

Координационная химия, 2020, T. 46, № 6, стр. 357-365
Синтез и спиновое состояние комплекса железа(II) с N,N'-дизамещенным 2,6-бис(пиразол-3-ил)пиридиновым лигандом
И. А. Никовский 1, А. В. Полежаев 1, 2, Д. Ю. Алешин 1, 3, Е. К. Мельникова 1, 4, Ю. В. Нелюбина 1, 5, *
1 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
2 Московский государственный технический университет им. Н.Э. Баумана
Москва, Россия
3 Российский химико-технологический университет им. Д.И. Менделеева
Москва, Россия
4 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
5 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
* E-mail: unelya@ineos.ac.ru
Поступила в редакцию 07.12.19
После доработки 27.01.2020
Принята к публикации 31.01.2020
Аннотация
При взаимодействии нового тридентатноголиганда 2,6-бис(5-трет-бутил-1-(2,6-дифторфенил)-1H-пиразол-3-ил)пиридина (L) с солью двухвалентного железа получен комплекс железа(II) [Fe(L)2](BF4)2 (I), выделенный в индивидуальном виде и охарактеризованый при помощи элементного анализа, спектроскопии ЯМР и рентгеновской дифракции. Согласно данным рентгеноструктурного анализа и метода Эванса, позволяющего из спектров ЯМР определять спиновое состояние парамагнитных соединений в растворе, ион железа(II) в комплексе I находится в высокоспиновом состоянии (S = 2 для Fe(II)) и не претерпевает температурно-индуцированного спинового перехода в диапазоне 120–345 К.
Некоторые комплексы переходных металлов могут существовать в двух спиновых состояниях и переключаться между ними при приложении подходящего внешнего воздействия [1], такого как изменение температуры, давления, облучение светом или приложение магнитного поля. Подобный спиновый переход приводит к изменению различных физических свойств, включая магнитный момент [1], цвет [2], диэлектрическую проницаемость [3] и электрическое сопротивление [4]. Это делает возможным создание на основе соответствующих комплексов переходных металлов различных молекулярных устройств и материалов, включая дисплеи, цвет которых переключается с помощью точечного нагрева и охлаждения [5], пленки в электролюминесцентных устройствах, в которых излучение света гасится за счет изменения его электрического сопротивления [6] и т.п.
Среди огромного многообразия молекулярных соединений, способных претерпевать спиновый переход при воздействии на них, в первую очередь, температуры [1], чаще всего такая способность встречается у комплексов железа(II) c ионом металла в (псевдо)октаэдрическом координационном окружении азотсодержащих гетероциклических лигандов [1]. Введение в соответствующие лиганды различных по природе заместителей, обеспечивающих необходимое для спинового перехода поле лигандов и способных к образованию прочных межмолекулярных взаимодействий (водородных связей, стекинг-взаимодействий и т.п.), которые обуславливают резкий спиновый переход с гистерезисом в кристаллическом образце [1], и систематический анализ влияния таких модификаций лиганда на спиновое состояние иона металла [7–16] лежат в основе направленного “молекулярного” дизайна [17] комплексов металлов с заданными параметрами спинового перехода.
2,6-Бис(пиразол-1-ил)пиридины [18] и изомерные им 2,6-бис(пиразол-3-ил)пиридины [19] – одни из самых популярных классов лигандов для указанной цели [17] благодаря широчайшим возможностям их химической функционализации [19] (схема 1 ). Это, в частности, позволило обнаружить зависимость спинового состояния иона железа(II) в комплексах металлов с 2,6-бис(пиразол-1-ил)пиридинами, содержащими заместители в различных положениях пиридинового и пиразол-1-ильного фрагментов, от стерических [17] и электронных [10] характеристик таких заместителей, что дает контроль над протеканием спинового перехода и его параметрами, в первую очередь температурой.
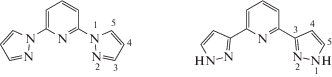
2,6-Бис(пиразол-1-ил)пиридин 2,6-Бис(пиразол-3-ил)пиридин
Схема 1 .
К сожалению, для комплексов с изомерными 2,6-бис(пиразол-3-ил)пиридинами подобных корреляций обнаружить до сих пор не удалось [17] из-за образуемых ими водородных связей N–H…X с противоионами или молекулами растворителя (схема 1 ), которые непредсказуемым образом влияют на спиновое состояние иона металла [20–24]. Единственным решением проблемы является поиск подходящего заместителя, введение которого в положении 1 пиразол-3-ильного кольца не помешает протеканию спинового перехода, что наблюдается для всех известных на настоящий момент комплексов железа(II) с 2,6-бис(пиразол-3-ил)пиридиновыми, 2,6-бис(пиразол-1-ил)пиридиновыми и другими тридентатными азотсодержащими лигандами [17] из-за стабилизации высокоспинового состояния иона металла объемными заместителями вблизи координирующих атомов азота.
Ранее в нашей лаборатории была получена серия бис(пиразол-3-ил)пиридиновых комплексов железа(II) с замещенными фенильными группами в положении 1 пиразол-3-ильного кольца, которые благодаря стерическому влиянию заместителей в орто-положении фенильных групп позволяют наблюдать температурно-индуцированный спиновый переход [25] при выборе в качестве соответствующих заместителей алкильных групп и атомов галогенов за исключением фтора. Для потенциального появления спинового перехода в этом случае в настоящей работе мы синтезировали новый 2,6-бис(пиразол-3-ил)пиридиновый лиганд, содержащий в положении 1 пиразол-3-ильного кольца дифторфенильную группу, а в положении 5 – электронодонорную и объемную трет-бутильную группу (схема 2 ). Аналогичная модификация положения 4 пиразол-1-ильного кольца 2,6-бис(пиразол-3-ил)пиридина ранее приводила к стабилизации низкоспинового состояния иона железа(II) в соответствующих комплексах [10, 17].
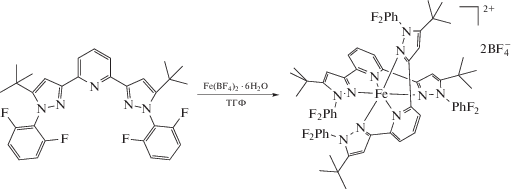
Схема 2 .
Введение стерически затрудненных заместителей одновременно в положениях 1 и 5 пиразол-3-ильного кольца 2,6-бис(пиразол-3-ил)пиридиновых лигандов ранее не проводилось в связи возможностью образования смеси региоизомеров (схема 3 ) при стандартном синтетическом подходе [26].
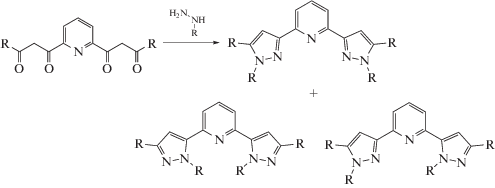
Схема 3 .
В настоящей работе мы получили необходимое 5-трет-бутил-1-фенилзамещенное производное 2,6-бис(пиразол-3-ил)пиридина – 2,6-бис(5-трет-бутил-1-(2,6-дифторфенил)-1H-пиразол-3-ил)пиридин (L) – и его комплекс [Fe(L)2](BF4)2 (I) с высокими выходами. Спиновое состояние иона железа(II) при наличии в N,N'-дизамещенном лиганде трет-бутильной группы в положении 5 пиразол-3-ильного кольца установлено по данным РСА монокристалла при 120 K. Возможность протекания спинового перехода в растворе комплекса I под действием температуры изучена с помощью стандартного для этих целей метода Эванса [27], основанного на многотемпературной спектроскопии ЯМР.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции, связанные с синтезом бис(пиразол-3-ил)пиридинового лиганда и его комплекса, выполняли на воздухе с использованием коммерчески доступных органических растворителей, перегнанных в атмосфере аргона. Гексагидраттетрафторборат железа Fe(BF4)2 ⋅ 6H2O (Sigma-Aldrich) использовали без дополнительной очистки. Этерификацию пиридин-2,6-дикарбоновой кислоты (Acros) этиловым спиртом проводили по методике [28] в присутствии серной кислоты. Анализ на углерод, азот и водород проводили на микроанализаторе CarloErba, модель 1106.
Синтез 1,1'-(пиридин-2,6-диил)бис(4,4-диметилпентан-1,3-диона). Раствор диэтилового эфира пиридин-2,6-дикарбоновой кислоты (2.80 г, 12.5 ммоль) в сухом ТГФ смешивали с NaH (1.22 г, 30.5 ммоль, 50%-ная суспензия в минеральном масле). К полученной суспензии по каплям добавляли трет-бутилметилкетон (3.40 мл, 27.5 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 4 ч, упаривали, твердый осадок промывали диэтиловым эфиром (2 × 30 мл), высушивали в вакууме, диспергировали в воде и доводили pH полученной суспензии до 5 добавлением 1 М соляной кислоты. Темно-желтый осадок отфильтровывали и высушивали в вакууме, затем растворяли в небольшом количестве горячего этанола и оставляли на 12 ч при –20°C. Образовавшиеся желтые кристаллы отделяли фильтрованием и высушивали в вакууме. Выход 2.87 г (69%).
NMR 1H (CDCl3; 400 МГц; δ, м.д.): 16.12, 15.89 (с., OH), 8.26–8.17 (м., m-Py–H, 2H), 8.01–7.96 (м., p-Py–H, 1H), 7.21, 6.91 и 4.51 (s., CH и СH2), 1.29, 1.27 и 1.25 (s., tBu, 18H). NMR 13C (100 МГц; CDCl3; смесь кето- и енольной форм; δ, м.д.): 210.4, 204.6 и 203.0 (CH), 196.5 и 182.5 (CH–COH), 181.0, 152.5, 152.1 и 152.0 (o-Py), 138.4 (p-Py), 125.6, 124.3 и 124.1 (m-Py), 92.9 и 92.8 (CH), 46.4 (CH2), 45.1, 40.4 и 40.0 (tBu), 27.4 и 26.7 (tBu).
Синтез 2,6-дифторфенилгидразина. 2,6-Дифторанилин (3.87 г, 30.0 ммоль) добавляли к 25 мл HCl (38%-ный водный раствор). К полученной суспензии, предварительно охлажденной до ‒10°C, по каплям прибавляли раствор нитрита натрия (2.17 г, 31.5 ммоль) в 5 мл воды, удерживая температуру ниже –5°C. Реакционную смесь перемешивали в течение 1 ч при –10°C. Затем к смеси по каплям добавляли раствор SnCl2 ⋅ 2H2O (16.85 г, 75 ммоль) в 30 мл концентрированной соляной кислоты при –5°C. Полученную суспензию перемешивали при комнатной температуре в течение 1 ч, осадок отфильтровывали и промывали 100 мл хлористого метилена. Образовавшийся желтый порошок высушивали в вакууме. Продукт реакции хранили в виде соли, которую переводили в форму свободного основания с помощью 1 M раствора гидроксида натрия в воде сразу перед использованием. Выход 3.30 г (61%).
NMR 1H (CDCl3; 300 МГц; δ, м.д.): 6.78–6.68 (м., 3H, m-Ph и p-Ph), 5.30 (уш.с., 1H, NH), 3.93 (уш.с., 2H, NH2). NMR 13C (DMSO-d6; 101 МГц; δ, м.д.): 153.73 (к., 1JC,F= 242,4 Гц, 3JC,F= 6.6 Гц, 2-Ph), 128.52 (т., 2JC,F= 12,7 Гц, 1-Ph), 120.46 (т., 4JC,F= = 7.2 Гц, 4-Ph), 111.58 (к., 2JC,F= 15,9 Гц, 4JC,F= 7.1 Гц, 3-Ph).
Синтез 2,6-бис(5-трет-бутил-1-(2,6-дифторфенил)-1H-пиразол-3-ил)пиридина (L). Диэтил 1,1'-(пиридин-2,6-диил)бис(4,4-диметилпентан-1,3-дион) (1 г, 3.02 ммоль) и 2,6-дифторфенилгидразин (1 г, 6.94 ммоль) растворяли в 10 мл ледяной уксусной кислоты и кипятили с обратным холодильником в течение 8 ч. Реакционную смесь охлаждали добавлением 50 мл измельченного льда. Белый осадок отфильтровывали, промывали водой и высушивали в вакууме. Для очистки полученный продукт растворяли в горячем этилацетате и добавляли гексан до появления осадка. Затем смесь выдерживали 12 ч при температуре ‒10°C. Образовавшиеся белые кристаллы отфильтровывали и высушивали в вакууме. Выход 546 мг (33%).
NMR 1H (CDCl3; 400 МГц; δ, м.д.): 1.28 (с., 18H, tBu), 7.06–7.09 (м., 6H, m-Ph–H+ Pyraz–CH), 7.44–7.51 (м., 2H, p-Ph–H), 7.70 (т.,3JH,H= = 7.7 Гц, 1H, Py), 7.90 (д., 3JH,H= 7.7 Гц, 2H, Py). NMR 13C (DMSO-d6; 101 МГц; δ, м.д.): 159.52 (к., 1JC,F= 254,92 Гц, 4JC,F= 3,3 Гц, 2-Ph), 156.06 (с., 5‑Pyraz), 152.75 (с., 2-Py), 151.53 (с., 3-Pyraz), 136.99 (с., 4-Py), 131.28 (т., 3JC,F= 9,9 Гц, 4-Ph), 119.39 (с., 3-Py), 112.16–112.21 (м., 1-Ph), 111.98–112.02 (м., 3-Ph), 103.49 (с., 4-Pyraz), 29.54 (с., 18H, tBu). NMR 19F (DMSO-d6; 376 МГц; δ. м.д.): –115.59 (с., 4F, PhF2).
Синтез комплекса I. Навески Fe(BF4)2 ⋅ 6H2O (0.0337 г, 0.1 ммоль) и L (0.109 г, 0.2 ммоль) перемешивали в ТГФ в течение 3 ч. Для очистки полученный раствор концентрировали и прикапывали гексан до появления осадка. Смесь выдерживали в течение 12 ч при температуре –10°C. Осадок отфильтровывали и высушивали в вакууме. Выход 226 мг (86%).
NMR 1H (CD3CN; 600 МГц; δ, м.д.): 0.61 (уш.с., 36H, tBu), 9.47 (уш.с., 8H, m-Ph-H), 13.24 (уш.с., 4H, p-Ph–H), 24.15 (уш.с., 2H, p-Py–H), 54.78 (уш.с., 4H, Pyraz–CH), 63.03 (уш.с., 4H, m-Py–H).
Спектры ЯМР 1H, 19F и13C регистрировали в ДМСО-d6, CDCl3 и CD3CN на спектрометрах Bruker Avance 300, 400 и 600 (с рабочими частотами для протонов 300.15, 400 и 600.22 МГц соответственно). Значения химических сдвигов в спектрах определяли относительно остаточного сигнала растворителя (1H 7.26, 13C 77.16 м.д. для CDCl3; 1H 2.5, 13C 39.52 м.д. для ДМСО-d6; 1H 1.94 м.д. для CD3CN). Спектры регистрировали с использованием следующих параметров. Для ЯМР 1Н – диапазон спектра 1000 м.д., время регистрации 0.1 с, длительность релаксационной задержки 0.1 с, длительность импульса 6.5 мкс, количество накоплений 1024; для ЯМР 13С{1H} – диапазон спектра 3000 м.д., время регистрации 0.1 с, длительность релаксационной задержки 0.1 с, длительность импульса 9 мкс, количество накоплений более 32 тысяч. Полученные спады свободной индукции в случае необходимости повышения соотношения сигнал/шум обрабатывали при помощи экспоненциального взвешивания с коэффициентом до 3 и 50 Гц в случае спектров ЯМР на ядрах 1H и 13C соответственно.
РСА. Монокристаллы комплекса I, полученные медленным испарением на воздухе из его раствора в ТГФ, проводили на дифрактометре Bruker APEX 2 CCD (MoKα-излучение, графитовый монохроматор, ω-сканирование). Структура расшифрована с использованием программы ShelXT [29] и уточнена в полноматричном МНК с помощью программы Olex2 [30] в анизотропном приближении по $F_{{hkl}}^{2}$. Положения атомов водорода рассчитаны геометрически, и они уточнены в изотропном приближении по модели наездника. Основные кристаллографические данные и параметры уточнения представлены в табл. 1.
Таблица 1.
Основные кристаллографические данные и параметры уточнения для I
| Параметр | Значение |
|---|---|
| М | 1324.65 |
| T, K | 120 |
| Сингония | Моноклинная |
| Пр. гр. | P21/n |
| Z | 4 |
| a, Å | 16.062(3) |
| b, Å | 21.690(5) |
| c, Å | 17.244(4) |
| β, град | 90.483(5) |
| V, Å3 | 6007(2) |
| ρ(выч.), г см–3 | 1.465 |
| μ, см–1 | 3.51 |
| F(000) | 2720 |
| 2θmax, град | 60 |
| Число измеренных отражений | 82 175 |
| Число независимых отражений (Rint) | 18 365 (0.0769) |
| Число отражений с I > 3σ(I) | 12 297 |
| Количество уточняемых параметров | 851 |
| GOОF | 1.010 |
| R1, wR2 (I > 2σ(I)) | 0.0478, 0.1014 |
| R1, wR2 (все данные) | 0.0869, 0.1181 |
| Δρmax/Δρmin, e Å–3 | 0.398/–0.346 |
Структурные данные для комплекса I депонированы в Кембриджском банке структурных данных (CCDC № 1968406; http://www.ccdc.cam.ac.uk/).
Метод Эванса. Температурную зависимость магнитной восприимчивости комплекса железа(II) в растворе ацетонитрила оценивали с помощью метода Эванса [27] в интервале 235–345 К с использованием ампулы для ЯМР с коаксиальной вставкой. Внутреннюю (контрольную) трубку заполняли ацетонитрилом-d3 с добавлением ~1% Me4Si, внешняя трубка содержала раствор парамагнитного комплекса (~1–5 мг/см3) в ацетонитриле-d3 с той же концентрацией Me4Si. Молярную магнитную восприимчивость (χМ) рассчитывали по разнице между химическим сдвигом Me4Si в чистом ацетонитриле-d3 и его сдвигом в растворе комплекса (Δδ, Гц) в ацетонитриле-d3 с использованием следующего уравнения:
(M – молярная масса комплекса железа(II), г/моль; ν0 – частота спектрометра, Гц; Sf – коэффициент формы магнита (4π/3); c – концентрация комплекса, г/см3; $\chi _{{\text{M}}}^{{{\text{dia}}}}$ – молярный диамагнитный вклад в парамагнитную восприимчивость, рассчитанный с использованием констант Паскаля [31]). Концентрацию с пересчитывали для каждой температуры в соответствии с изменением плотности растворителя (ρ): cT= mкρ/mр-р, где mкρ – масса комплекса, mр-р – масса раствора.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для синтеза лиганда L исходный 1,1'-(пиридин-2,6-диил)бис(4,4-диметилпентан-1,3-дион) получали по методике [32] конденсацией Кляйзена между диэтил-2,6-пиридиндикарбоксилатом и пинаконом под действием гидрида натрия в растворе ТГФ. Конденсация полученного дикетона и 2,6-дифторфенилгидразина с последующей циклизацией в одну стадию в уксусной кислоте приводила к смеси региоизомеров с преобладанием требуемого бис(пиразол-3-ил)пиридина L (L1 на схеме 4 ). Благодаря разной растворимости двух региоизомеров данный продукт легко выделяется в чистом виде перекристаллизацией смеси в этилацетат–гексан (соотношение 1 : 1). Интересно отметить, что при таком синтетическом подходе практически не наблюдалось образования смешанного региоизомера (схема 3 ).
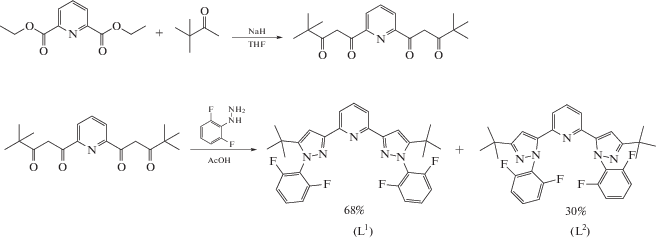
Схема 4 .
Для синтеза соответствующего комплекса железа(II) I проводили реакцию бис(пиразол-3-ил)пиридина L с гидратом тетрафторбората железа(II) в ТГФ при комнатной температуре (cхема 3). Комплекс выделен в индивидуальном виде и охарактеризован методами элементного анализа, спектроскопии ЯМР, а также РCА.
Несмотря на наличие электронодонорного заместителя в положении 5 пиразол-3-ильного кольца в лиганде L, что в случае комплексов с изомерными бис(пиразол-3-ил)пиридинами приводило к стабилизации низкоспинового состояния иона железа(II), комплекс I в кристалле находится в высокоспиновом состоянии согласно рентгенодифракционным данным при 120 К. Об этом свидетельствуют расстояния Fe–N с атомами азота двух бис(пиразол-3-ил)пиридиновых лигандов (табл. 2), типичные для высокоспиновых комплексов железа(II) с азотсодержащими гетероциклами (2.0–2.2 Å [1]). Кроме того, координационный полиэдр (КП) FeN6, который в низкоспиновом состоянии представляет собой октаэдр, искажен в сторону тригональной призмы [33]. Соответствующие значения углов N(Py)MN(Py) и θ между среднеквадратичными плоскостями двух лигандов, равные 90° и 180° в случае идеального октаэдра, составляют 86.802(15)° и 176.80(6)°. Для сравнения аналогичные значения для описанных ранее комплексов железа(II) [34] с N,N'-дифенилзамещенными бис(пиразол-3-ил)пиридинами лежали в диапазонах 67.5(3)°–68.3(3)° и 176.1(2)°–180° соответственно.
Таблица 2.
Основные геометрические параметры для комплекса I по данным РСА при 120 К*
| Параметр | Значение |
|---|---|
| M–N(Py), Å | 2.1507(14)/2.1527(15) |
| M–N(Pz), Å | 2.1749(15)–2.2043(15) |
| θ, град | 86.802(15) |
| N(Py)MN(Py), град | 176.80(6) |
| α, град | 85.56(7)–88.82(7) |
| S(TP-6) | 10.780 |
| S(OC-6) | 5.460 |
* θ – двугранный угол между среднеквадратичными плоскостями 2,6-бис(пиразол-3-ил)пиридиновых лигандов, атомы N(Py) и N(Pz) соответствуют атомам азота пиридинового и пиразол-3-ильного фрагментов, α – углу поворота дифторфенильных заместителей относительно плоскости пиразол-3-ильного кольца. S(TP-6) и S(OC-6) – отклонения формы координационного полиэдра MN6 от идеальной тригональной призмы (TP-6) и идеального октаэдра (OC-6) соответственно.
Менее выраженное отклонение формы КП в комплексе I от октаэдрического, по-видимому, вызвано наличием у него объемных орто-дифторфенильных групп в положении 1 пиразол-3-ильного цикла, которые за счет стерических эффектов орто-заместителей поворачиваются относительно плоскости последнего (рис. 1), стабилизируя (псевдо)октаэдрическую молекулярную геометрию комплекса I. Действительно, углы поворота орто-замещенных фенильных групп в нем составляют 85.56(7)°–88.82(7)°, что заметно превышает аналогичные значения (42.7(2)°–66.4(2)°) в комплексах железа(II) с N,N'-дифенилзамещенными бис(пиразол-3-ил)пиридиновымилигандами [34].
Рис. 1.
Общий вид комплекса I (а) и его проекция в перпендикулярном направлении в представлении атомов эллипсоидами тепловых колебаний (б) (p = 50%). Тетрафторборат-анионы и атомы водорода не показаны, нумерация атомов приведена только для гетероатомов.
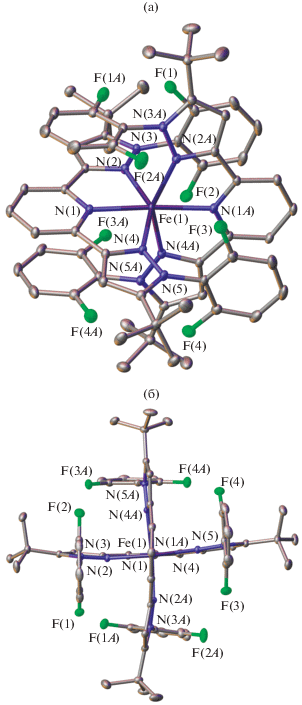
Рис. 2.
Графическое представление “мер симметрии” S(TP-6) и S(OC-6), описывающих отклонение формы полиэдра FeN6 в кристалле I (•) от идеальной тригональной призмы (TP-6) и идеального октаэдра (OC-6) соответственно. Черная линия представляет собой путь наименьшего искажения геометрии полиэдра при переходе между указанными многогранниками.
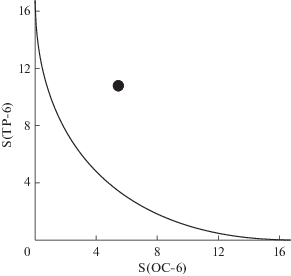
Рис. 3.
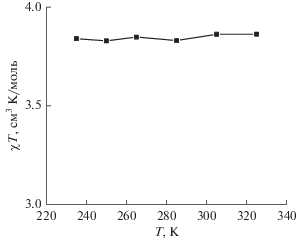
Температурная зависимость магнитной восприимчивости комплекса I в растворе дейтероацетонитрила по данным спектроскопии ЯМР (метод Эванса).
Тем не менее это не приводит к отсутствию в комплексе I тригонально-призматического искажения КП, характерного для высокоспиновых комплексов железа(II) с би- и тридентатными лигандами [35]. Графически его можно представить в виде так называемых “мер симметрии” [35], описывающих отклонения формы КП FeN6 от идеального октаэдра (S(OC-6)) и идеальной тригональной призмы (S(TP-6)). Чем эти значения меньше, тем лучше форма КП описывается соответствующим многогранником (pис. 2). В комплексе I величины октаэдрической S(OC-6) и тригонально-призматической S(TP-6) “мер симметрии”, оцененные на основе рентгенодифракционных данных с использованием программы Shape 2.1 [35], составляют 5.460 и 10.780 соответственно (табл. 2). Они попадают в диапазон “мер симметрии” S(OC-6) и S(TP-6) для высокоспиновых комплексов железа(II) [34] с N,N'-дифенилзамещенными бис(пиразол-3-ил)пиридинами, в которых координационное окружение иона металла заметно искажено в сторону тригональной призмы, что не позволяет ему переходить в низкоспиновое состояние [36].
Таким образом, данные РСА для комплекса I указывают на то, что в окружении двух бис(пиразол-3-ил)пиридиновых лигандов L с объемными дифторфенильными заместителями в положении 1 пиразол-3-ильного кольца [17] ион железа(II) находится в высокоспиновом состоянии в кристалле при 120 K.
В таком же спиновом состоянии комплекс I находится в диапазоне 235–345 К в растворе ацетонитрила, что однозначно подтверждается данными метода Эванса [27], основанного на использовании спектроскопии ЯМР. Указанный метод – самый доступный способ измерения χМ раствора, из-за чего он активно применяется для поиска корреляций структура–свойство в отсутствие эффектов кристаллической упаковки, зачатую приводящих к блокированию иона металла в одном спиновом состоянии [36], и последующего направленного дизайна комплексов металлов с заданными параметрами спинового перехода [17]. Для измерения χМ раствора, изменяющейся при добавлении к нему парамагнитного соединения, такого как комплекс железа(II) в высокоспиновом состоянии (см. Экспериментальную часть), в методе Эванса одновременно регистрируют спектры ЯМР для раствора стандартного соединения, обычно тетраметилсилана (ТМС), в присутствии парамагнитного комплекса и в отсутствии оного. Для этого в стандартную ампулу, содержащую раствор соответствующего комплекса и ТМС в известной концентрации, помещают специальную коаксиальную вставку, содержащую раствор ТМС в том же растворителе, в котором при охлаждении не будет наблюдаться образования осадка, что является одним из ограничений метода Эванса. Разница в значениях химического сдвига ТМС в спектре ЯМР, зарегистрированном от этих двух растворов, позволяет рассчитать χМ раствора исследуемого парамагнитного соединения и тем самым однозначно установить спиновое состояние иона металла при определенной температуре или же в диапазоне температур, доступных для выбранного растворителя.
Согласно полученным таким образом данным, значения χТ для раствора комплекса I в ацетонитриле при изменении температуры в пределах 235–345 K (pис. 3) практически не отклоняются от 3.8 см3 моль–1 K, что однозначно подтверждает высокоспиновое состояние иона железа(II) (S = 2) во всем диапазоне температур. Наблюдаемая стабилизация высокоспинового состояния в растворе, в котором отсутствуют эффекты кристаллической упаковки, свидетельствует о “внутримолекулярной” природе данного явления, по всей видимости, вызванного наличием в лиганде объемных орто-замещенных фенильных групп вблизи координирующих атомов азота [17]. Это, в свою очередь, указывает на недостаточное электронное и стерическое влияние трет-бутильных заместителей в положении 5 пиразол-3-ильного кольца, что в результате приводит к отсутствию температурно-индуцированного спинового перехода в комплексе I.
Таким образом, мы синтезировали и охарактеризовали новый комплекс железа(II) с N,N'-замещенным 2,6-бис(пиразол-3-ил)пиридиновым лигандом, содержащим объемные заместители одновременно в положениях 1 и 5 пиразол-3-ильного кольца. Полученные для него низкотемпературные рентгенодифракционные данные, в первую очередь длины связей M–N и тригонально-призматическое искажение координационного полиэдра иона железа(II), однозначно указывают на то, что он находится в высокоспиновом состоянии (S = 2) в кристалле даже при 120 К. Отсутствие температурно-индуцируемого спинового перехода в растворе в диапазоне 235–345 K подтверждается данными спектроскопии ЯМР (метода Эванса). Таким образом, введение электронодонорного и стерически затрудненного заместителя (трет-бутильной группы) в положении 5 пиразол-3-ильного кольца 2,6-бис(пиразол-3-ил)пиридина при наличии в нем объемного N-заместителя не оказывает влияния на спиновое состояние иона железа(II) в соответствующем комплексе, который остается высокоспиновым и в кристаллическом состоянии, и в растворе ацетонитрила.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Spin-Crossover Materials: Properties and Applications / Ed. Halcrow M.A. John Wiley & Sons, Ltd., 2013.
König E. // Progress in Inorganic Chemistry, 2007. P. 527.
Bousseksou A., Molnár G., Demont P., Menegotto J. // J. Mat. Chem. 2003. V. 13. № 9. P. 2069.
Matsuda M., Tajima H. // Chem. Lett. 2007. V. 36. № 6. P. 700.
Kahn O., Martinez C. J. // Science. 1998. V. 279. № 5347. P. 44.
Matsuda M., Isozaki H., Tajima H. // Thin Solid Films. 2008. V. 517. № 4. P. 1465.
Phan H., Hrudka J.J., Igimbayeva D. et al. // J. Am. Chem. Soc. 2017. V. 139. № 18. P. 6437.
Rodríguez-Jiménez S., Yang M., Stewart I. et al. // J. Am. Chem. Soc. 2017. V. 139. № 50. P. 18392.
Kimura A., Ishida T. // ACS Omega. 2018. V. 3. № 6. P. 6737.
Kershaw Cook L.J., Kulmaczewski R., Mohammed R. et al. // Angew. Chem. Int. Ed. 2016. V. 55. № 13. P. 4327.
Elhaïk J., Evans D.J., Kilner C.A., Halcrow M.A. // Dalton Trans. 2005. № 9. P. 1693.
McPherson J.N., Elton T.E., Colbran S.B. // Inorg. Chem. 2018. V. 57. № 19. P. 12312.
Santoro A., Kershaw Cook L.J., Kulmaczewski R. et al. // Inorg. Chem. 2015. V. 54. № 2. P. 682.
Nakano K., Suemura N., Yoneda K. et al. // Dalton Trans. 2005. № 4. P. 740.
Kitchen J.A., Olguín J., Kulmaczewski R. et al. // Inorg. Chem. 2013. V. 52. № 19. P. 11185.
Hoselton M., Wilson L.J., Drago R.S. // J. Am. Chem. Soc. 1975. V. 97. № 7. P. 1722.
Halcrow M.A. // Crystals. 2016. V. 6. № 5. P. 58.
Halcrow M.A. // Coord. Chem. Rev. 2009. V. 253. № 21. P. 2493.
Halcrow M.A. // Coord. Chem. Rev. 2005. V. 249. № 25. P. 2880.
Scudder M.L., Craig D.C., Goodwin H.A. // CrystEngComm. 2005. V. 7. № 107. P. 642.
Clemente-León M., Coronado E., Giménez-López M.C., Romero F.M. // Inorg. Chem. 2007. V. 46. № 26. P. 11266.
Coronado E., Gimenez-Lopez M.C., Gimenez-Saiz C., Romero F.M. // CrystEngComm. 2009. V. 11. № 10. P. 2198.
Bartual-Murgui C., Codina C., Roubeau O., Aromí G. // Chem. Eur. J. 2016. V. 22. № 36. P. 12767.
Jornet-Molla V., Gimenez-Saiz C., Romero F.M. // Crystals. 2018. V. 8. P. 439.
Nikovskiy I., Polezhaev A., Novikov V. et al. // Chem. Eur. J. 2020. https://doi.org/10.1002/chem.202000047
van der Valk P., Potvin P.G. // J. Org. Chem. 1994. V. 59. № 7. P. 1766.
Evans D. F. // J. Chem. Soc. (Resumed) 1959. P. 2003.
Polezhaev A.V., Chen C.-H., Kinne A.S. et al. // Inorg. Chem. 2017. V. 56. № 16. P. 9505.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Cryst. 2009. V. 42. P. 339.
Bain G.A., Berry J.F. // J. Chem. Educ. 2008. V. 85. № 4. P. 532.
Korzekwa J., Scheurer A., Heinemann F.W., Meyer K. // Dalton Trans. 2017. V. 46. № 40. P. 13811.
Alvarez S. // J. Am. Chem. Soc. 2003. V. 125. №22. P. 6795.
Nelyubina Y.V., Polezhaev A.V., Pavlov A.A. et al. // Magnetochemistry. 2018. V. 4. № 4. P. 46.
Alvarez S. // Chem. Rev. 2015. V. 115. P. 13447.
Kershaw Cook L., Mohammed R., Sherborne G. et al. // Coord. Chem. Rev. 2015. V. 289–290. P. 2.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия