

Координационная химия, 2021, T. 47, № 1, стр. 3-12
Комплексы железа(III) на основе N-бензилиден-2-гидрокси-3,5-ди-трет-бутиланилина
И. В. Ершова 1, А. С. Богомяков 2, С. П. Кубрин 3, А. В. Черкасов 1, А. В. Пискунов 1, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Международный томографический центр СО РАН
Новосибирск, Россия
3 Научно-исследовательский институт физики ЮФУ
Ростов-на-Дону, Россия
* E-mail: pial@iomc.ras.ru
Поступила в редакцию 18.04.2020
После доработки 28.05.2020
Принята к публикации 05.06.2020
Аннотация
Синтезирована и детально охарактеризована серия пятикоординационных комплексов железа(III), содержащих в координационной сфере два бидентатно-координированных основания Шиффа (N‑бензилиден-2-гидрокси-3,5-ди-трет-бутиланилина, (ImPh)H), а также атом галогена/азидную группу – (ImPh)2FeX (X = Cl (I), Br (II), I (III), N3 (IV)). Согласно измерениям магнитной восприимчивости поликристаллических образцов I–IV, а также данным мессбауэровской спектроскопии в интервале 2–300 К ион металла находится в высокоспиновом состоянии (HS Fe3+, d5, SFe = 5/2). Соединения I–IV устойчивы в кристаллическом состоянии в отсутствие кислорода и влаги воздуха, однако в растворе претерпевают симметризацию, приводящую к образованию трис-лигандного комплекса (ImPh)3Fe (V), который также был синтезирован по реакции (ImPh)H и FeCl3 (3 : 1). В отличие от пятикоординационных комплексов I–IV, в шестикоординационном комплексе V ион железа(III) является низкоспиновым (LS Fe3+, d5, SFe = 1/2). Молекулярное строение комплекса I установлено методом РСА (CIF file CCDC № 1996527).
Координационные соединения трехвалентного железа вызывают пристальный интерес исследователей по всему миру, поскольку, наряду с комплексами железа(II), являются наиболее распространенным классом соединений, демонстрирующих эффект спин-кроссовера (spin-crossover, SCO) [1–3]. Явление SCO заключается в переключении спинового состояния комплекса при изменении температуры, давления [4], реже – под действием светового облучения [5–7] или магнитного поля [8]. Процесс переключения между высокоспиновым (high spin, HS) и низкоспиновым (low spin, LS) состояниями комплекса сопровождается изменением его физических свойств (магнитного момента, цвета, диэлектрической константы и электрического сопротивления), ввиду чего SCO-активные соединения являются крайне перспективными кандидатами для использования в качестве молекулярных магнитных устройств, сенсоров, а также компонентов мультифункциональных материалов [9]. Активный поиск SCO-систем проводится не только экспериментальными методами, но и в ходе теоретических исследований [10–15]. В отличие от комплексов железа(II) соединения железа(III) в большинстве случаев устойчивы к окислению на воздухе, что делает их более привлекательными объектами для потенциального использования в будущих устройствах. На сегодняшний день наиболее многочисленными и детально изученными SCO-активными комплексами Fe(III) являются производные на основе мультидентатных оснований Шиффа: симметричных и асимметричных лигандов на основе салицилальдегида или ацетилацетона [2, 3]. Однако вне зависимости от дентатности указанных лигандов (три- (N2O, SNO), тетра- (N2O2), пента- (N3O2) или гексадентатные (N6, N4O2) основания Шиффа) заполнение координационной сферы иона Fe3+ происходит таким образом, что он находится в октаэдрическом координационном окружении, при котором спиновый переход происходит между HS (SFe= 5/2) и LS (SFe = 1/2) состояниями. Исключение составляют пятикоординационные нитрозильные производные железа(III) (координационной центр FeN3O2), содержащие в координационной сфере тетрадентатный лиганд N2O2, а также молекулу NO [16–22]. Для данного класса соединений характерным является спиновый переход между низко- и среднеспиновым (intermediate spin, IS) состояниями LS (SFe = = 1/2) ↔ IS (SFe = 3/2). Для пятикоординационных порфириновых производных железа(III) характерна лабильная природа спинового состояния иона металла (HS–IS) [23]; редкий для данных систем спиновый переход HS (SFe = 5/2) ↔ IS (SFe = 3/2) был детектирован в комплексе [FeIII(OMTArP) (H2O)]ClO4, где OMTArP – периферически замещенный октаметилтетрафенилпорфинин [24]. Другим классом SCO-активных пятикоординационных комплексов железа(III) являются его бис-о-иминобензосемихиноновые производные – (ImSQR)2FeIIIX, где ImSQ – моновосстановленная форма R-фенил замещенного о-иминобензохинона, X – неорганический анион) [25–29]. Тонкая настройка спинового состояния иона Fe3+ (HS (SFe = 5/2) или IS (SFe = 3/2)) посредством вариации как заместителя X (X = Hal, N3, NCO, NCS), так и за счет введения заместителей R (H, Me, OMe) в редокс-активный ImSQ-лиганд делает возможным протекание в (ImSQR)2FeIIIX спинового перехода HS (SFe = 5/2) ↔ IS (SFe = 3/2). Согласно недавнему теоретическому исследованию серии тетрагонально-пирамидальных комплексов (ImSQH)2FeX (X = галогенид/псевдогалогенид), наблюдаемый спиновый переход обусловлен комбинацией свойств O,N-хелатных ImSQ-лигандов и апикального лиганда X [30].
Цель настоящего исследования – экспериментальное подтверждение принципиальной роли анион-радикальных ImSQ-лигандов для осуществления спин-кроссовера в пятикоординационных производных с координационным ядром FeN2O2X. Для этого была синтезирована серия пятикоординационных производных железа(III) – (ImPh)2FeX (X = Hal, N3) на основе диамагнитного аналога ImSQ-лиганда – N-бензилиден-2-гидрокси-3,5-ди-трет-бутиланилина ((ImPh)H).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу комплексов железа проводили в условиях отсутствия кислорода и влаги воздуха. Использованные в работе растворители очищали согласно рекомендациям [31]. В работе применяли коммерческие реактивы (Aldrich): хлорид железа(III), бромид калия, иодид калия, азид натрия, триэтиламин. Лиганд (ImPh)H синтезировали согласно методике, описанной в [32]. Элементный анализ выполняли на элементном анализаторе Elementar Vario EL cube. Масс-спектры соединений регистрировали на масс-спектрометре Polaris Q/TraceGC Ultra, масс-анализатор – ионная ловушка, энергия ионизирующих электронов 70 эВ, температура ионного источника 250°C, температура образца 50–450°С. ИК-спектры регистрировали на Фурье-спектрометре FSM-1201 в вазелиновом масле (диапазон: 4000–400 см–1) в кюветах из KBr. Электронные спектры поглощения (ЭСП) комплексов записывали на Perkin-Elmer Lambda 25 UV/Vis спектрометре (диапазон: 220–1100 нм) при комнатной температуре. Магнитную восприимчивость (χ) поликристаллических образцов измеряли на SQUID-магнетометре MPMSXL фирмы “Quantum Design” в интервале 2–300 K при напряженности магнитного поля 5 кЭ. Парамагнитные составляющие магнитной восприимчивости определяли с учетом диамагнитного вклада, оцененного из констант Паскаля. Эффективный магнитный момент вычисляли по формуле μэфф = [3kχT/ (NA$\mu _{{\text{B}}}^{2}$)]1/2, где NA, μB и k – число Авогадро, магнетон Бора и постоянная Больцмана соответственно. Мессбауэровские спектры регистрировали на MS1104Em спектрометре. В качестве источника гамма-излучения использовали 57Co в родиевой матрице. При охлаждении образцы помещали в камеру гелиевого криостата CCS-850. Величины изомерного сдвига δ приведены относительно α‑Fe.
Синтез комплекса (ImPh)2FeCl (I). Навески FeCl3 (0.13 г, 0.81 ммоль) и (ImPh)H (0.5 г, 1.62 ммоль) растворяли в 15 мл THF. Добавление к полученной гомогенной реакционной смеси избытка Et3N (0.3 мл) сопровождалось моментальным изменением ее цвета на красно-коричневый и выпадением светлого осадка. Реакционную смесь выдерживали 15 мин при комнатной температуре, после чего удаляли растворитель при пониженном давлении. Сухой остаток растворяли в толуоле (15 мл) и полученный раствор фильтровали от [Et3NH]Cl на стеклянном фильтре Шотта № 4. Комплекс I кристаллизовали из гексана (10 мл) при выдерживании раствора при –18°С в течение суток. Выход темного коричнево-зеленого высушенного на воздухе кристаллического осадка I составил 0.21 г (37%).
EI-MS (m/z): 708 (100%), 709 (51%), 710 (42%), 711 (18%) [M]+. ЭСП (CH2Cl2) нм (ε, л моль–1 см–1): 230 (96 279), 237 пл (92 673), 262 (90 912), 267 (87 942), 302 пл (44 163), 638 пл (1574). ИК-спектр (ν, см–1): 1594 с, 1575 ср, 1551 сл, 1409 ср, 1362 с, 1295 ср, 1272 с, 1255 о.с, 1224 с, 1203 ср, 1182 сл, 1160 ср, 1130 сл, 1028 сл, 976 сл, 957 ср, 916 сл, 886 сл, 857 сл, 833 с, 812 сл, 772 сл, 755 с, 691 с, 668 сл, 644 сл, 573 с, 559 сл, 540 сл, 495 сл, 478 сл.
При перекристаллизации комплекса (ImPh)2FeCl (I) из н-гексана получили кристаллы I ⋅ 1.25C6H14, пригодные для РСА.
Синтез комплекса (ImPh)2FeBr (II). К навеске KBr (0.42 г, 3.5 ммоль) в метаноле (10 мл) приливали коричнево-зеленый раствор I (0.25 г, 0.35 моль) в THF (10 мл). Полученную гетерогенную реакционную смесь перемешивали при комнатной температуре в течение 3 сут, при этом ее цвет менялся на бордово-фиолетовый. Растворители удаляли при пониженном давлении, сухой остаток растворяли в гексане (15 мл). Реакционную смесь отделяли от галогенидов калия фильтрацией. Комплекс II выделяли из реакционной смеси в виде темно-коричневого кристаллического осадка при выдерживании сконцентрированного фильтрата при –18°С в течение суток. Выход 0.08 г (31%).
EI-MS (m/z): 752 (76), 753 (100), 754 (43%) [M]+. ЭСП (CH2Cl2) нм (ε, л моль–1 см–1): 274 (23 786), 369 (11 856), 653 (2105). ИК-спектр (ν, см–1): 1594 с, 1575 с, 1551 ср, 1445 о.с, 1409 с, 1364 с, 1321 сл, 1295 ср, 1274 с, 1255 о.с, 1222 с, 1205 ср, 1186 сл, 1160 ср, 1127 сл, 1025 сл, 976 сл, 959 ср, 921 сл, 914 сл, 886 сл, 864 сл, 836 с, 812 сл, 772 ср, 755 с, 689 с, 668 сл, 646 сл, 620 сл, 575 с, 540 ср, 497 сл, 483 сл.
Синтез комплекса (ImPh)2FeI (III). К навеске KI (0.59 г, 3.5 ммоль) в метаноле (10 мл) приливали коричнево-зеленый раствор I (0.25 г, 0.35 моль) в THF (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение суток, при этом осадок KI растворялся, а цвет раствора менялся на бордово-красный. Растворители удаляли при пониженном давлении, сухой остаток растворяли в толуоле (15 мл) и отделяли реакционную смесь от галогенидов калия фильтрацией. Толуол из фильтрата удаляли при пониженном давлении, сухой остаток растворяли в пентане (10 мл) и выдерживали при –18°С в течение суток. Комплекс III выделили из реакционной смеси в виде темно-коричневого кристаллического осадка. Выход 0.12 г (42%).
EI-MS (m/z): 800 (100), 801 (57), 802 (47%) [M]+. ЭСП (CH2Cl2) нм (ε, л моль–1 см–1): 232 (27 074), 270 (27 626), 308 пл (22 233), 367 (11 734), 659 (2035). ИК-спектр (ν, см–1): 1593 ср, 1572 ср, 1548 сл, 1411 ср, 1364 с, 1322 сл, 1296 ср, 1271 с, 1254 с, 1224 ср, 1205 ср, 1186 сл, 1161 сл, 1126 сл, 1027 сл, 976 сл, 957 сл, 915 сл, 885 сл, 864 сл, 857 сл, 836 с, 810 сл, 773 сл, 756 с, 691 ср, 668 сл, 647 сл, 618 сл, 574 ср, 539 сл, 497 сл, 483 сл.
Синтез комплекса (ImPh)2FeN3 (IV). К навеске NaN3 (0.23 г, 3.5 ммоль) приливали коричнево-зеленый раствор I (0.25 г, 0.35 моль) в THF (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение суток, при этом ее цвет менялся на бордово-красный. Растворители удаляли при пониженном давлении, сухой остаток растворяли в толуоле (15 мл) и отделяли реакционную смесь от осадков NaCl и NaN3 фильтрацией. В фильтрате удаляли растворитель при пониженном давлении, сухой остаток растворяли в гексане (10 мл) и выдерживали при –18°С в течение недели. Комплекс IV выделили из реакционной смеси в виде темно-коричневого кристаллического осадка. Выход 0.10 г (39%).
EI-MS (m/z): 713 (100%), 714 (78%), 715 (17%) [M]+. ЭСП (CH2Cl2) нм (ε, л моль–1 см–1): 231 (83 736), 276 (31 478), 357 (13 819), 635 пл (1511). ИК-спектр (ν, см–1): 2094 о.с, 1594 с, 1575 ср, 1551 сл, 1409 с, 1362 с, 1298 ср, 1272 с, 1255 о.с, 1222 с, 1203 ср, 1182 сл, 1160 ср, 1130 сл, 1025 сл, 980 сл, 959 сл, 914 сл, 886 сл, 859 сл, 833 с, 812 сл, 772 сл, 755 с, 694 ср, 687 ср, 668 сл, 644 сл, 620 сл, 594 сл, 573 с, 540 сл, 495 сл, 478 сл.
Синтез комплекса (ImPh)3Fe (V) аналогичен синтезу комплекса I, но при стехиометрическом соотношении стартовых реагентов 1 : 3 (0.09 г FeCl3 (0.54 ммоль), 0.5 г (ImPh)H (1.62 ммоль)). Комплекс V выделяли из реакционной смеси в виде оранжево-коричневого мелкокристаллического осадка при выдерживании сконцентрированного раствора в гексане при –18°С в течение суток. Выход 0.11 г (21%).
EI-MS (m/z): 981 (100), 982 (71), 983 (13%) [M]+. ЭСП (CH2Cl2) нм (ε, л моль–1 см–1): 253 (40 896), 306 (31 934), 403 пл (14 243), 485 пл (7632). ИК-спектр (ν, см–1): 1596 с, 1575 ср, 1549 сл, 1497 ср, 1411 с, 1359 с, 1284 ос, 1260 ос, 1236 с, 1222 с, 1203 с, 1182 ср, 1158 с, 1130 сл, 1025 сл, 1002 сл, 985 сл, 968 ср, 952 ср, 931 сл, 916 сл, 883 ср, 864 с, 833 ос, 810 ср, 777 сл, 750 ос, 691 с, 668 ср, 641 сл, 620 сл, 604 сл, 563 с, 554 с, 537 с, 495 ср, 480 ср.
РСА. Сбор дифракционных данных монокристалла I · 1.25C6H14 (0.50 × 0.31 × 0.13 мм) проведен на дифрактометре Rigaku OD Xcalibur (ω-сканирование, МоКα-излучение, λ = 0.71073 Å, T = 298 K, 2θ = 54.2°). Измерение и интегрирование экспериментальных наборов интенсивностей, учет поглощения и уточнение структуры проведены с использованием программных пакетов CrysAlisPro [33] и SHELX [34]. Соединение I (C42H52ClFeN2O2 ⋅ ⋅ 1.25C6H14) кристаллизуется в пространственной группе P21/n (a = 16.0380(6), b = 13.6024(5), с = = 24.0414(9) Å, β = 109.103(4)°, V = 4955.9(3) Å3, Z = 4, ρрасч = 0.949 г/см3, μ = 0.386 мм–1). Измерено 68 869 отражений, 10 896 независимых отражений (Rint = 0.0427) были использованы для решения структуры и последующего уточнения 550 параметров полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для неводородных атомов. Водородные атомы в I помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв (U(H)изо = 1.5U(C)экв для метильных групп). Инструкции DFIX, ISOR и RIGU использованы для ограничения геометрических характеристик разупорядоченных трет-бутильных групп и анизотропных параметров атомных смещений при уточнении структуры. Вклад сольватных молекул гексана в структурную модель I учтен с помощью процедуры SQUEEZE (Platon) [35]. После финального уточнения wR2 = = 0.1152 и S(F 2) = 1.005 для всех отражений (R1 = = 0.0400 для всех отражений, удовлетворяющих условию I > 2σ(I)). Остаточные максимум и ми-нимум электронной плотности составили 0.26/–0.16 e/Å3.
Структура I зарегистрирована в Кембриджском банке структурных данных (CCDC № 1996527; ccdc.cam.ac.uk/structures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В рамках поставленной цели исследования, призванной оценить роль радикальных лигандов в осуществлении процессов спин-кроссовера в пятикоординационных производных железа(III), было необходимо синтезировать схожие по строению соединения металла, содержащие диамагнитные моноанионные O,N-хелатирующие лиганды, максимально близко воспроизводящие координационный узел бис-о-иминобензосемихинолятов типа (ImSQR)2FeIIIX, где ImSQ – моновосстановленная форма R-фенил замещенного о-иминобензохинона, X – неорганический анион (схема 1 ) [25–29].
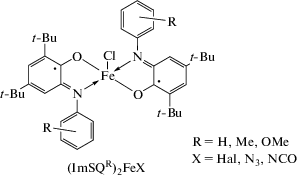
Схема 1 .
В качестве лигандной системы было выбрано основание Шиффа, образующееся в ходе конденсации 4,6-ди-трет-бутил-о-аминофенола с бенз-альдегидом. Последующая обменная реакция между (ImPh)H и безводным FeCl3 в стехиометрическом соотношении 2 : 1 в анаэробных условиях и в присутствии основания приводит к образованию целевого комплекса (ImPh)2FeCl (I) (схема 2 ).
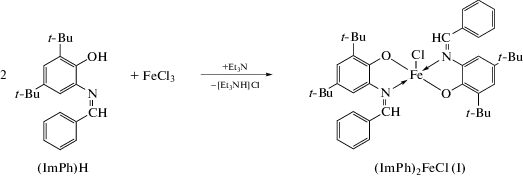
Схема 2 .
Производные (ImPh)2FeX (X = Br (II), I (III), N3 (IV)) были получены при взаимодействии I с десятикратным избытком KBr, KI и NaN3 соответственно (схема 3 ).
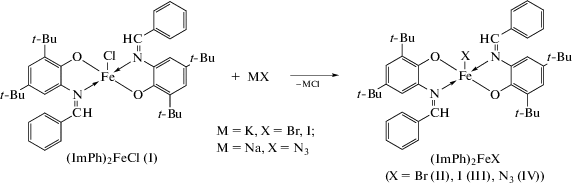
Схема 3 .
Соединения I–IV выделены из реакционных смесей в виде темных кристаллических осадков, устойчивых в условиях отсутствия кислорода и влаги воздуха. Высокая растворимость I–IV в большинстве органических растворителей обуславливает низкие препаративные выходы комплексов (30–40%); кроме того, продолжительное нахождение I–IV в растворе приводит к симметризации и образованию трис-лигандного оранжевого мелкокристаллического комплекса (ImPh)3Fe (V). Комплекс V также был получен встречным синтезом при взаимодействии FeCl3 с 3 экв. (ImPh)H (схема 4 ).
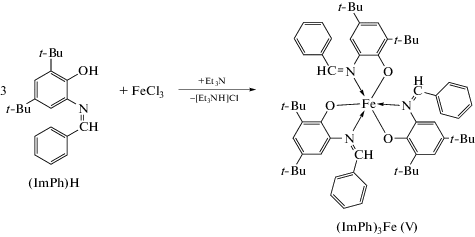
Схема 4 .
На ИК-спектрах I–V отсутствуют полосы в области 3100–3500 см–1, присутствующие в некоординированном (ImPh)H-лиганде и отвечающие валентным колебаниям ν(OH), что однозначно указывает на образование металлокомплексов. О бидентатной координации ImPh-лигандов на металл свидетельствует смещение полосы, отвечающей колебаниям связи C=N (ν(C=N) 1623 см–1 в некоординированном (ImPh)H, 1593–1594 см–1 в I–IV, 1596 см–1 в V). Кроме того, на ИК-спектре IV присутствует интенсивная полоса при 2090 см–1, отвечающая валентным колебаниям азидной группы ν(N3). На масс-спектрах I–IV помимо пика молекулярного иона [M]+ (см. экспериментальную часть) также присутствует интенсивный пик, отвечающий фрагменту [(ImPh)2Fe]+ – [M–X]+ (EI-MS (m/z): 672 (100), 673 (47), 674 (14%)). Наличие интенсивного пика [M–X]+ наряду с [M]+ указывает на легкое элиминирование заместителя X в условиях регистрации масс-спектров. Ранее такое поведение наблюдалось при анализе масс-спектров схожих пятикоординационных бис-о-иминобензосемихиноновых производных трехвалентных металлов (ImSQ)2MX (M = Al, Ga, Mn, Fe) [28, 29, 36–38]. Наличие атома галогена в I–III, как и его отсутствие в V, также контролировалось с помощью метода, предложенного Бельштейном [39].
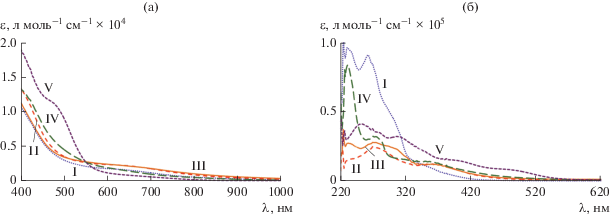
ЭСП комплексов I–V были зарегистрированы в хлористом метилене при 25°С в диапазоне 200–1000 нм (рис. 1). ЭСП комплексов I–V в УФ-диапазоне и на границе с видимой областью содержат высокоинтенсивные полосы поглощения (рис. 1б); видимая область спектра в случае I–IV представлена слабоинтенсивной уширенной полосой в области 500–900 нм, отсутствующей в ЭСП комплекса V (рис. 1а). Необходимо отметить, что родственные бис-о-иминобензосемихиноляты типа (ImSQR)2FeIIIX [25–29], в отличие от I–V, демонстрируют очень интенсивную полосу поглощения в ближней ИК-области (700–850 нм), что вызвано анион-радикальной природой O,N-хелатирующих лигандов, обуславливающих возникновение переноса заряда лиганд–лиганд (LLCT) в ЭСП.
Рентгеноструктурное исследование показало, что (ImPh)2FeCl (I) представляет собой пятикоординационный комплекс, в котором катион металла связан с двумя атомами кислорода и двумя атомами азота ImPh-лигандов и одним атомом хлора (рис. 2а). Значения избранных длин связей и углов комплексa представлены в табл. 1. Наличие двух асимметричных хелатных колец в (ImPh)2FeCl предполагает хиральность металлоцентра. Согласно данным РСА, комплекс кристаллизуется в центросимметричной пространственной группе P21/n, при этом в независимой области кристаллической ячейки располагается одна молекула комплекса в общем положении. Таким образом, в кристаллической ячейке представлены оба изомера I (рис. 2б) в соотношении 1 : 1. Необходимо отметить, что в кристаллической ячейке также присутствуют сольватные молекулы гексана: на четыре молекулы комплекса приходится пять молекул растворителя. Другими словами, комплекс (ImPh)2FeCl кристаллизуется в виде I ⋅ 1.25C6H14.
Рис. 2.
а – молекулярная структура комплекса I ⋅ 1.25C6H14 (атомы водорода опущены для ясности); б – фрагменты структур двух изомеров I ⋅ 1.25C6H14 (тепловые эллипсоиды приведены с 50%-ной вероятностью).
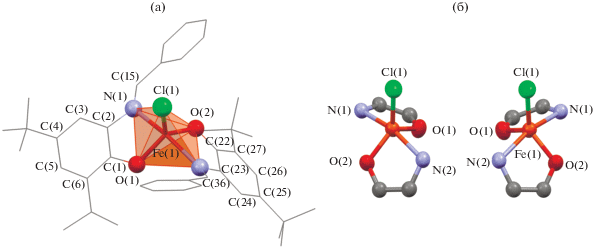
Таблица 1.
Основные длины связей (Å) и валентные углы (град) в комплексе I ⋅ 1.25C6H14
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| Fe(1)–O(1) | 1.868(2) | C(1)–C(6) | 1.411(2) |
| Fe(1)–O(2) | 1.871(2) | C(2)–C(3) | 1.396(2) |
| Fe(1)–N(1) | 2.187(2) | C(3)–C(4) | 1.379(2) |
| Fe(1)–N(2) | 2.213(2) | C(4)–C(5) | 1.398(3) |
| Fe(1)–Cl(1) | 2.2294(6) | C(5)–C(6) | 1.387(2) |
| O(1)–C(1) | 1.347(2) | C(22)–C(27) | 1.403(2) |
| O(2)–C(22) | 1.343(2) | C(22)–C(23) | 1.401(2) |
| N(1)–C(2) | 1.436(2) | C(23)–C(24) | 1.385(2) |
| N(2)–C(23) | 1.426(2) | C(24)–C(25) | 1.382(3) |
| N(1)–C(15) | 1.287(2) | C(25)–C(26) | 1.397(3) |
| N(2)–C(36) | 1.287(2) | C(26)–C(27) | 1.389(3) |
| Угол | ω, град | Угол | ω, град |
| O(1)Fe(1)O(2) | 118.62(6) | N(1)Fe(1)N(2) | 163.13(5) |
| O(1)Fe(1)N(1) | 80.57(5) | O(1)Fe(1)Cl(1) | 117.60(5) |
| O(2)Fe(1)N(1) | 90.54(5) | O(2)Fe(1)Cl(1) | 123.78(4) |
| O(1)Fe(1)N(2) | 92.54(5) | N(1)Fe(1)Cl(1) | 98.62(4) |
| O(2)Fe(1)N(2) | 79.20(5) | N(2)Fe(1)Cl(1) | 98.22(4) |
При анализе координационной геометрии пятикоординационных комплексов и степени их искажения часто используется геометрический параметр τ, значение которого в идеальной тетрагональной пирамиде равно нулю, а в идеальной тригональной бипирамиде – единице [40]. В I τ = 0.74 и геометрия координационного полиэдра FeN2O2Cl представляет собой искаженную тригональную бипирамиду, в экваториальной плоскости которой располагаются атомы кислорода и хлора, а в аксиальных положениях находятся атомы азота. Примечательно, что в бис-о-иминобензосемихиноновых производных (ImSQ)2FeX значение τ находится в диапазоне 0–0.55 [25–29]. Таким образом, переход от анион-радикальных ImSQ-лигандов к близким по строению диамагнитным ImPh-лигандам сопровождается изменением типа координационной геометрии в образующихся комплексах от тетрагональной пирамиды к тригональной бипирамиде.
Значения длин связей С–O (1.343(2)–1.347(2) Å), C–N (1.426(2)–1.436(2) Å), CH=N (1.287(2) Å) находятся в интервалах, характерных для одинарных C–O (1.32–1.37 Å), C–N (1.41–1.45 Å) и двойных CH=N (1.27–1.30 Å) связей в комплексах металлов с лигандами данного типа [41–44]. Длины связей C–С в ароматических кольцах С(1–6), С(22–27) в ImPh-лигандах (1.379(2)–1.411(2) Å, средн. 1.39 Å) близки к таковым в молекуле бензола (1.40 Å). Значения длин связей Fe–O (1.868(2), 1.871(2) Å) меньше суммы ковалентных радиусов соответствующих элементов (rков(FeIII) = 1.36 Å, rков(O) = = 0.73 Å [45]), что указывает на ковалентный тип связывания. Напротив, расстояния Fe–N (2.187(2), 2.213(2) Å) существенно превышают сумму ковалентных радиусов соответствующих атомов (rков(FeIII) = 1.36 Å, rков(N) = 0.74 Å [45]); таким образом, взаимодействия Fe…N в I можно охарактеризовать как донорно-акцепторные.
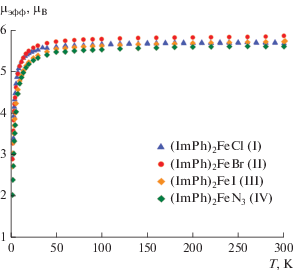
Магнитная восприимчивость кристаллических образцов I–IV была измерена в интервале 2–300 К. Температурные зависимости эффективного магнитного момента μэфф(T) для серии комплексов (ImPh)2FeX (X = Cl (I), Br (II), I (III), N3 (IV)) представлены на рис. 3 и имеют сходный характер. Для пятикоординационных комплексов I–IV высокотемпературные значения μэфф (5.69, 5.88, 5.74 и 5.62 μB в I–IV соответственно) близки к теоретическому чисто спиновому значению 5.92 μB, рассчитанному для системы, содержащей ион железа(III) в высокоспиновом состоянии (d5, HS, SFe = 5/2). При понижении температуры до 30 К значение μэфф изменяется незначительно, однако резко уменьшается при дальнейшем охлаждении и при температуре 2 К достигает значений 2.92, 2.89, 2.72 и 2.03 μB в I–IV соответственно. Уменьшение величины μэфф в интервале 30–2 К обусловлено наличием межмолекулярного обменного взаимодействия в кристаллических образцах I–IV.
Рис. 3.
Температурные зависимости эффективного магнитного момента для комплексов (ImPh)2FeX (X = Cl (I), Br (II), I (III), N3 (IV)).
Кристаллические образцы I–IV также были охарактеризованы методом мессбауэровской спектроскопии. Мессбауэровские спектры комплексов II и V приведены на рис. 4, параметры спектров для I–IV просуммированы в табл. 2. Понижение температуры с 300 до 14 К не приводит к появлению существенных трансформаций в структуре спектров, указывающих на изменение спинового состояния иона Fe3+, что находится в согласии с данными магнетохимических измерений. Величины изомерного сдвига (δ) в пятикоординационных производных I–IV (0.30–0.52 мм с–1) близки по значению к таковым для пятикоординационных бис-о-иминобензосемихиноновых производных (ImSQMe)2FeX, содержащих высокоспиновый ион Fe3+ (HS, SFe = 5/2), в то же время величины квадрупольного расщепления (ΔEQ) в I–IV составляют 1.66–1.82 мм с–1, что несколько превышает значения ΔEQ в упомянутых (ImSQMe)2FeX комплексах (1.08–1.31 мм с–1) [29]. Мессбауэровский спектр шестикоординационного комплекса (ImPh)3Fe (V) (рис. 4) отличается от спектров пятикоординационных комплексов (ImPh)2FeX (I–IV) и имеет параметры изомерного сдвига и квадрупольного расщепления (0.29–0.41 и 1.96–1.98 мм с–1 соответственно), характерные для октаэдрических низкоспиновых производных железа(III) [2, 3]. Это находится в согласии с измерениями магнитной восприимчивости для соединения V – значение эффективного магнитного момента при 14 К составляет 1.72 μB, что отвечает одному иону Fe3+ в низкоспиновом (LS, SFe = 1/2) состоянии.
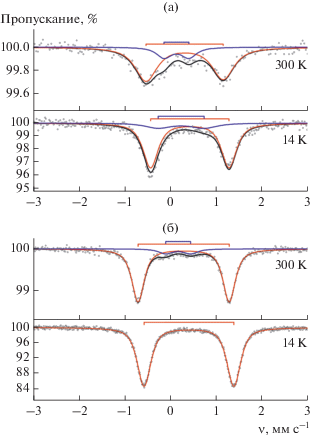
Таблица 2.
Параметры мессбауэровских спектров для комплексов I–V*
| Комплекс | T, K | δ, мм с–1 | ΔEQ, мм с–1 |
|---|---|---|---|
| I | 300 | 0.36 | 1.72 |
| 14 | 0.52 | 1.76 | |
| II | 300 | 0.32 | 1.68 |
| 14 | 0.43 | 1.72 | |
| III | 300 | 0.30 | 1.68 |
| 14 | 0.46 | 1.66 | |
| IV | 300 | 0.33 | 1.70 |
| 14 | 0.52 | 1.82 | |
| V | 300 | 0.29 | 1.98 |
| 14 | 0.41 | 1.96 |
Таким образом, в ходе настоящего исследования была синтезирована серия пятикоординационных бис-о-иминофенолятных производных железа(III) (ImPh)2FeX (X = Cl (I), Br (II), I (III), N3 (IV)) – структурных аналогов пятикоординационных бис-о-иминобензосемихиноновых производных железа(III). Несмотря на идентичность координационного ядра (FeN2O2X) в сериях соединений (ImPh)2FeX и (ImSQ)2FeX, строение их координационного полиэдра существенно отличается. При координации на металл иминофенолятных лигандов реализуется два типа связывания: ковалентное (Fe–O) и донорно-акцепторное (Fe–N), что приводит к формированию искаженной тригонально-бипирамидальной геометрии в образующихся комплексах (ImPh)2FeX. Напротив, характер связей Fe–O и Fe–N, обусловленный гораздо большей делокализацией отрицательного заряда в анион-радикальном ImSQ-лиганде, способствует формированию идеальной/искаженной тетраго-нально-пирамидальной геометрии в (ImSQ)2FeX. Согласно совокупности данных физико-химических методов исследования ион Fe3+ в иминофенолятных комплексах I–IV находится в высокоспиновом состоянии во всем интервале 2–300 К. Полученный результат позволяет сделать вывод о том, что анион-радикальный характер O,N-хелатирующих ImSQ-лигандов в координационной сфере Fe3+ играет ключевую роль для протекания спин-кроссовера в пятикоординационных производных железа(III), обладающих координационным ядром FeN2O2X [25–30].
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
van Koningsbruggen P.J., Maeda Y., Oshio H. // Top. Curr. Chem. 2004. V. 233. P. 259.
Nihei M., Shiga T., Maeda Y., Oshio H. // Coord. Chem. Rev. 2007. V. 251. P. 2606.
Harding D.J., Harding P., Phonsri W. // Coord. Chem. Rev. 2016. V. 313. P. 38.
Ksenofontov V., Gaspar A.B., Gütlich P. // Top. Curr. Chem. 2004. V. 235. P. 23.
Hauser A. // Top. Curr. Chem. 2004. V. 234. P. 155.
Létard J.-F. // J. Mater. Chem. 2006. V. 16. P. 2550.
Cannizzo A., Milne C.J., Consani C. et al. // Coord. Chem. Rev. 2010. V. 254. P. 2677.
Bousseksou A., Varret F., Goiran M. et al. // Top. Curr. Chem. 2004. V. 235. P. 65.
Halcrow M.A. Spin-Crossover Materials, Properties and Applications. N.Y.: John Wiley & Sons, Ltd., 2013.
Starikova A., Starikov A.G., Minkin V.I. // Russ. Chem. Bull. 2016. V. 65. P. 1464.
Starikova A.A., Starikov A.G., Minkin V.I. // Russ. J. Coord. Chem. 2017. V. 43. P. 718. https://doi.org/10.1134/S1070328417110094
Starikov A.G., Chegerev M.G., Starikova A.A., Minkin V.I. // Russ. J. Coord. Chem. 2019. V. 45. P. 675. https://doi.org/10.1134/S1070328419090082
Starikov A.G., Starikova A.A., Chegerev M.G., Minkin V.I. // Russ. Chem. Bull. 2019. V. 68. P. 725.
Starikova A.A., Chegerev M.G., Starikov A.G., Minkin V.I. // J. Comput. Chem. 2019. V. 40. P. 2284.
Starikova A.A., Metelitsa E.A., Starikov A.G. // J. Struct. Chem. 2019. V. 60. P. 1219.
Earnshaw A., King E.A., Larkworthy L.F. // Chem. Commun. 1965. V. P. 180.
Earnshaw A., King E.A., Larkworthy L.F. // J. Chem. Soc. A. 1969. V. P. 2459.
Haller K.J., Johnson P.L., Feltham R.D. et al. // Inorg. Chim. Acta. 1979. V. 33. P. 119.
Fitzsimmons B.W., Larkworthy L.F., Rogers K.A. // Inorg. Chim. Acta. 1980. V. 44. P. L53.
Wells F.V., McCann S.W., Wickman H.H. et al. // Inorg. Chem. 1982. V. 21. P. 2306.
Koenig E., Ritter G., Waigel J. et al. // Inorg. Chem. 1987. V. 26. P. 1563.
Weber B., Görls H., Rudolph M., Jäger E.-G. // Inorg. Chim. Acta. 2002. V. 337. P. 247.
Nakamura M. // Coord. Chem. Rev. 2006. V. 250. P. 2271.
Ohgo Y., Chiba Y., Hashizume D. et al. // Chem. Commun. 2006. P. 1935.
Chun H., Weyhermüller T., Bill E., Wieghardt K. // Angew. Chem. Int. Ed. 2001. V. 40. P. 2489.
Chun H., Bill E., Weyhermüller T., Wieghardt K. // Inorg. Chem. 2003. V. 42. P. 5612.
Abakumov G.A., Cherkasov V.K., Bubnov M.P. et al. // Russ. Chem. Bull. Int. Ed. 2006. V. 55. P. 44.
Piskunov A.V., Pashanova K.I., Ershova I.V. et al. // J. Mol. Struct. 2018. V. 1165. P. 51.
Ershova I.V., Bogomyakov A.S., Kubrin S.P. et al. // Inorg. Chim. Acta. 2020. V. 503. P. 119402.
Credendino L., Sproules S. // Asian J. Org. Chem. 2019. V. 9. P. 421.
Perrin D.D., Armarego W.L.F., Perrin D.R. Purification of Laboratory Chemicals. Oxford: Pergamon Press, 1980.
Yasuhiko S., Norio K., Terunori F. // Chem. Lett. 2002. V. 31. P. 358.
Rigaku Oxford Diffraction. CrysAlisPro software system. Version 1.171.38.46. Wroclaw (Poland): Rigaku Corporation, 2015.
Sheldrick G.M. // Acta Crystfllogr. C. 2015. V. 71. P. 3.
Spek A.L. // Acta Crystfllogr. C. 2015. V. 71. P. 9.
Piskunov A.V., Ershova I.V., Bogomyakov A.S. et al. // Inorg. Chem. 2015. V. 54. P. 6090.
Piskunov A.V., Ershova I.V., Bogomyakov A.S., Fukin G.K. // Inorg. Chem. Commun. 2016. V. 66. P. 94.
Ershova I.V., Bogomyakov A.S., Fukin G.K., Piskunov A.V. // Eur. J. Inorg. Chem. 2019. V. 2019. P. 938.
Beilstein F. // Ber. Dtsch. Chem. Ges. 1872. V. 5. P. 620.
Addison A.W., Rao T.N., Reedijk J. et al. // Dalton Trans. 1984. P. 1349.
Mukherjee S., Weyhermüller T., Bothe E., Chaudhuri P. // Eur. J. Inorg. Chem. 2003. V. 2003. P. 1956.
Suzuki Y., Tanaka H., Oshiki T. et al. // Chem. Asian J. 2006. V. 1. P. 878.
Zhang L., Luo X., Gao W. et al. // Organometallics. 2013. V. 32. P. 6277.
Safaei E., Alaji Z., Panahi F. et al. // New J. Chem. 2018. V. 42. P. 7230.
Бацанов С.С. // Журн. неорган. химии. 1991. Т. 36. С. 3015 (Batsanov S.S. // Russ. J. Inorg. Chem. 1991. V. 36. P. 1694).
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия