3(24) | P21/n | a = 15.904(3), b = 15.891(3),
c 16.450(4) Å, α = 90°, β = 93.22(2)°, γ = 90°, Z = 4 |
30 | RT |
| Fe(3,6-DBSQ)3 (25) | Pbca | a = 18.277(5), b = 11.634(3),
c = 39.903(10) Å, α = 90°, β = 90°, γ = 90°, Z = 8 |
31 | RT | |
| Ni | Ni(3,6-DBSQ)2(3,6-DBQ)·C6H12 (2) | Ccca | a = 20.092(6), b = 24.335(8),
c = 18.340(4) Å, α = 90°, β = 90°, γ = 90°, Z = 8 |
12 | RT |
| Ru | Ru(3,5-DBQ)-cis (26) | P | a = 10.210(3), b = 11.812(4),
c = 19.052(6) Å, α = 77.78(3)°, β = 81.23°, γ = 85.05°, Z = 2 |
32 | 198 K |
| Ru(3,5-DBQ)-trans | P21/c | a = 19.357(9), b = 10.579(6),
c = 20.669(11) Å, α = 90°, β = 97.73(4)°, γ = 90°, Z = 4 |
32 | RT | |
| Os | Os(3,5-DBQ)3 (27) | P21/c | a = 19.278(7), b = 10.487(6),
c 20.689(9) Å, α = 90°, β = 98.11(3)°, γ = 90°, Z = 4 |
32 | 213 K |
| Rh | Rh(3,5-DBSQ)3 (28) | Cc | a = 11.2499(16), b = 33.145(5),
c = 24.000(3) Å, α = 90°, β = 102.819(10)°, γ = 90°, Z = 8 |
34 | RT |
1) В молекулах трис-диоксоленовых комплексов, имеющих осевую симметрию, лиганд, расположенный на оси, всегда немного скручен относительно ее. Другие два имеют изломы по линии O…O. Это же наблюдение относится к большинству остальных комплексов, но излому по линии О…О может сопутствовать скручивание.
2) Большинство комплексов характеризуются искаженной октаэдрической геометрией внутренней координационной сферы. Во многих таких комплексах угол скручивания составляет 40° ± 5°. Остальные имеют геометрию, близкую к тригональной призме.
3) Комплексы, имеющие геометрию тригональной призмы, (9) и (22), имеют ярко выраженную слоистую (2D) упаковку, в которой молекулярные оси (или псевдооси (21)) симметрии третьего порядка коллинеарны. Исключение составляет комплекс (6).
4) В 2011 г. мы описали комплекс индия (6) с тригонально-призматической геометрией [17]. Позднее в 2012 г. опубликованы два комплекса кобальта (22) и (21), один с геометрией идеальной, другой – слабо искаженной призмы [5]. Чуть позже был случайно получен комплекс (23) такого же состава, как (22), но с геометрией искаженного октаэдра, содержащий гостевую молекулу н-гексана (настоящая работа). Возникло предположение, что наличие или отсутствие гостевых молекул растворителя в кристаллической решетке соединения влияет на образование межмолекулярных взаимодействий и, соответственно, определяет геометрию комплекса. Поэтому был получен ряд соединений III группы: (7), (8) и (8a), (9) [20], а также комплексы хрома: (13), (14), (22) (настоящая работа). При этом некоторые из них были специально кристаллизованы из разных растворителей, в том числе из таких, молекулы которых слишком велики для внедрения в решетку комплекса (например, (8)). Однако предположение не подтвердилось. Геометрия внутренней координационной сферы определяется ионным радиусом (как в случае с индием 0.92 Å) или межмолекулярными контактами (как в случае с кобальтом).
Магнитные свойства комплексов – закономерное следствие особенностей геометрии их координационного полиэдра и зарядового распределения. Например, в комплексе кадмия (1) магнетизм определяется двумя спинами на лигандах, которые слабо взаимодействуют друг с другом, в отличие от комплексов (4) и (5), где взаимодействие между лигандами носит ферромагнитный характер. Причина, вероятно, в симметрии. В комплексе кадмия лиганды сильно искажены, а координационный полиэдр далек от октаэдра. Как следствие низкой симметрии, магнитные орбитали лигандов имеют ненулевые интегралы перекрывания. В то же время в комплексах алюминия и галлия координационное окружение металла близко к октаэдру. Орбитали не имеют возможностей для перекрывания и локализованные на них неспаренные электроны взаимодействуют ферромагнитно. Соединения индия (6) и (9) с координационной геометрией тригональной призмы демонстрируют антиферромагнитное взаимодействие лигандов, поскольку в тригональной призме два из трех лигандов и две орбитали металла принадлежат к одному неприводимому представлению, что приводит к их эффективному смешиванию. Комплексы хрома (12) и железа (25) обнаруживают сильное антиферромагнитное взаимодействие металл–лиганд. Причина та же, три неспаренных электрона в t2g-блоке атомных орбиталей в октаэдре эффективно взаимодействуют с неспаренными электронами лигандов, имеющими ту же симметрию. Однако встречаются и неоднозначности в интерпретации магнитных свойств, например в комплексах кобальта (21) и (22). Как уже отмечалось, их магнитные свойства интерпретировались в рамках двух моделей, каждая из которых имеет право на существование.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Рентгенодифракционные данные собраны на автоматических дифрактометрах SMARTAPEX (для (13), (14), (15), (23)) и Agilent Xcalibur E (для (4)) (ω- и φ-сканирование, MoKα-излучение, λ = = 0.71073 Å). Экспериментальные наборы интенсивностей интегрированы с помощью программ SAINT Plus [39] и CrysAlisPro [40] для (13)–(15), (23) и (4) соответственно. Все структуры решены прямым методом и уточнены полноматричным методом наименьших квадратов по $F_{{hkl}}^{2}$ с использованием программного пакета SHELXTL [41] в анизотропном приближении для неводородных атомов. Водородные атомы помещены в геометрически рассчитанные положения и уточнены изотропно. Учет поглощения произведен в программах SADABS [42] для (13)–(15), (23) и SCALE3 ABSPACK [43] для (4). Кристаллы (23) и (13) содержат сольватные молекулы н-гексана и толуола соответственно, разупорядоченные по двум общим положениям. В кристалле (23) целая молекула комплекса кобальта разупорядочена по двум положениям с заселенностями ~0.87 и 0.13. Одна из групп t-Bu в (14) найдена разупорядоченной по двум положениям. Также в кристалле (14) обнаружена сольватная молекула диизоамилового эфира в частном положении (на поворотной оси C2). Соотношение молекул эфира и комплекса хрома в (14) составляет 1 : 2. Основные кристаллографические данные и параметры рентгеноструктурных экспериментов приведены в табл. 2.
Таблица 2.
Кристаллографические данные и детали уточнения структур комплексов (4), (13)–(15), (23)
| Параметр | Значение | ||||
|---|---|---|---|---|---|
| (4) | (13) | (14) | (15) | (23) | |
| Формула | C42H60AlO6 | C61H86CrN6O6 | C59H89CrN6O6.5 | C48H66CrO12 | C60H92CoN6O6 |
| М | 687.88 | 1051.36 | 1038.36 | 887.01 | 1052.32 |
| Температура, K | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| Длина волны, Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Сингония | Моноклинная | Моноклинная | Моноклинная | Моноклинная | Ромбическая |
| Пр. гр. | C2/c | P21/c | C2/c | P21/c | Pbca |
| Параметры ячейки: | |||||
| a, Å | 21.8759(12) | 15.0706(11) | 15.0692(13) | 21.0214(8) | 19.5724(18) |
| b, Å | 19.1976(9) | 26.400(2) | 23.844(2) | 19.7460(7) | 21.9542(19) |
| c, Å | 10.1066(5) | 14.2269(11) | 19.0140(16) | 11.8423(4) | 26.825(3) |
| β, град | 95.189(4) | 91.525(2) | 112.880(2) | 103.577(1) | |
| Объем, Å3 | 4227.0(4) | 5658.3(7) | 6294.5(9) | 4778.2(3) | 11526.6(18) |
| Z; ρ, г/см3 | 4; 1.081 | 4; 1.234 | 4; 1.096 | 4; 1.233 | 8; 1.213 |
| µ, мм–1 | 0.089 | 0.258 | 0.231 | 0.298 | 0.352 |
| θ, град | 3.32–26.00 | 2.09–25.00 | 2.33–25.00 | 1.99–26.00 | 1.86–30.05 |
| Число измеренных/независимых отражений | 13 762/4150 | 44 536/9918 | 24 526/5468 | 40 706/9357 | 84 460/16140 |
| Rint | 0.0466 | 0.0945 | 0.0724 | 0.0398 | 0.0934 |
| GOOF | 1.033 | 0.947 | 1.004 | 1.032 | 1.115 |
| R1, wR2 (I > 2σ(I)) | 0.0486, 0.1121 |
0.0537, 0.1118 |
0.0899, 0.2331 |
0.0463, 0.1143 |
0.1080, 0.2476 |
| R1, wR2 (все отражения) | 0.0748, 0.1221 |
0.1113, 0.1279 |
0.1330, 0.2626 |
0.0656, 0.1225 |
0.1635, 0.2798 |
| Остаточная электронная плотность (Δρmax/Δρmin), e Å3 | 0.388/–0.216 | 0.752/–0.447 | 1.213/–0.506 | 1.252/–0.397 | 1.080/–0.957 |
Структуры зарегистрированы в Кембриджском банке структурных данных (CCDC № 1956614 (23), 1956615 (4), 1956616 (15), 1956617 (13), 1956618 (14); ccdc.cam.ac.uk/getstructures).
Синтез комплекса (23). К раствору 0.155 г (0.16 ммоль) трис-(4,5-N,N'-пиперазино-3,6-ди-трет-бутил-о-семихинолята)кобальта(III) в ~20 мл диэтилового эфира приливали раствор 0.024 г (0.08 ммоль) 2,2'-(пиридин-2,6-диил)бис(N1,N1,N3,N3- тетраметилпропан-1,3-диаминa в ~15 мл н-гептана. Полученный раствор концентрировали до объема 15 мл. После упаривания раствор выдерживали при комнатной температуре в течение суток. Выпавший мелкокристаллический порошок темно-зеленого цвета отфильтровывали, промывали холодным гептаном и сушили в вакууме. Выход – несколько кристаллов.
Синтез комплексов (13)–(15). Раствор соответствующего о-хинона (0.5 ммоль) в ТГФ (15 мл) приливали к металлическому натрию (2.3 г, 0.1 моль) и перемешивали до полного исчезновения исходной окраски хинона и получения бледно-желтого раствора катехолата натрия. Полученный раствор отделяли от избытка металла и приливали его к раствору такого же о-хинона (0.5 ммоль) в 10 мл ТГФ. Полученный сине-фиолетовый раствор приливали при постоянном перемешивании к раствору бис-тетрагидрофуранататрихлорида хрома(III) CrCl3 · 2ТГФ (0.33 ммоль) в ТГФ (5 мл). Реакционная смесь мгновенно окрашивалась в малиновый цвет. Растворитель ТГФ заменяли на гексан, фильтровали. Фильтрат концентрировали до 20% от исходного объема и выдерживали при –18°С в течение суток, что приводило к образованию мелкокристаллических комплексов. Выход комплексов (13) 0.3 г (86%) и (15) 0.25 г (84.5%) соответственно. Комплекс (14) получали перекристаллизацией соединения (13) из ди-изо-амилового эфира.
ИК-спектр (ν, см–1): 1518 с, 1483 ср, 1415 с, 1400 с, 1360 ср, 1340 с, 1317 с, 1228 сл, 1216 ср, 1174 ср, 1111 с, 1067 сл, 1031 сл, 989 с, 963 с, 930 сл, 891 с, 877 сл, 795 сл, 784 ср, 707 сл, 665 ср.
ИК-спектр (ν, см–1): 1515 ср, 1480 ср, 1430 ср, 1402 с, 1392 ср, 1365 ср, 1360 ср, 1350 ср, 1340 ср, 1314 ср, 1294 сл, 1224 ср, 1217 ср, 1212 ср, 1196 сл, 1180 ср, 1122 ср, 1075 сл, 1057 с, 1050 с, 1000 с, 983 с, 935 сл, 898 сл, 867 сл, 853 с, 815 ср, 796 сл, 773 с, 682 сл.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Pierpont C.G. // Coord. Chem. Rev. 2001. V. 219–221. P. 415.
Pierpont C.G., Kelly J.K. PATAI’S Chemistry of Functional Groups. John Wiley & Sons, Ltd., 2013. P. 1.
Buchanan R.M., Pierpont C.G. // Inorg. Chem. 1979. V. 18. P. 3439.
Lange C.W., Conklin B.J., Pierpont C.G. // Inorg. Chem. 1994. V. 33. P. 1276.
Bubnov M.P., Skorodumova N.A., Baranov E.V. et al. // Inorg. Chim. Acta. 2012. V. 392. P. 84.
Sofen S.R., Ware D.C., Cooper S.R. // Inorg. Chem. 1979. V. 18. P. 234.
Pierpont C. G., Buchanan R. M. // Coord. Chem. Rev. 1981. V. 38. P. 45.
Brown S. N. // Inorg. Chem. 2012. V. 51. P. 1251.
Пискунов А.В., Ладо А.В., Абакумов Г.А. и др. // Изв. АН. Сер. хим. 2007. № 1. С. 92.
Bellan E.V., Poddel’sky A.I., Protasenko N.A. et al. // Inorg. Chem. Commun. 2014. V. 50. P. 1.
Stiefel E.I., Brown G.F. // Inorg. Chem. 1972. V. 11. P. 434.
Lange C.W., Pierpont C.G. // Inorg. Chim. Acta. 1997. V. 263. P. 219.
Adams D.M., Rheingold A.L., Dei A. et al. // Angew. Chem. Int. Ed. 1993. V. 32. P. 391.
Ozarowski A., McGarvey B.R., El-Hadad A. et al. // Inorg. Chem. 1993. V. 32. P. 841.
Das C., Shukla P., Sorace L. et al. // Dalton Trans. 2017. V. 46. P. 1439.
Пискунов А.В., Малеева А.В., Фукин Г.К. и др. // Коорд. химия. 2010. Т. 36. № 3. С. 163 (Piskunov A.V., Maleeva A.V., Fukin G.K. et al. // Russ. J. Coord. Chem. 2010. V. 36. № 3. P. 161). https://doi.org/10.1134/S1070328410030012
Piskunov A.V., Maleeva A.V., Fukin G.K. et al. // Dalton Trans. 2011. V. 40. P. 718.
Бокий Г.Б. Введение в кристаллохимию. М.: Изд-во МГУ, 1954. 491 с.
Ilyakina E.V., Poddel’sky A.I., Piskunov A.V. et al. // New J. Chem. 2012. V. 36. P. 1944.
Piskunov A.V., Meshcheryakova I.N., Maleeva A.V. et al. // Eur. J. Inorg. Chem. 2014. P. 3252.
Kuropatov V., Klementieva S., Fukin G. et al. // Tetrahedron. 2010. V. 66. P. 7605.
Shurygina M.P., Druzhkov N.O., Arsen’ev M.V. et al. // Russ. J. Org. Chem. 2011. V. 47. P. 490.
Fukin G.K., Cherkasov A.V., Shurygina M.P. et al. // Struct. Chem. 2010. V. 21. P. 607.
Morris A.M., Pierpont C.G., Finke R.G. // Inorg. Chem. 2009. V. 48. P. 3496.
Sofen S.R., Ware D.C., Cooper S.R. et al. // Inorg. Chem. 1979. V. 18. P. 234.
Kapre R.R., Bothe E., Weyhermuller T. et al. // Inorg. Chem. 2007. V. 46. P. 7827.
Attia A.S., Pierpont C.G. // Inorg. Chem. 1998. V. 37. P. 3051.
Bubnov M., Teplova I., Kozhanov K. et al. // Inorg. Chim. Acta 2019. V. 486. P. 113.
DeLearie L.A., Pierpont C.G. // J. Am. Chem. Soc. 1986. V. 108. P. 6393.
Delearie L.A., Haltiwanger R.C., Pierpont C.G. // Inorg. Chem. 1987. V. 26. P. 817.
Delearie L.A., Haltiwanger R.C., Pierpont C.G. // J. Am. Chem. Soc. 1989. V. 111. P. 4324.
Gerber T.I.A., Luzipo D., Mayer P. // J. Coord. Chem. 2004. V. 57. № 10. P. 893.
Boone S.R., Purser G.H., Chang H.R. et al. // J. Am. Chem. Soc. 1989. V. 111. P. 2292.
Attia A.S., Conklin B.J., Lange C.W. et al. // Inorg. Chem. 1996. V. 35. P. 1033.
Bhattacharya S., Boone S.R., Fox G.A. et al. // J. Am. Chem. Soc. 1990. V. 112. P. 1088.
Hursthouse M.B., Fram T., New L. // Transition Met. Chem. 1978. V. 3. P. 255.
Sengupta P., Bhattacharya S. // J. Indian Chem. Soc. 2013. V. 90. № 2. P. 169.
Adams D.M., Dei A., Rheingold A.L. et al. // J. Am. Chem. Soc. 1993. V. 115. P. 8221.
SAINT Plus. Data Reduction and Correction Program. Version 6.45a. Madison (WI, USA): Bruker AXS, 2003.
Data Collection, Reduction and Correction Program. CrysAlisPro – Software Package. Agilent Technologies, 2012.
Sheldrick G.M. SHELXTL. Version 6.14. Structure Determination Software Suite. Madison (WI, USA): Bruker AXS, 2003.
Sheldrick G.M. SADABS. Version 2.01. Bruker Siemens Area Detector Absorption Correction Program. Madison (WI, USA): Bruker AXS, 1998.
SCALE3 ABSPACK: Empirical Absorption Correction. CrysAlisPro – Software Package. Agilent Technologies, 2012.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия