

Координационная химия, 2020, T. 46, № 7, стр. 402-409
Влияние строения карбоксилатных пористых координационных полимеров – стационарных фаз для жидкостной хроматографии – на эффективность разделения производных анилина
Д. А. Лагошняк 1, А. М. Мишура 1, М. Н. Курмач 1, А. С. Литвиненко 1, Н. В. Грабовая 1, К. С. Гавриленко 2, 3, О. В. Манойленко 2, С. В. Колотилов 1, *
1 Институт физической химии им. Л.В. Писаржевского НАН Украины
Киев, Украина
2 Научно-производственное предприятие “Енамин”
Киев, Украина
3 Научно-образовательный химико-биологический центр
Киевского национального университета им. Т. Шевченко
Киев, Украина
* E-mail: svk001@mail.ru
Поступила в редакцию 20.11.2019
После доработки 13.02.2020
Принята к публикации 17.02.2020
Аннотация
Изучена возможность хроматографического разделения изомерных нитроанилинов и фторзамещенных аналогов с использованием пористых координационных полимеров [Al(OH)(Fum)]n (I) (Fum2– = анион транс-1,2-этилендикарбоновой кислоты), [Al(OH)(Bdc)]n (II) (Bdc2– = анион 1,4-бензолдикарбоновой кислоты), [Cu3(Btc)2(H2O)3] (III) (Btc3– = анион 1,3,5-бензолтрикарбоновой кислоты) и [Cr3(O)(OH)(H2O)2(Bdc)3] (IV) как неподвижных фаз в смеси неполярных растворителей. Показано, что разделение аминов достигалось только в случае [Al(OH)(Fum)]n. Проанализирована возможная связь факторов разделения аминов с размерами их молекул, значениями полярности и липофильности. Среди рассмотренных факторов только разница значений липофильности оказывает заметное влияние на эффективность.
Фторсодержащие амины широко используются как активные вещества в агрохимии [1] и фармацевтике [2, 3]. Разделение таких аминов, а также аналогов, не содержащих фтор, представляет важную задачу химического производства, в частности тонкого органического синтеза. Такая задача часто возникает при получении фторзамещенных анилинов путем замены аминогруппы на фтор методом Шимана, при которой может образовываться значительное количество побочного продукта, содержащего атом водорода на месте аминогруппы [4]. Для разделения таких веществ используют методы, основанные на кристаллизации аминов или их солей, селективной сорбции аминов [5], а во многих случаях наиболее результативное разделение смесей аминов достигается при использовании хроматографии [6]. Поиск новых носителей для хроматографического разделения фторсодержащих аминов и аналогов, не содержащих фтор, а также определение факторов, влияющих на эффективность разделения таких смесей, – одна из важных задач современной химии [7]. Как сорбенты или стационарные фазы для хроматографии могут быть использованы пористые координационные полимеры (ПКП), эффективность которых как носителей для хроматографии показана в ряде работ [8–11].
Цель настоящей работы – исследование влияния химического состава и строения ПКП на возможность и эффективность хроматографического разделения изомерных нитроанилинов и их фторзамещенных аналогов.
В качестве объектов исследования были выбраны ПКП алюминия [Al(OH)(Fum)]n (I) и [Al(OH)(Bdc)]n (II), относящиеся к структурному типу MIL-53(Al) (Fum2– = анион транс-1,2-этилендикарбоновой кислоты, 1,4-Bdc2– = анион 1,4-бензолдикарбоновой кислоты); [Cu3(Btc)2(H2O)3] (III), известный как HKUST-1, где Btc3– = анион 1,3,5-бензолтрикарбоновой кислоты) и [Cr3(O)(OH)- (H2O)2(Bdc)3]n (IV), относящийся к структурному типу MIL-101(Cr). Для сравнения было исследовано разделение аминов на силикагеле Silica-60. Как амины использовали 2-амино-1-нитробензол (1), 3-амино-1-нитробензол (2), 4-амино-1-нитробензол (3), 2-амино-3-фтор-1-нитробензол (4), 2-амино-4-фтор-1-нитробензол (5), 2-амино-5-фтор-1-нитробензол (6); их структурные формулы представлены на схеме 1 . В работе проанализировано влияние строения и физических свойств исследуемых аминов на возможность разделения их смесей.
Схема 1 .
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Коммерчески доступные реактивы (Aldrich, НПП “Енамин”) использовали без предварительной очистки. Метил-трет-бутиловый эфир для хроматографии очищали для удаления следов влаги путем перегонки над LiAlH4, н-гексан использовали без дополнительной очистки.
Синтез [Al(OH)(Fum)]n (I) и [Al(OH)(Bdc)]n (II), [Cr3(O)(OH)(H2O)2(Bdc)3]n (IV) выполняли по описанным ранее методикам [12–14]. Для удаления примесей H2Bdc образец II нагревали в вакууме при 320°С в течение 2 сут, как описано в [13], образец IV последовательно промывали N,N-диметилформамидом и горячим этанолом, как описано в [14].
| Найдено для I · 4H2O, %: | C 20.7; | Н 4.95. | |
| Для C4H11O9Al | |||
| вычислено, %: | C 20.9; | Н 4.82; | N 0.0. |
| Найдено для II · 1.5H2O, %: | C 41.1; | Н 3.15; | N 0.0. |
| Для C8H8O6.5Al | |||
| вычислено, %: | C 40.9; | Н 3.43; | N 0.0. |
| Найдено для IV · 28H2O, %: | C 23.9; | Н 5.95; | N 0.08. |
| Для C24H73O44Cr3 | |||
| вычислено, %: | C 23.6; | Н 6.02; | N 0.0. |
Синтез [Cu3(Btc)2(H2O)3] (III) проводили путем электрохимического растворения меди в растворе H3BTC по модифицированной методике [15]: две медные пластины (1.5 × 2.8 см) обрабатывали 25%-ной азотной кислотой в течение 10 с, промывали дистиллированной водой, изопропанолом и помещали в раствор 1.2 г NEt4BF4 и 1.6 г H3Btc в 50 мл 96%-ного EtOH при 60°С. Электролиз проводили в гальваностатическом режиме с плотностью тока 0.11–0.13 A/см2 (ток потенциостата I = 1.4–1.5 A, напряжение U = 6.0–6.5 В) в течение 2 ч, что соответствует 3 Ф/моль H3Btc, источник тока подключали по двухэлектродной схеме. Образовавшийся продукт отфильтровывали и трижды промывали горячим этанолом, сушили 12 ч на воздухе и 2 ч в сушильном шкафу при 80°С. Получили порошок голубого цвета. Выход 1.43 г (83% по H3Btc).
Химическую чистоту полученных соединений проверяли методом CHN-анализа с использованием анализатора Carlo Erba 1106.
Фазовую чистоту ПКП I–IV подтверждали с помощью рентгенофазового анализа (РФА) с использованием дифрактометра Bruker D8 Advance (CuKα-излучение, λCu = 0.1542 Å). Характеристики пористой структуры ПКП определяли путем анализа изотерм адсорбции азота, измеренных при 78 К с использованием прибора Sorptomatic 1990 [16]. Перед измерениями образцы десольватировали путем нагревания в вакууме (10–3 торр) при 150°С.
Для разделения смесей нитроанилинов стеклянную хроматографическую колонку диаметром 1 см наполняли ПКП I–IV на высоту 10 см (высота после “утрамбовки” ПКП под слоем смеси растворителей, используемых в качестве подвижной фазы). Масса ПКП составляла 9.2–9.5 г. Все ПКП перед использованием не десольватировали. В качестве подвижной фазы (элюента) использовали смесь метил-трет-бутиловый эфир (МТБЭ)–н-гексан (70 : 30), которую пропускали без дополнительного давления. Для эксперимента носитель диспергировали 5 мин ультразвуком в подвижной фазе и порциями наливали суспензию в колонку, периодически постукивали по стенке, до тех пор, пока столбик носителя не достигал 10 см. ПКП IV перед заполнением колонки промывали чистым МТБЭ под действием ультразвука 10 мин. Смесь аминов, содержащую по 1–2 мг вещества, вводили в колонку в растворе в хлористом метилене (50 мкл), после чего элюировали смесью МТБЭ–гексан. Эксперимент проводили на воздухе без дополнительной защиты от влаги. Идентификацию аминов на выходе из колонки осуществляли на основании сравнения их Rf с Rf стандартов (чистых аминов) при тонкослойной хроматографии (ТСХ) на силикагеле Nagel, элюент – смесь МТБЭ–гексан (70 : 30).
Коэффициент липофильности исследованных аминов (lg(P)) рассчитывали по программе ACD/Log P DB 10.04 [17], дипольные моменты и размеры молекул – методом квантовой механики MP2 (теория возмущений Мёллера–Плессета 2-го порядка) с использованием программы ORCA 4.1.2 [18], для описания электронных орбиталей использовали базисный набор def2-TZVPP (а также вспомогательные базисные наборы def2/J и def2-TZVPP/C для алгоритмов RI [19] и COSX [20], применяемых для ускорения расчетов). В качестве исходной геометрии молекулы принимали геометрию, полученную путем оптимизации методом молекулярной механики (силовое поле MMFF94) по программе Avogadro 1.1.1. Исходную геометрию подвергали искажению (значение каждой координаты каждого атома было изменено на случайную величину в пределах ±0.05 Å) с целью снижения вероятности попадания в седловую точку при оптимизации, после чего молекулы оптимизировали методом MP2. Расчет дипольного момента производили для равновесной геометрии также методом MP2 (на основе распределения электронной плотности с учетом поправок MP2). Выбор метода MP2 обусловлен данными литературы, обосновывающими его достаточную точность для расчета дипольного момента [21] наряду с приемлемыми затратами вычислительных ресурсов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Соединения I–IV представляют собой 3D-координационные полимеры с непрерывными порами – каналами в кристаллических решетках [12, 13, 22, 23].
Для определения фазовой чистоты ПКП I–IV и конформации, которую принимают ПКП I и II, был проведен РФА образцов в виде сольватов, находящихся в равновесии с воздухом. Такой поход был выбран с учетом того, что исследования разделения аминов проводили из растворов, а в таких экспериментах ПКП не десольватировали. Положения рефлексов на полученных дифрактограммах (рис. 1) хорошо согласуются с ожидаемыми для I – модификации с параметрами: пр. гр. P21/c, a = 6.842(3), b = 12.088(2), c = 4.21(1) Å, β = 122.55(6)° [12], II – модификации с параметрами: пр. гр. C2/c, a = 19.685(4), b = 7.849(1), c = 6.782(1) Å, β = 104.90(2)° [24], III с параметрами пр. гр. Fm$\bar {3}$m, a = 26.2879(3) Å [25]) и IV с параметрами: пр. гр. Fd$\bar {3}$m, a = 88.869 Å [23]. В случае I рефлексы существенно уширены, для IV относительные интенсивности рефлексов в области 2θ < 12° намного ниже ожидаемых. Такие отличия экспериментальных дифрактограмм от рассчитанных обусловлены, по-видимому, разупорядочением кристаллических решеток I и IV.
Рис. 1.
Экспериментальные и теоретические дифрактограммы для I (а), II (б), III (в), IV (г). Теоретические дифрактограммы рассчитаны для структур с параметрами, указанными в тексте.
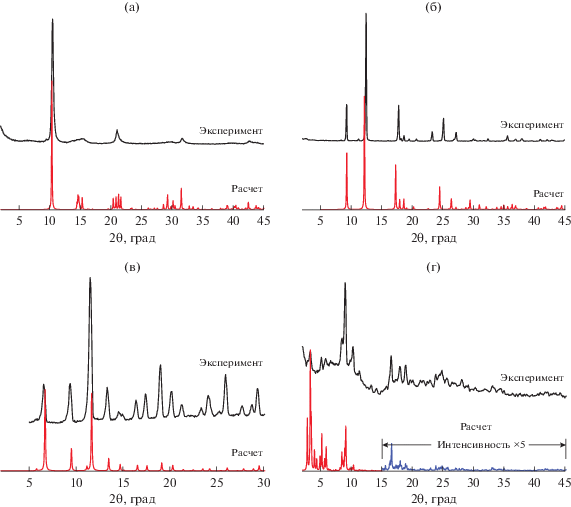
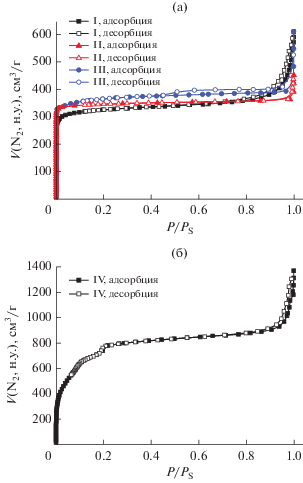
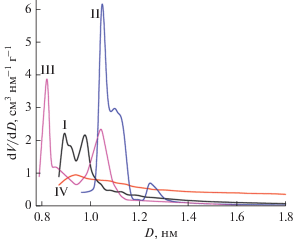
Рис. 3.
Распределение микропор ПКП I–IV по диаметру, рассчитанное из изотерм адсорбции азота по модели Сайто–Фоли.
Полимерные сетки ПКП I и II лабильны. Строение II легко меняется в зависимости от наличия молекул-гостей, а размер пор этого ПКП может принимать значения от 8.5 × 8.5 Å2 (ПКП с незаполненными порами) до 2.6 × 13.6 Å2 (ПКП с захваченной молекулой воды) [13]. Лабильность структуры I проявляется при высоких давлениях [26], но наличие молекул-гостей не оказывает существенного влияния на строение кристаллической решетки этого соединения [12]. Тем не менее в литературе приводятся разные значения размеров пор I – от 5.7 × 6.0 Å2 [12] до 7.3 × 7.7 Å2 [26]. Полимерные сетки ПКП III и IV конформационно-жесткие, размер пор составляет 9 × 9 Å2 в случае III [22] и 29–34 Å для IV [23]. Эти оценки размера пор основаны на измерении расстояний между атомами, находящимися на противоположных сторонах поры, с учетом их ван-дер-ваальсовых радиусов. Недостаток такого подхода заключается в неоднозначности выбора атомов, используемых для оценки. По данным РФА, ПКП I сохраняет конфигурацию с “раскрытыми” порами, тогда как II принимает “сложенную” конформацию.
Альтернативным подходом может быть оценка размера пор с использованием программы PL-ATON [27] по зависимости доступного для молекул-гостей объема от диаметра молекулы-зонда. Размером поры можно считать максимальный диаметр молекулы-зонда, для которого доступный объем еще не равен нулю. Недостаток такого подхода заключается в том, что он не позволяет непосредственно оценить размер “окна”, ведущего в пору, и, как следствие, значение полученного размера поры не гарантирует ее доступность для молекул близкого размера. В соответствии с оценкой, проведенной с использованием программы PLATON, диаметр пор в I составляет 5.5 ± ± 0.1 Å, в IV – 30.6 ± 0.1 Å, что хорошо согласуется с приведенными выше значениями. В ПКП II есть поры диаметром 3.0 ± 0.1 Å. Для “развернутой” модификации II (с параметрами пр. гр. Imcm, a = 16.772(8), b = 13.145(5), c = 6.847(5) Å [28]), переход в которую теоретически не исключен при сорбции нитроанилина, подобная оценка дает значение диаметра пор на уровне 7.2 ± 0.1 Å. В случае III оценка размера пор, проведенная с использованием молекул-зондов в программе PLATON, составляет 13.6 ± 0.1 Å, что превышает размер пор, определенный как расстояние между противоположными атомами в поре. Такое отличие может объясняться большим отличием между размером “окна”, ведущего в пору, и размером самой полости.
Характеристики пористой структуры ПКП I–IV определяли путем анализа изотерм адсорбции азота (pис. 2). Значения площади поверхности (SБЭТ) исследованных ПКП лежат в пределах от 1290 до 3160 м2/г, объем микропор (VDR) по Дубинину–Радушкевичу – в пределах от 0.498 до 0.710 см3/г, полный объем пор (VT) – от 0.560 до 1.464 см3/г, что сопоставимо с приведенными в литературе данными [12, 23, 29–31]. Размер микропор, определенный методом Сайто–Фоли [32], в случае I и II заметно превышает размер пор, определенный по кристаллографическим данным (для II сравнивали с формой с пр. гр. Imcm), в случае III и IV – наоборот, размер меньше (pис. 3, табл. 1). Одной из причин отличия размеров микропор, определенных по данным адсорбции азота и по кристаллографическим данным, может быть погрешность, связанная с использованием функции, параметризированной не для ПКП (использовали функцию для адсорбции азота на цеолите [33]). В то же время применение такого же подхода в случае пивалатных ПКП дало значения размера пор, близкое к определенному по структурным данным [34]. Таким образом, отличие в случае I и II обусловлено, скорее, разупорядочением их кристаллических решеток, связанных с “дыханием”, а не с ограниченной пригодностью функции, описывающей адсорбцию азота. В случае III распределение, по-видимому, отражает наличие “окон” и “полостей” (бимодальность распределения пор по диаметру для III согласуется с описанной в [30]), а в случае IV максимум на кривой распределения пор по диаметру выражен слабо и погрешность определения размера пор, по-видимому, высокая. Для сопоставления размеров пор ПКП и молекул разделяемых аминов в данном случае корректнее использовать значения диаметра пор, рассчитанные по кристаллографическим данным для сольватированных кристаллов (которые соответствуют экспериментально полученным дифрактограммам), потому что адсорбция аминов происходит из растворов, а данные адсорбции азота отражают строение тщательно десольватированных образцов. Как отмечалось выше, десольватация II ведет к существенному изменению его структуры.
Таблица 1.
Характеристики пористой структуры ПКП по данным адсорбции азота
| ПКП* | SБЭТ, м2/г | Vпор, см3/г** | Vмикро, см3/г*** | Dпор(Сайто–Фоли), Å**** |
|---|---|---|---|---|
| I | 1290 | 0.645 | 0.498 | 9.0 |
| II | 1430 | 0.560 | 0.535 | 10.5 |
| III | 1435 | 0.605 | 0.533 | 8.2 |
| IV | 3160 | 1.464 | 0.710 | 9.5 |
Таким образом, в соответствии с приведенными выше оценками, размер пор в I сопоставим с размером молекул исследуемых аминов (не менее 3.9 × 7.7 Å, табл. 2), но, как будет показано ниже, такие поры для исследуемых субстратов недоступны либо энергетический барьер заполнения пор такими субстратами высокий [35]. Поры в II для исследуемых аминов недоступны, если в процессе сорбции II не перейдет в конформацию с “раскрытыми” порами (аналогично ситуации, описанной в [29]). В то же время поры в III и IV потенциально доступны для аминов.
Таблица 2.
Некоторые физические свойства изомерных нитроанилинов и фторнитроанилинов аминов
| Название | Размер молекул, Å3 | Полярность, Db | lg(P) |
|---|---|---|---|
| 2-Амино-1-нитробензол (1) | 7.7 × 8.7 × 3.9 | 4.14 | 1.83 ± 0.26 |
| 3-Амино-1-нитробензол (2) | 7.9 × 8.6 × 3.9 | 5.2 | 1.37 ± 0.24 |
| 4-Амино-1-нитробензол (3) | 6.8 × 9.5 × 3.9 | 6.1 | 1.39 ± 0.24 |
| 2-Амино-3-фтор-1-нитробензол (4) | 8.8 × 8.2 × 4.2 | 4.23 | 2.07 ± 0.37 |
| 2-Амино-4-фтор-1-нитробензол (5) | 7.7 × 9.2 × 3.9 | 2.96 | 2.08 ± 0.37 |
| 2-Амино-5-фтор-1-нитробензол (6) | 7.7 × 8.7 × 3.9 | 4.21 | 1.94 ± 0.36 |
В настоящей работе для элюирования аминов была использована смесь метил-трет-бутиловый эфир–н-гексан, выбор которых связан с желанием минимизировать возможность образования водородных связей растворителя с ПКП и координации растворителя к ионам металла в ПКП (особенно в III), что важно для минимизации конкуренции растворителя за центры адсорбции в ПКП. Метил-трет-бутиловый эфир был добавлен к н-гексану для повышения полярности элюента и растворимости в нем исследуемых нитроанилинов. B выбранной смеси растворителей вероятность протонирования аминов очень мала, что важно для корректного расчета меры гидрофобности lg(P) (табл. 2).
В эксперименте по разделению м-нитроанилина (2) и п-нитроанилина (3) на колонке с I первым выходит амин (3), его выход из колонки начинается приблизительно через 60 мин после начала эксперимента и заканчивается через 90 мин (здесь и далее времена указываются от начала эксперимента с точностью до ±5 мин). На 110-й мин начинается выход изомера (2), который полностью выходит на ~170-й минуте. В аналогичном эксперименте с использованием колонки с ПКП IV эти же амины проходят через нее за ~180 мин, в случае колонки с ПКП III – за ~60 мин, что сопоставимо с временем прохождения через колонку с ПКП I. Oднако в случае колонок, заполненных III или IV, разделения аминов (2) и (3) не достигалось. В случае II амины проходят через колонку за ~90 мин и разделения также не наблюдалось. Разное время выхода аминов из колонок с разными ПКП, по-видимому, в первую очередь связано с разной проницаемостью носителя – способностью жидкости протекать через частицы твердой фазы, но также может быть обусловлено и отличием в их сорбционной способности по отношению к аминам. В то же время, принимая во внимание близкие значения времени выхода аминов в случае колонок с ПКП I и IV, можно сделать вывод о том, что низкая эффективность разделения в случае IV не связана с отличием в проницаемости слоя ПКП.
Следует учитывать, что проницаемость ПКП может меняться в зависимости от времени из-за уплотнения частиц, изменения их морфологии при действии растворителя. Для исключения влияния этих факторов количественные характеристики эффективности разделения аминов оценивали по величине фактора разделения α, который определяли как отношение времен начала выхода соответствующих аминов [36] (здесь и далее для удобства α рассчитывается как отношение большего времени к меньшему). Такой подход используется для характеризации эффективности разделения веществ и исключения действия случайных факторов [37]. Для разделения (2) и (3) на колонке с ПКП I значение α составляет 1.8.
В отличие от колонки с ПКП I, при разделении (2) и (3) на колонке с силикагелем первым выходит (2) через ~20 мин и заканчивается на 50-й минуте. На 70-й мин начинается выход III, который заканчивается на 80-й минуте. Таким образом, порядок выхода аминов в смеси (2) и (3) на силикагеле “инвертируется” по сравнению с порядком выхода этих аминов на ПКП I, что может быть связано с существенной разницей гидрофильности/гидрофобности поверхности таких носителей; такое отличие будет обсуждаться ниже. Эффективность разделения (2) и (3) на колонке с силикагелем по критерию α превышает аналогичный показатель для I и составляет 3.5.
Учитывая низкую эффективность разделения (2) и (3) на колонках с ПКП II–IV, эксперименты по разделению фторсодержащих аминов проводили только на колонках с I и для сравнения c силикагелем. Результаты исследования приведены в табл. 3. С наиболее высокой эффективностью (α = 3) проходило разделение смеси (1) и (5). В то же время смесь аминов (4) и (6) не разделяется. В случаях разделения пар аминов 2/3, 1/6 и 4/5 порядок выхода этих веществ на колонке с силикагелем отличается от наблюдаемого порядка выхода на ПКП I. Изменение порядка аминов на колонке с ПКП по сравнению с силикагелем выхода можно объяснить различием природы поверхности. Таким образом, важно отметить, что выводы о связи эффективности разделения аминов и природы поверхности носителя справедливы только для ПКП подобного состава с близким химическим строением поверхности.
Таблица 3.
Результаты разделения замещенных нитроанилинов на ПКП I
| Выход амина из колонки | tнач(1-й), мин | tкон(1-й), мин | tнач(2-й), мин | tкон(2-й), мин | α* | Порядок выхода на колонке с силикагелем | |
|---|---|---|---|---|---|---|---|
| 1-й | 2-й | ||||||
| (3) | (2) | 60 | 90 | 110 | 170 | 1.8 | Обратный |
| (5) | (1) | 360 | 1080 | 1080 | 1440 | 3 | Совпадает |
| (6) | (1) | 360 | 1440 | 720 | 2 | Обратный | |
| (5) | (4) | 120 | 330 | 270 | 2.3 | Обратный | |
| (6) | (4) | 360 | 1440 | 360 | 1 | Совпадает | |
Поскольку лучший результат удалось достичь для смеси (1) и (5), была исследована адсорбция этих веществ на ПКП в смеси н-гексан–МТБЭ. Образцы ПКП предварительно не десольватировали для наилучшего соответствия условиям хроматографического разделения. Сорбционная емкость ПКП I в отношении (1) превышает емкость в отношении (5), что соответствует результатам эксперимента по хроматографическому разделению смеси аминов – первым из колонки выходит то вещество, которое хуже сорбируется. Однако следует отметить низкую сорбционную емкость ПКП I в отношении этих аминов, которая не превышает 1 × 10–6 моля на грамм сорбента. По-видимому, сорбция аминов происходит только на поверхности ПКП, а заполнение пор аминами, если и происходит, не играет ключевой роли в процессе хроматографического разделения. Этот вывод согласуется с результатами разделения аминов (1) и (3) на ПКП I–IV: среди исследованных ПКП соединения III и IV содержат поры, размер которых заведомо превышает размер пор исследованных аминов (табл. 2), однако разделения аминов на колонках с этими ПКП не происходит.
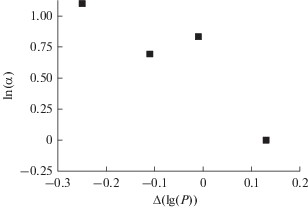
Для определения факторов, влияющих на эффективность хроматографического разделения исследованных аминов на ПКП, было проведено сравнение результатов экспериментов по разделению аминов с их физическими свойствами (такими, как полярность, размер молекулы и липофильность, которая в некоторой мере характеризует гидрофобность вещества [38]). Не удалось найти непосредственной связи величин α и разницы или отношения дипольных моментов разделяемых аминов, отличием в размере молекул аминов. В то же время наблюдается симбатная зависимость между величинами ln(α) и Δlg(P), которое определяется как lg(P)1 – lg(P)2 (pис. 4). Такая зависимость согласуется с теоретически ожидаемой: связь фактора разделения с разницей энергий взаимодействия вещества с носителем (ΔG) описывается выражением ΔΔG = –RT ln(α) [39]. Если предположить, что равновесная константа распределения вещества между носителем и элюентом K пропорциональна константе его распределения между октанолом и водой, которая служит основой определения параметра липофильности lgP, и учитывая, что ΔG = –RT lnK, можно получить выражение ΔΔG = –RT(lnK1 – lnK2) = –RT(lgP1 – ‒lgP2)B, где B – константа. Тогда ln(α) = B(lgP1 – – lgP2). Аналогичное выражение c поправкой на объем подвижной и стационарной фаз приведено в [40].
Таким образом, среди трех исследованных ПКП разделение нитроанилинов наблюдалось только на колонке с ПКП I. Такое разделение не связано с заполнением пор аминами, что подтверждается данными адсорбции аминов из растворов и согласуется с результатами оценки размера пор. ПКП II–IV оказались неэффективными для разделения изученных аминов при использовании в качестве стационарных фаз для жидкостной хроматографии несмотря на доступность пор в ПКП III и IV. Эффективность разделения аминов в при использовании ПКП I связана, по-видимому, с особенностями поверхности этого ПКП, что подтверждается отличием в порядке движения аминов при хроматографировании на ПКП и на силикагеле. Среди проанализированных свойств аминов – размера молекул, полярности и липофильности – только последняя характеристика, по-видимому, оказывает заметное влияние на эффективность их разделения.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Theodoridis G. // Adv. Flour. Sci. 2006. V. 2. P. 121.
Zhou Y., Wang J., Gu Z.S. et al. // Chem. Rev. 2016. V. 116. P. 422.
Shah P., Westwell A. // J. Enz. Inhib. Med. Chem. 2007. V. 22. P. 527.
Furuya T., Klein J., Ritter T. // Synthesis. 2010. V. 11. P. 1804.
Valkova L., Borovkov N., Koifman O. et al. // Biosensors Bioelectronics. 2004. V. 20. P. 1177.
McCalley D.V. // J. Chromatogr. A. 2010. V. 1217. P. 858.
Kagan M., Chlenov M., Melnikov S. et al. // J. Chromatogr. A. 2008. V. 1194. P. 80.
Han S., Wei Y., Valente C. et al. // J. Am. Chem. Soc. 2010. V. 132. P. 16358.
Yusuf K., Agel A., ALOthman Z. // J. Chromatogr. A. 2014. V. 1348. P. 1.
Сацкая Ю.А., Комарова Н.П., Гавриленко К.С. и др. // Теорет. и эксперим. химия. 2015. Т. 51. С. 41.
Сацкая Ю.А., Комарова Н.П., Гавриленко К.С. и др. // Изв. АН. Сер. хим. 2015. С. 630.
Alvarez E., Guillou N., Martineau C. et al. // Angew. Chem. Int. Ed. 2015. V. 54. P. 3664.
Loiseau T., Serre C., Huguenard C. et al. // Chem. Eur. J. 2004. V. 10. P. 1373.
Chowdhury P., Bikkina C., Gumma S. // J. Phys. Chem. C. 2009. V. 113. P. 6616.
Domenech A., Garcia H., Domenech-Carbo M.T. // Electrochem. Commun. 2006. V. 8. P. 1830.
Brunauer S., Emmett P., Teller E. // J. Am. Chem. Soc. 1938. V. 60. P. 309.
ACD/LogP DB. Version 10.00. Advanced Chemistry Development, Inc., Toronto. https://www.acdlabs.com.
Neese F. // WIREs Comput. Mol. Sci. 2017. V. 8. P. e1327.
Weigend F., Häser M., Patzelt H., Ahlrichs R. // Chem. Phys. Lett. 1998. V. 294. P. 143.
Neese F., Wennmohs F., Hansen A., Becker U. // Chem. Phys. 2009. V. 356. P. 98.
Bak K., Gauss J., Helgaker T. et al. // Chem. Phys. Lett. 2000. V. 319. P. 563.
Chui S. // Science. 1999. V. 283. P. 1148.
Ferey G., Mellot-Draznieks C., Serre C. et al. // Science. 2005. V. 310. P. 2040.
Serre C., Millange F., Thouvenot C. et al. // J. Am. Chem. Soc. 2002. V. 124. P. 13519.
Xiang S., Zhou W., Gallegos J. et al. // J. Am. Chem. Soc. 2009. V. 131. P. 12415.
Yot P.G., Vanduyfhuys L., Alvarez E. et al. // Chem. Sci. 2016. V. 7. P. 446.
Van der Sluis P., Spek A.L. // Acta Crystallogr. A. 1990. V. 46. P. 194.
Mulder F., Assfour B., Huot J. et al. // J. Phys. Chem. 2010. V. 114. P. 10648.
Xiao Y., Han T., Xiao G. et al. // Langmuir. 2014. V. 30. P. 12229.
Krawiec P., Kramer M., Sabo M. et al. // Adv. Eng. Mater. 2006. V. 8. P. 293.
Zhao T., Jeremias F., Boldog I. et al. // Dalton Trans. 2015. V. 44. P. 16791.
Saito A., Foley H. // Am. Inst. Chem. Eng. 1991. V. 37. P. 429.
Ross S., Olivier J.P. On Physical Adsorption. N.Y.: Wiley and Sons, 1964.
Sotnik S.A., Polunin R.A., Kiskin M.A. et al. // Inorg. Chem. 2015. V. 54. P. 5169.
Zauzolkova N., Dobrokhotova Z., Lermontov A. et al. // J. Solid State Chem. 2013. V. 197. P. 379.
Кельнер Р., Мерме Ж.-М., Отто М., Видмер Г. // Аналит. химия. 2004. Т. 1. С. 235.
Айвазов Б.В. Введение в хроматографию: учебное пособие. М.: Высш. школа, 1983. 240 с.
Valko K., Bevan C., Reynolds D. // Anal. Chem. 1997. V. 69. P. 2022.
Tetrahedron Organic Chemistry Series / Eds. Gawley R.E., Aubé J. 1996. V. 14. P. 45.
Valkó K. // J. Chromatogr. A. 2004. V. 1037. P. 299.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия