

Координационная химия, 2021, T. 47, № 1, стр. 30-36
Фазовый переход в кристалле тетраметоксисилана по данным in situ рентгенодифракционного исследования
А. Ф. Смольяков 1, Ю. В. Нелюбина 1, 2, *
1 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
2 Московский государственный технический университет им. Баумана
Москва, Россия
* E-mail: unelya@ineos.ac.ru
Поступила в редакцию 28.05.2020
После доработки 08.06.2020
Принята к публикации 15.06.2020
Аннотация
С помощью оригинальной установки для in situ кристаллизации получены монокристаллы тетра-метоксисилана (I), являющегося жидкостью при комнатной температуре. Исследование I методом РСА при двух разных температурах (CIF files CCDC № 2006156 и 2006157 для 150 и 200 К соответственно) позволило обнаружить у данного соединения фазовый переход, сопровождающийся разупорядочением метоксигрупп. Разработанная установка, отличающаяся от коммерческих аналогов необычайно низкой стоимостью и возможностью модификации всех составных частей, открывает широкие возможности роста кристаллов непосредственно на дифрактометре для определения структуры различных элементоорганических соединений и, возможно, комплексов металлов, находящихся в жидком или даже газообразном состоянии.
Рентгеноструктурный анализ является основным методом определения структуры новых химических соединений при наличии для них монокристаллов подходящего качества [1]. К сожалению, данное требование обычно невыполнимо для соединений, представляющих собой жидкости при комнатной температуре. Из-за высокой молекулярной подвижности, определяющей их низкие барьеры вращения вокруг связей и деформации валентных углов [2], и отсутствия прочных межмолекулярных взаимодействий такие соединения кристаллизуются при низкой температуре с образованием рыхлой кристаллической упаковки с частичным разупорядочением отдельных молекулярных фрагментов [3–5]. В результате получаемые для них данные РСА зачастую страдают от погрешностей эксперимента, в первую очередь, – низкой полноты массивы и точности определения геометрических параметров [3, 4].
Имеющиеся в литературе примеры рентгенодифракционных исследований веществ, являющихся жидкостями при комнатной температуре [5–8], как правило, посвящены органическим соединениям. Причиной этому могут быть трудности [9], связанные с очисткой элементоорганических соединений и комплексов металлов от примесей, их чувствительностью к влаге и кислороду воздуха, а также их более высокая вязкость, заметно осложняющая процесс кристаллизации. Одним из таких классов соединений являются простые органосиланы и силоксаны [10], широко применяемые в промышленности и медицине в качестве инертных загустителей, гидрофобизаторов и смазочных материалов [10]. К сожалению, при комнатной температуре они являются вязкими жидкостями [10], что препятствует установлению их структуры методом РСА и в результате – определению широкого спектра их физико-химических свойств [11].
Решением данной проблемы может быть рост кристаллов таких соединений непосредственно на рентгеновском дифрактометре [4], который предполагает их охлаждение–нагревание при помощи специальных установок для зонной плавки [5, 12]. Начиная с пионерских работ по in situ кристаллизации бензола и циклогексана [13, 14], подобный подход позволял не только определять кристаллическое строение жидкостей [4], но и следить за ходом реакций [5], а также изучать сам процесс кристаллизации [12]. Для этого исследуемое соединение в жидком состоянии помещали в стеклянный капилляр, который устанавливали на специальную гониометрическую головку, снабженную направляющим столиком и находящуюся непосредственно на рентгеновском дифрактометре [4].
Ранее мы успешно осуществили in situ кристаллизацию жидкого диметилдиметоксисилана [15] с помощью разработанной нами оригинальной установки для in situ роста кристаллов, отличающейся от своего коммерческого аналога OHCD (Optical Heating and Crystallization Device) [16] заметно более низкой стоимостью и возможностью оперативной модификации всех ее составных частей.
В настоящей работе аналогичным образом мы получили монокристаллы тераметоксисилана (I). Строение данного соединения [17] было установлено в ходе РСА при двух разных температурах (150 и 200 К), что позволило обнаружить в его кристалле фазовый переход, сопровождающийся разупорядочением метоксильных групп.
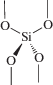
(I)
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали жидкий тетраметоксисилан (Sigma-Aldrich) без дополнительной очистки. Соответствующие монокристаллы, качество которых было пригодным для их последующего РСА, получали методом зонной плавки с помощью разработанной нами установки для in situ кристаллизации (рис. 1). Она включает в себя гониометрическую головку для рентгеновского дифрактометра, сконструированную из доступных компонентов, с возможностью снятия направляющего столика. Указанная конструктивная особенность гониометрической головки (рис. 1) позволяет ориентировать капилляр, заполненный исследуемым веществом, под разными углами по отношению к столику и потоку азота, выходящему из сопла низкотемпературной приставки. При этом удается добиться существенного увеличения диапазона углов сканирования в ходе рентгенодифракционного эксперимента, что необходимо для сбора соответствующих данных с высокой избыточностью.

После получения монокристалла исследуемого вещества методом зонной плавки в капилляре, помещенном на гониометрическую головку, оператор на персональном компьютере вводит все необходимые параметры в специально разработанную программу, обрабатывающую полученные данные и отправляющую их микроконтроллеру Arduino по протоколу беспроводных персональных сетей Bluetooth. Микроконтроллер Arduino генерирует управляющие импульсы, сообщая драйверу шагового двигателя установки на какой угол, с какой скоростью и в каком направлении вращать мотор. Драйвер управляет мотором, следуя полученным инструкциям. Такая последовательность передачи данных и отсутствие зависимостей между компонентами позволяет не только дистанционно управлять разработанной нами установкой для in situ роста кристаллов, но и заменять ее отдельные составляющие “на лету” без риска возникновения неполадок.
РСА монокристалла I проведен при температурах 150 и 200 К на дифрактометре Bruker APEX2 CCD (MoKα-излучение, графитовый монохроматор, ω-сканирование). Структура I расшифрована с использованием программы ShelXT [18] и уточнена в полноматричном МНК с помощью программы Olex2 [19] в анизотропном приближении по $F_{{hkl}}^{{\text{2}}}.$ Положения атомов водорода рассчитывали геометрически и уточняли их в изотропном приближении по модели наездника. Основные кристаллографические данные и параметры уточнения для соединения I представлены в табл. 1.
Таблица 1.
Основные кристаллографические данные и параметры уточнения I при 150 и 200 К
| Параметр | Значение | |
|---|---|---|
| Температура | 150 К | 200 К |
| Брутто-формула | C4H12O4Si | C4H12O4Si |
| М | 152.23 | 152.23 |
| T, K | 150 | 200 |
| Сингония | Моноклинная | Моноклинная |
| Пр. гр. | P2/n | C2/c |
| Z | 4 | 4 |
| a, Å | 11.4881(12) | 13.039(3) |
| b, Å | 5.8655(6) | 5.9999(18) |
| c, Å | 12.7824(12) | 11.852(3) |
| α, град | 90 | 90 |
| β, град | 116.249(3) | 116.476(9) |
| γ, град | 90 | 90 |
| V, Å3 | 772.50(14) | 830.0(4) |
| ρ(выч.), г см–3 | 1.309 | 1.218 |
| μ, см–1 | 2.56 | 2.38 |
| F(000) | 328 | 328 |
| 2θmax, град | 52 | 52 |
| Число измеренных отражений | 4662 | 2358 |
| Число независимых отражений (Rint) | 1613 | 787 |
| Число отражений с I > 2σ(I) | 1438 | 673 |
| Количество уточняемых параметров | 88 | 44 |
| R1, wR2 (I > 2σ(I)) | 0.0333, 0.0885 | 0.1700, 0.5017 |
| R1, wR2 (все данные) | 0.0371, 0.0912 | 0.1766, 0.5147 |
| GOOF | 1.072 | 2.511 |
| Δρmax/Δρmin, e Å–3 | 0.276/–0.262 | 0.788/–0.781 |
Структурные параметры для I депонированы в Кембриджском банке структурных данных (№ 2006156 и 2006157 для 150 и 200 К соответственно; http://www.ccdc.cam.ac.uk/).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Монокристалл подходящего качества для коммерчески доступного (Sigma-Aldrich) тетраметоксисилана I, представляющего собой вязкую жидкость при комнатной температуре, нам удалось получить только после нескольких попыток его in situ кристаллизации в стеклянном капилляре методом зонной плавки в диапазоне 150–200 К. При этом ключевым параметром оказалась скорость охлаждения–нагревания капилляра, которая в оптимальном случае составляла 60 К/ч. Последующий РСА указанного монокристалла однозначно подтвердил структуру тетраметоксисилана I (рис. 2), чего не удавалось сделать ранее [17]. Более того, качество его монокристалла, выращенного с использованием изготовленной нами установки для in situ кристаллизации, позволило получить рентгенодифракционные данные при двух разных температурах: 150 и 200 K. Обнаруженное различие параметров элементарной ячейки при 150 и 200 K (табл. 1) свидетельствовало о протекании фазового перехода в соответствующем диапазоне температур. Хотя во всех случаях тетра-метоксисилан I образует моноклинные кристаллы, при 150 K они принадлежат пространственной группе P2/n, при 200 K – пространственной группе C2/c.
Рис. 2.
Общий вид двух симметрически независимых молекул соединения I в представлении атомов эллипсоидами тепловых колебаний (p = 50%) при температуре 150 К.
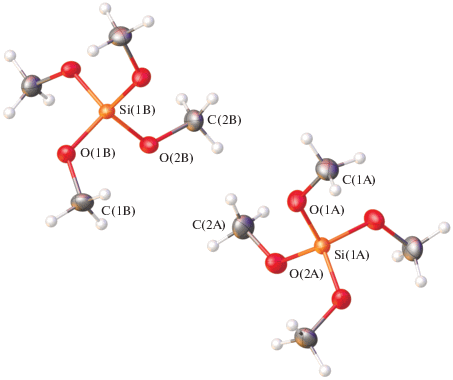
Согласно полученным рентгенодифракционным данным, при температуре 150 К в элементарной ячейке присутствуют две симметрически независимые молекулы тетраметоксисилана I (рис. 2), занимающие в кристалле частное положение – ось симметрии второго порядка, проходящую через атомы кремния и середину отрезков O(1)…O(1) и O(2)…O(2). Отметим, что изученный нами ранее диметилдиметоксисилан [15], также являющийся жидкостью при комнатной температуре, кристаллизуется в пространственной группе C2/c с молекулой на кристаллографической оси симметрии второго порядка. В обоих случаях атом кремния находится в тетраэдрическом окружении (табл. 2), которое незначительно искажается при переходе от I к диметилдиметоксисилану [15] из-за замены двух метоксигрупп на метильные группы. Графически это можно представить в виде так называемых “мер симметрии” [20], описывающих отклонения формы полиэдра SiO4 в I и SiO2С2 в диметилдиметоксисилане [15] от идеального тетраэдра (S(T-4)) и идеального плоского квадрата (S(SP-4)). Чем соответствующие значения меньше, тем лучше форма полиэдра описывается соответствующим многогранником. В соединении I величины тетраэдрической S(T-4) и плоско-квадратной S(SP-4) “мер симметрии”, оцененные на основе рентгенодифракционных данных при 150 К с использованием программы Shape 2.1 [20], составляют 0.178, 29.418 и 0.190, 29.297 для двух независимых молекул тетраметоксисилана I (табл. 2). Для сравнения аналогичные значения в диметилдиметоксисилане [15] достигают 0.544 и 31.204 соответственно.
Таблица 2.
Основные геометрические параметры для ранее изученного диметилдиметоксисилана [15] и тетра-метоксисилана I по данным РСА при 150 и 200 К*
| Параметр | Диметилдиметоксисилан | 150 К | 200 К |
|---|---|---|---|
| Si–O, Å | 1.6391(11) | 1.6107(12)–1.6161(11) | 1.524(15)–1.649(14) |
| Si–С, Å | 1.8468(19) | ||
| O–С, Å | 1.420(2) | 1.4258(19)–1.430(2) | 1.382(15)–1.522(14) |
| OSiO, град | 110.92(7) | 106.85(6)–114.60(9) | 103.4(11)–120.5(11) |
| OSiС, град | 111.82(8) | ||
| СSiС, град | 113.98(10) | ||
| S(T-4) | 0.544 | 0.178, 0.190 | 0.101, 0.285 |
| S(SP-4) | 31.204 | 29.418, 29.297 | 32.875, 29.426 |
При нагревании кристалла I до температуры 200 К (табл. 1) происходит переход от пространственной группы P2/n к группе C2/c, в которой единственная независимая молекула тетраметоксисилана лежит на оси симметрии второго порядка, также проходящей через атом кремния и середину отрезков O(1)…O(1) и O(2)…O(2). При этом наблюдается существенная перестройка кристаллической упаковки тетраметоксисилана I (рис. 3). Из-за отсутствия у него эффективных доноров и акцепторов протона при 150 К она образована за счет контактов С–Н…О (с минимальным расстоянием С…О 3.455(3) Å), тогда как при 200 К за ее формирование уже отвечают более слабые взаимодействия С–Н…Н (с минимальным расстоянием С…Н 3.123(4) Å). Подобная перестройка кристаллической структуры при нагревании сопровождается ожидаемым увеличением объема элементарной ячейки, приходящегося на одну молекулу тетраметоксисилана, с 193.12 до 207.5 Å3 (табл. 1).
Рис. 3.
Фрагмент кристаллической упаковки соединения I: а – 150 К, б – 200 К (показаны обе компоненты разупорядоченных метоксигрупп).
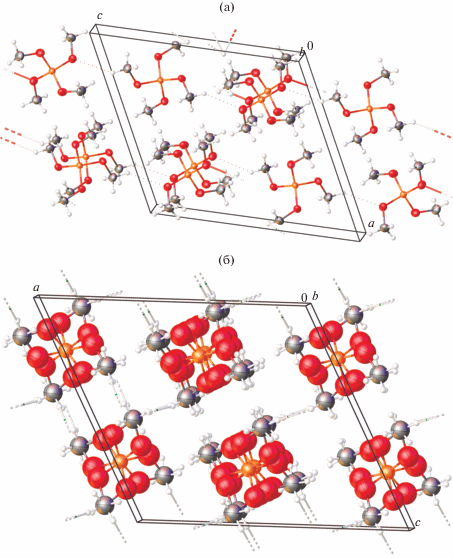
Дополнительное отличие от низкотемпературной фазы заключается в том, что при 200 K атомы кислорода метоксильных групп оказываются разупорядоченными по двум положениям с одинаковой заселенностью 50 : 50 (рис. 4), по-видимому, вследствие высокой симметрии и слабости межмолекулярных взаимодействий, приводящих к суперпозиции указанных групп. Следует отметить, что уточнение координат и тепловых параметров атомов в разупорядоченных заместителях при атоме кремния очень затруднительно, поэтому их уточняли с большим количеством ограничений. В пределах вызванных этим погрешностей эксперимента и уточняемой модели атом кремния сохраняет свое ковалентное окружение (табл. 2), которое остается очень близким к тетраэдрическому. Действительно, величины тетраэдрической S(T-4) и плоско-квадратной S(SP-4) “мер симметрии”, оцененные для обеих компонент разупорядоченной молекулы при 200 К, составляют 0.101, 32.875 и 0.285, 29.426 соответственно (табл. 2).
Рис. 4.
Общий вид соединения I в представлении атомов эллипсоидами тепловых колебаний (p = 50%) при температуре 200 К. Показаны обе компоненты разупорядоченных метоксигрупп.
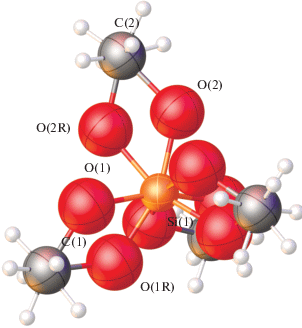
Таким образом, с помощью оригинальной установки для in situ кристаллизации (рис. 1) нами получены монокристаллы тетраметоксисилана, являющегося вязкой жидкостью при комнатной температуре. Их рентгеноструктурный анализ при 150 и 200 К позволил не только однозначно установить структуру данного соединения, но и обнаружить наблюдающийся у него в данном диапазоне температур фазовый переход, сопровождающийся разупорядочением метоксильных групп. К сожалению, нам не удалось повторно провести рентгенодифракционное исследование при 150 К из-за технических сбоев в работе низкотемпературной приставки, приведших к расплавлению соответствующего монокристалла в вязкую жидкость. Последующие попытки вырастить из нее монокристаллы аналогичного качества, подходящего для рентгеноструктурного анализа, не увенчались успехом. Несмотря на это, разработанная нами установка, отличающаяся от своего коммерческого аналога OHCD (Optical Heating and Crystallization Device) [16] значительно более низкой стоимостью и возможностью оперативной модификации ее различных компонентов, открывает широкие перспективы реализации походов in situ кристаллизации для элементоорганических соединений и, возможно, комплексов металлов, находящихся в жидком или даже газообразном состоянии [3] (в последнем случае – при условии их запаивания в кварцевые капилляры).
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Brooks-Bartlett J.C., Garman E.F. // Interdiscipl. Sci. Rev. 2015. V. 40. № 3. P. 244.
Davey R.J., Garside J. From Molecules to Crystallizers. An Introduction to Crystallization. Oxford: Oxford Univ. Press, 2001.
Howard J.A.K., Probert M.R. // Science. 2014. V. 343. № 6175. P. 1098.
Roland B. // Z. Krist. 2014. V. 229. № 9. P. 595.
Dey D., Chopra D. // Resonance. 2014. V. 19. № 12. P. 1104.
Antipin M.Y. // Russ. Chem. Rev. 1990. V. 59. № 7. P. 607.
Otálora F., Gavira J.A., Ng J.D. et al. // Progr. Biophys. Mol. Biol. 2009. V. 101. № 1. P. 26.
McPherson A., Cudney B. // Acta Crystallogr. F. 2014. V. 70. № 11. P. 1445.
Elschenbroich C. Organometallics. 3rd ed. Completely Revised and Extended. Weinheim: WILEY-VCH Verlag GmbH & Co., 2003.
Organosilanes: Properties, Performance and Applications / Eds. Wyman E.B., Skief M.C. Hauppauge: Nova Science Publishers, 2010.
Корлюков А.А., Нелюбина Ю.В. // Успехи химии. 2019. Т. 88. № 7. С. 677.
Eyer A., Nitsche R., Zimmermann H. // J. Cryst. Growth. 1979. V. 47. № 2. P. 219.
Hassel O., Kringstad H. // Tids. Kjemi Bergvesen. Met. 1930. V. 10. P. 128.
Bacon G.E., Curry N.A., Wilson S.A. // Proc. R. Soc. Lond. A. 1964. V. 279. P. 98.
Fedyanin I.V., Smol’yakov A.F., Lyssenko K.A. // Mendeleev Commun. 2019. V. 29. № 5. P. 531.
Choudhury A.R., Yufit D.S., Howard J.A.K. // Z. Kristallogr. 2014. V. 229. P. 625.
Eulitz Z. // Z. Kristallogr., Kristallgeom., Kristallphys., Kristallchem. 1931. V. 80. P. 204.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Cryst. 2009. V. 42. P. 339.
Alvarez S. // Chem. Rev. 2015. V. 115. P. 13447.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия