

Координационная химия, 2021, T. 47, № 12, стр. 719-788
Синтез, строение и применение органических соединений висмута
В. В. Шарутин 1, *, А. И. Поддельский 2, **, О. К. Шарутина 1
1 Южно-Уральский государственный университет
(национальный исследовательский университет)
Челябинск, Россия
2 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: vvsharutin@rambler.ru
** E-mail: aip@iomc.ras.ru
Поступила в редакцию 16.02.2021
После доработки 21.04.2021
Принята к публикации 24.04.2021
Аннотация
Настоящий обзор, включающий в себя описание современных достижений в области методов синтеза, исследовании особенностей строения и возможности практического использования органических соединений висмута, основан на анализе литературы, опубликованной за период с 2010 по 2020 г. Некоторые более ранние работы представлены в обзоре из-за их важности. Библиография – 190 ссылок.
ОГЛАВЛЕНИЕ
Введение 719
Триорганильные соединения висмута 719
Производные висмута общей формулы RBiX2 и R2BiX 732
Реакции отщепления органических заместителей от триорганилвисмута 732
Иные методы синтеза производных висмута RBiX2 и R2BiX 742
Синтез соединений висмута с полидентатными арильными лигандами 752
Арильные производные висмута(V) 778
Заключение 785
Список литературы 785
ВВЕДЕНИЕ
С момента открытия в 1975 г. возможности использования органических соединений висмута в тонком органическом синтезе [1], число публикаций, посвященных разработке методов синтеза, исследованию реакционной способности и особенностей строения висмуторганических производных, значительно возросло. Атом висмута в своих органических соединениях может быть непосредственно связан с 1, 2, 3, 4, 5 или 6 органическими радикалами, кроме того, известно много типов производных висмута, в которых имеет место замещение одного или нескольких органических заместителей на атомы галогенов или другие электроотрицательные группы. Разнообразие типов висмуторганических соединений привело к значительному расширению исследований в этой области [2], наблюдаемому за последние годы. Непосредственным поводом для этого явились непрекращающиеся попытки найти для висмуторганических соединений возможности их более широкого использования в химии и медицине. Ключевое положение в химии органических производных висмута занимают соединения R3Bi, среди которых подавляющая часть − арильные производные. Из них могут быть получены многочисленные соединения висмута несимметричного строения (RBiX2 и R2BiX) и производные пятивалентного висмута.
ТРИОРГАНИЛЬНЫЕ СОЕДИНЕНИЯ ВИСМУТА
Триорганильные производные висмута R3Bi, как правило, с высоким выходом синтезируют из магний- или литийорганических соединений. Так, бензильные производные висмута получены по реакции дихлорида арилвисмута Ar'BiCl2 [Ar' = = 2,6-(Me2NCH2)2C6H3] с бензилмагнийхлоридом в тетрагидрофуране с образованием Ar'Bi(η1-CH2Ph)2 с высоким выходом (схема 1 ) [3].
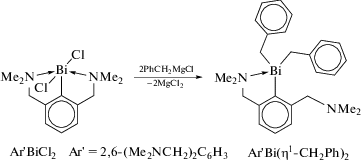
Схема 1 .
Аналогично синтезировали трибензилвисмут Bi(η1-CH2Ph)3. Рентгеновская кристаллография и спектроскопические исследования подтверждают η1-связь бензильных лигандов в данных соединениях. В первом соединении Ar'Bi(η1-CH2Ph)2 присутствует лишь один короткий контакт Bi⋅⋅⋅N (3.058(4) Å), расстояния Bi−CAlk (2.299(4) и 2.340(4) Å) длиннее, чем в Bi(η1-CH2Ph)3 (2.289(4), 2.291(4) и 2.295(4) Å) и триметилвисмуте (2.23(2), 2.26(2) и 2.288(16) Å) [4]. Отметим особенность структуры последнего, в кристалле которого молекулы ассоциированы в димеры (расстояние Bi⋅⋅⋅Bi 3.899(1) Å).
Триаллилвисмут All3Bi, синтезированый из аллилмагнийбромида и трихлорида висмута, был предложен в качестве инициатора контролируемой радикальной полимеризации стирола [5]. Протонолиз All3Bi достаточно сильной кислотой Бренстеда [PhNMe2H]+[B(C6H3Cl2)4]− позволил выделить комплекс висмута [All2Bi(THF)2]+[B(C6H3Cl2)4]− в виде твердого вещества желтого цвета (схема 2 ), в катионе которого атом висмута имеет бис-феноидальную координационную геометрию с апикально расположенными молекулами ТГФ.
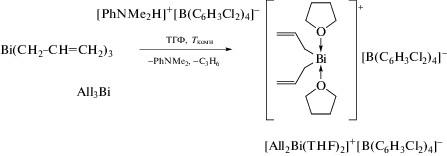
Схема 2 .
Трис(метилаллил)висмут, полученный из реактива Гриньяра и трихлорида висмута, также был структурно охарактеризован. По данным РСА, в этом соединении алильные лиганды координируются с атомом висмута по η1-типу, причем средняя длина связи Bi−C (2.32(2) Å) близка к аналогичной связи для другого охарактеризованного соединения висмута, содержащего аллильный лиганд − {2,6-(Me2NCH2)2C6H3}2(η1-All)Bi [6]. Комплекс [All2Bi(THF)2]+[B(C6H3Cl2)4]− количественно реагирует с двумя эквивалентами альдегида в ТГФ при температуре окружающей среды с образованием продукта карбометаллирования [(AllCH(Ar)O)2Bi(THF)2]+[B(C6H3Cl2)4]− с выходом до 99% (cхема 3).
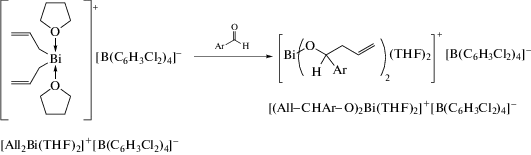
Схема 3 .
Триорганилвисмут Bi(CH2C6H4Cl-2)3, полученный из трихлорида висмута и алкилмагнийбромида, охарактеризован с помощью рентгеноструктурного анализа, который выявил формирование двумерной сетки в результате π-координационных взаимодействий висмут–арен с расстояниями 3.659 Å (висмут-ареноцентроид) и 3.869 Å (ареновые центроиды) соответственно (cхема 4) [7].
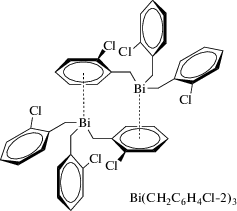
Схема 4 .
С целью сравнения с координационной химией лантанидов были синтезированы и структурно охарактеризованы соединения висмута несимметричного строения Ar2BiR, содержащие N,C,N-лиганды 2,6-(Me2NCH2)2C6H3 [6]. В частности, добавлением раствора аллилмагнийхлорида в тетрагидрофуране к хлориду диарилвисмута в ТГФ был синтезирован аллильный комплекс висмута (2,6-(Me2NCH2)2C6H3)2Bi(All), в котором аллильный лиганд связанс атомом металла по η1-типу; один N,C,N-лиганд является тридентатным, а второй образует только одну координационную связь Bi⋅⋅⋅N (cхема 5).
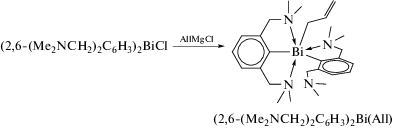
Схема 5 .
Из пентафторэтиллития и треххлористого висмута в эфире при пониженной температуре получали трис(пентафторэтил)висмут Bi(CF2−CF3)3 (cхема 6), в котором длины связей Bi−C (2.331(5), 2.338(5), 2.351(5) Å) [8] максимальны в ряду производных R3Bi из-за повышения электроотрицательности алкильных заместителей при атоме висмута.
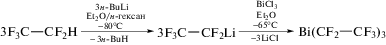
Схема 6 .
Стабильный висмабензол был синтезирован из алюминийорганического производного (cхема 7) [9].
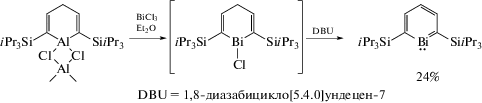
Схема 7 .
Предполагаемая ароматичность этого тяжелого бензола, включающего элемент шестого периода, была исследована методами РСА, ЯМР, УФ-спектроскопии, а также теоретическими расчетами. Структурный анализ полученного висмабензола выявил плоское кольцо, содержащие ненасыщенные связи Bi−C и C−C. Также исследована реакция висмабензоласдиметилацетилендикарбоксилатом (cхема 8),врезультате которой был синтезирован висма-[2.2.2]-бициклооктадиен, охарактеризованный методом РСА.
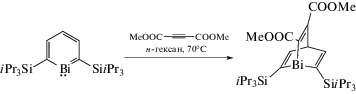
Схема 8 .
Висмуторганическое производное пропеллеровидного орто-замещенного фтортрифенилсилана (висмасилатриптицен) синтезировали из хлорида висмута и трис(2-литийфенил)фторсилана, который получали по реакции литирования трис(2-бромфенил)фторсилана или трис(2-иодфенил)фторсилана бутиллитием в гексане (cхема 9) [10].
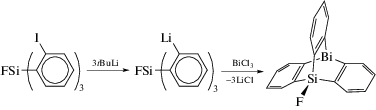
Схема 9 .
Из данных РСА следовало, что взаимодействия между атомами кремния и висмута не наблюдалось несмотря на их большие размеры.
Висмуторганические соединения с N,C-арильным лигандом LBi(Ph)Cl и LMPh2 были получены c хорошим выходом из дихлорида ариллития LBiCl2 (L = o-(СH=N-Dipp)C6H4) и фениллития в соотношении 1 : 1 или 1 : 2 (cхема 10) [11].
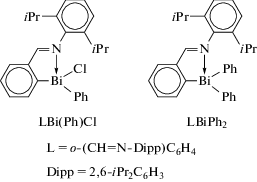
Схема 10 .
Несимметричные соединения (C6F5)2BiR и [2,4,6-(C6F5)3C6H2]2BiR, где R = 2-(Me2NCH2)C6H4, были получены по реакции RBiBr2 с C6F5MgBr и 2,4,6-(C6F5)3C6H2Li, соответственно, при мольном соотношении 1 : 2 [12]. Бромиды R(C6F5)BiBr, R(Mes)BiBr и R(Ph)BiBr были получены из эквимолярных количеств RBiBr2 и C6F5MgBr, MesMgBr или PhMgBr или из PhBiBr2 и RLi в мольном соотношении 1 : 1. Во всех соединениях, содержащих диметиламинометильную группу в арильном лиганде, присутствует весьма прочная координационная связь Bi⋅⋅⋅N (2.511(9)−3.334(16) Å).
Продуктом реакции RLi (R = 2-(Et2NCH2)C6H4) с BiCl3 в мольном соотношении 3 : 1 является триарилвисмут (2-(Et2NCH2)C6H4)3Bi (схема 11 ) [13].
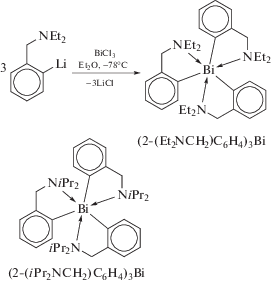
Схема 11 .
По аналогичной схеме был получен (2-iPr2NCH2−C6H4)3Bi, в котором координационный полиэдр центрального атома представляет искаженный октаэдр (Bi−C 2.272(3), 2.276(3), 2.279(3) Å и тесные контакты Bi⋅⋅⋅N 3.052(3), 3.021(3), 3.074(2) Å) (схема 11 ) [14].
Производные (iPr2P−Ace)3Bi и (iPr2P−Ace)2BiPh (Ace = аценафтен-5,6-диил)былисинтезированы из хлорида висмута иариллития[15] по следующей схеме (схема 12 ).
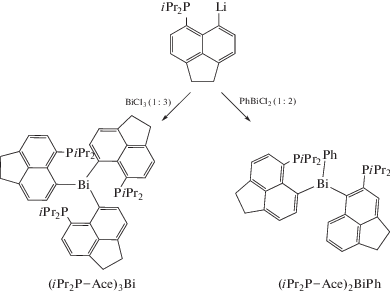
Схема 12 .
В [16] описан синтез нейтрального трис-2-пиридильного Bi-содержащего лиганда (6-Me-2-Рy)3Bi, который при действии [Cu(MeCN)4]+[PF6]− в ра-створе ацетонитрила превращается в комплекс меди {[(6-Me-2-Рy)3Bi]Cu(MeCN)}+[PF6]− (схема 13 ).
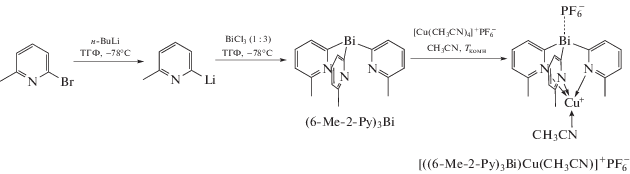
Схема 13 .
Прибавление к {[(6-Me-2-Рy)3Bi]Cu(MeCN)}+-[PF6]− хлорида тетрабутиламмония приводит к количественному образованию димерного комплекса [(6-Me-2-Рy)Bi(6-Me-2-Рy)2CuCl]2 (схема 14 ).
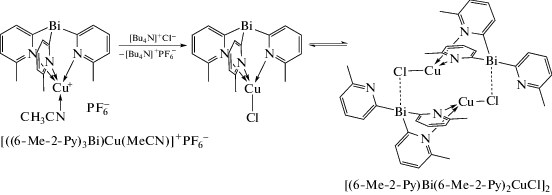
Схема 14 .
Реакция димезитил-1,8-нафталиндиилбората лития с хлоридом дифенилвисмута приводит к образованию 1-(дифенилвисмут)-8-(димезитилбор)нафталиндиила Mes2B(1,8-Napht)BiPh2 (схема 15 ) [17].
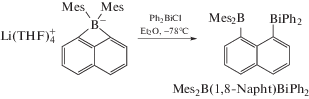
Схема 15 .
Из данных РСА следует, что 1,8-нафталиндиильный остов обеспечивает короткие расстояния Bi → B (3.330 Å). За счет взаимодействия p(Bi) → → p(B) стабильность комплекса возрастает на 6.32 ккал/моль.
Соединения висмута (o-PPh2−C6H4)2BiX (X = = Me, C6F5) были синтезированы из хлорида ди-арилвисмута и органиллития (схема 16 ) [18].
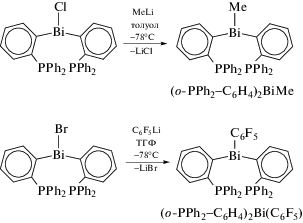
Схема 16 .
Авторы [19] из хлоридов дифенилфосфора и дифенилвисмута синтезировали Р,Bi-содержащий ксантеновый лиганд Xan(PPh2)(BiPh2) (схема 17 ).
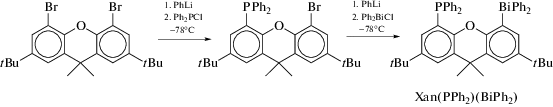
Схема 17.
Атомы фосфора и висмута в Xan(PPh2)(BiPh2) имеют тетраэдрическое окружение, однако расстояние между ними (4.2096(15) Å) незначительно меньше суммы их ван-дер-ваальсовых радиусов 4.3 Å [20].
Новое соединение висмута (п-Tol)2BiR (схема 18 ), содержащий амидный фрагмент, было синтезировано и структурно охарактеризовано [21].
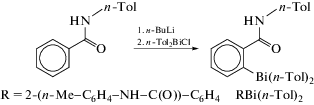
Схема 18 .
Из данных РСА следует наличие внутримолекулярных взаимодействий между висмутом и карбонильным атомом кислорода. Центральный атом имеет псевдотригонально-бипирамидальную координацию. Соединение показало сильную антипролиферативную активность во всех протестированных клеточных линиях. В частности, комплекс был более чувствительным, чем аналогичное соединение сурьмы.
Комлекс с N,C,N-пинцерным лигандом − [2‑(диметиламинометил)фенил]-бис(4-метилфенил)висмут (2-Me2NСH2C6H4)Bi(п-Tol)2 получали из хлорида бис(пара-толил)висмута и о-литий-N-диметилбензиламина [22]. В молекулах соединения наблюдаются внутримолекулярные контакты Bi⋅⋅⋅N (2.902(4) Å).
Соединения висмута [2-{E(CH2CH2)2NCH2}C6H4]3Bi, где E = O или MeN, синтезировали по реакции соответствующего орто-литиевого производного с трихлоридом висмута в мольном соотношени 3 : 1 [23]. Для R3Bi внутримолекулярные взаимодействия N⋅⋅⋅Bi средней силы (3.170(7) Å для [2-{O(CH2CH2)2NCH2}C6H4]3Bi и 3.211(5) Å для [2‑{MeN(CH2CH2)2NCH2}C6H4]3Bi) приводят к искажению октаэдрического (C,N)3Bi-ядра. Шестичленные кольца морфолина и пиперазина в этих комплексах принимают конформацию стула, которая препятствует внутримолекулярной координации атомов кислорода или азота.
Из замещенного ферроцениллития и хлорида дифенилвисмута получили Ph2Bi(2-Me2NCH2−Fc') (где 2-Me2NCH2−Fc' − ферроценильный заместитель (2-Me2NCH2−C5H3)FeCp, содержащий дополнительную функциональную группу Me2NCH2 во втором положении ферроценильного кольца, связанного с атомом висмута), квартенизацией которого иодистым метилом получили ферроценилвисмутинсодержащую соль аммония {[2-(Me3N+CH2)Fc]}[Ph2Bi(2-Me3N+CH2−Fc')][I]− (cхема 19) [24].
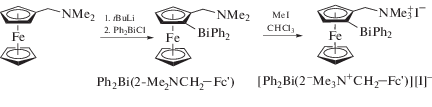
Схема 19 .
Молекулярное строение висмутинов Ph2Bi(2-Me2NCH2−Fc') и [Ph2Bi(2-Me3N+CH2−Fc')][I]− в кристаллическом состоянии было определено с помощью рентгеновской кристаллографии. В соединении Ph2Bi(2-Me2NCH2−Fc') не наблюдалось гипервалентного взаимодействия Bi⋅⋅⋅N.
N,C,N-хелатированные хлориды висмута(III) LBiCl2, где L = 2,6-(tBu-N=CH)2C6H3 и 2,6-(2',6'-Me2C6H3−N=CH)2C6H3) получали из производных лития и трихлорида висмута (схема 20 ) [25]. Обработкаполученныхдихлоридов арилвисмута LBiCl2 алкил- или фениллитием (схема 20 ) приводит к образованию триорганильных соединений висмута LBiR'2, в которых не наблюдаются внутримолекулярные контакты Bi⋅⋅⋅N.
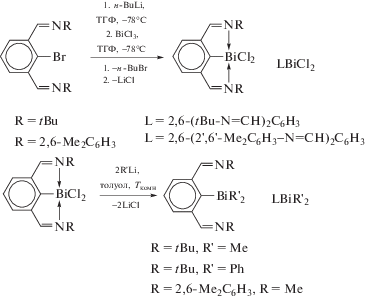
Схема 20 .
Вопросы синтеза триорганильных соединений висмута непосредственно связаны с поиском их возможного использования в тонком органическом синтезе [26]. С этой целью исследована реакционная способность ряда стерически затрудненных соединений триарилвисмута Ar3Bi (Ar3Bi = = (2-MeOC6H4)3Bi, (2-MeC6H4)3Bi, Mes3Bi, (2,6-Me2C6H3)3Bi,(1-Napht)3Bi,[2,4-(MeO)2C6H3]3Bi) в реакцияхкросс-сочетанияс арилиодидами или арилбромидами в присутствии Pd/Cu-содержащих соединений (схема 21 ).
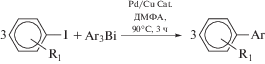
Схема 21 .
Ожидается, что это исследованиеоткроетперспективы для дальнейшего примененияэтихреагентов висмута в тонкоморганических синтезе.
О подобном использовании функционализированных производных триарилвисмута в реакциях C-, N- и O-арилирования, катализируемых соединениями палладия и меди (схема 22 ), сообщали авторы [27].
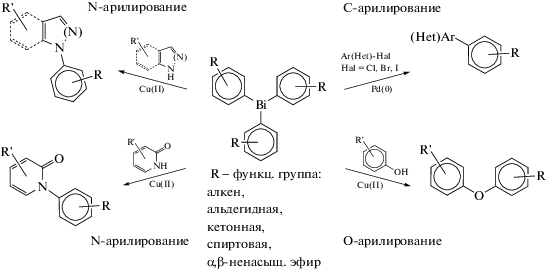
Схема 22 .
Сообщается о катализируемыми соединениями палладия реакциях кросс-сочетания трифенилвисмута или функционализированного триарилвисмута (например, схема 23 ) с галогензамещенными пиридинами, пиразинами и пиридазинами, содержащими реакционноспособные фрагменты [28].
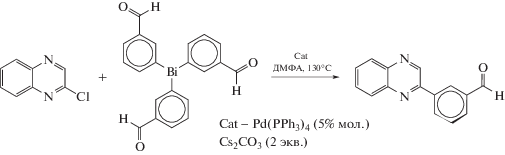
Схема 23 .
Реакции протекают в мягких условиях с превосходными выходами целевых продуктов.
Разработан хемоселективный медный катализатор реакции O-арилирования (1R,2R)-N-BOC-2-амино-1-(4-нитрофенил)-1,3-пропандиолов с использованием триарилвисмута, где BOC = = трет-бутоксикарбонильная группа (схема 24 ). Методпозволяетпереносить орто-, мета- и пара-замещенныеарильныегруппы, имеет хорошую толерантность к функциональным группам и приводит к арилированию первичного спирта [29].
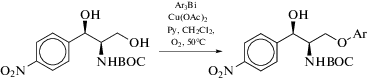
Схема 24 .
Кроме триарильных производных висмута с атомами азота в арильных заместителях описан синтез подобных комплексов с иными потенциальными координирующими центрами, такими как атом кислорода метоксигрупп. Так, трис(2-бром-5-метоксифенил)висмут синтезирован из трихлорида висмута и 5-бром-2-метоксифениллития, полученного металлированием пара-броманизола фениллитием в эфире [30, 31].
Другие триорганильные соединения висмута (схема 25 ), в которых два атома висмута соединены между собой мостиковыми кислородсодержащими лигандами, такими как O{(CH2)2BiPh2}2, MeN(CH2−2-C6H4BiR2)2 и S(CH2−2-C6H4BiR2)2 (R = Me, Ph), были получены и охарактеризованы методом РСА [32].
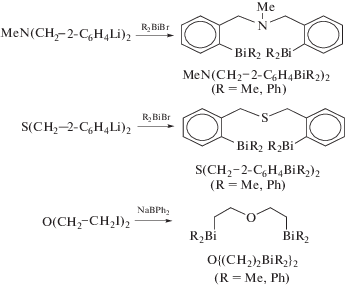
Схема 25 .
Показано, что в структурах O{(CH2)2BiPh2}2 и S(CH2−2-C6H4BiPh2)2 наблюдаются гипервалентные взаимодействия между атомами O или S и атомами висмута (Bi⋅⋅⋅O 3.203(3), 3.126(3) Å при сумме ван-дер-ваальсовых радиусов висмута и кислорода 3.52 Å и Bi⋅⋅⋅S 3.3254(12), 3.3013(12) Å при сумме ван-дер-ваальсовых радиусов висмута и серы 3.8 Å) [20].
Новый фотосенсибилизатор [L+BiR][PF6]− на основе замещенного триарилвисмута LBiPh (схема 26 ) синтезирован из дихлорида фенилвисмута и литиевого производного бис(4-диметиламино-2-бромфенил)-2-толилметана путем его литирования втор-BuLi с последующим окислением п-хлоранилом образующегося LBiPh (схема 26 ). Целевое соединение получили с выходом 4% из LBiPh в виде соли гексафторфосфата [L+BiR][PF6]− [33].
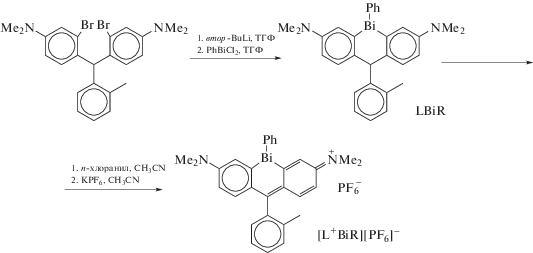
Схема 26 .
Ряд новых фосфоресценцирующих материалов − соединений висмута с гетероциклическими лигандами − получали из β,β-дилитиобитиофенов и дигалогенида фенилвисмута (схема 27 ) [34].
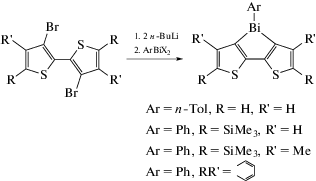
Схема 27 .
Новое соединение висмута − [(2-ди-п-толилбисмутанофенил)диазенил]пирролидин (cхема 28) − синтезировано из хлорида ди(п-толил)висмута и соответствующего соединения лития, полученного литированием 1-[(2-иодфенил)диазенил]пирролидина бутиллитием в тетрагидрофуране, и протестировано на биологическую активность в отношении линий опухолевых клеток человека [35].
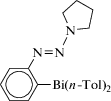
Схема 28 .
Показано, что соединение обладает сильным антипролиферативным эффектом.
Предложен новый способ синтеза триорганильных соединений висмута Bi(2-C4H2X-5-R)3 из силанолов и алкоксидов или амидов висмута (схема 29 ) [36, 37].
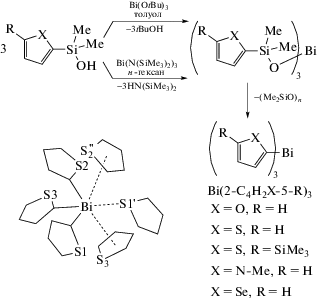
Схема 29 .
В полученных соединениях Bi(2-C4H2X-5-R)3 (X = O, R = H; X = S, R = H; X = S, R = SiMe3; X = = NMe, R = H; X = Se, R = H) и Bi(3-C4H3S)3, по данным РСА, наблюдаются межмолекулярные Bi⋯π-гетероареновые взаимодействия, например, как показано на схеме 29 .
Производные триарилвисмута [2-(ArS)C6H4]nBiAr3 − n с орто-тиоарильным заместителем удобно синтезировать путем вставки бензина в связь висмута с серой в соединения (ArS)nBiAr3 − n (n = 1, 2) (схема 30 ) [38].
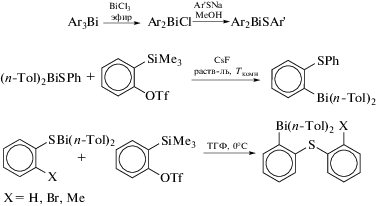
Схема 30 .
Связь Bi−C в [2-(2-BrC6H4S)C6H4]nBiAr3 – n разрывается в присутствии Pd-катализатора, при этом образуется с хорошим выходом дибензотиофен. Рентгеноструктурное исследование 2-(2-BrC6H4S)C6H4Bi(п-Tol)2 показывает присутствие внутримолекулярного контакта S⋅⋅⋅Bi (3.397(2) Å), что значительно меньше суммы ван-дер-ваальсовых радиусов данных элементов(4.2 Å [20]). Дегидробензолможетвнедрятьсясразупо двум связям Bi−S, чтоприводиткобразованию функционализированныхтриарильных соединений висмута, синтезкоторыхиными методами достаточно сложен (схема 31 ).
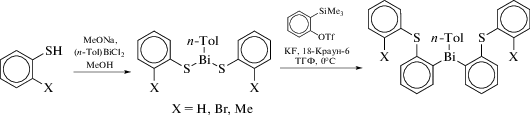
Схема 31 .
Несколько гетероциклических соединений висмута C4R4BiAr (R = Et, Ph; Ar = Ph, Mes) и L(Thi)2BiMes были синтезированы с помощью эффективного переноса металлоцикла с участием легкодоступных цирконоциклов и изучены их люминесцентные свойства (схема 32 ) [39].
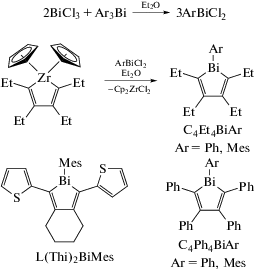
Схема 32 .
Азид-алкиновое циклоприсоединение, катализируемое соединением меди(I), в настоящее время широко используется в качестве надежного метода ковалентного соединения различных строительных блоков [40]. Использование висмуторганического ацетиленида R1−C≡C−BiAr2 устраняет нежелательные реакции протодегалогенирования, и целевой продукт N3C2(R1R2)−BiAr2 выделяется из реакционной смеси с выходом до 99% (схема 33 ). На его основе описано получение различных функционализированных N-гетероциклических производных (схема 34 ).
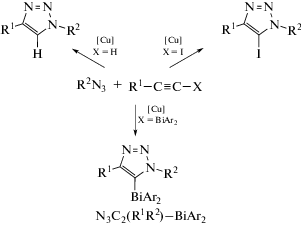
Схема 33 .
Схема 34 .
В [41] сообщается о получении ряда фосфоресцирующих висмутсодержащих полимеров и блок-сополимеров с арилированными норборненами (схема 35 ).
Схема 35 .
Полученные полимеры с высокой молекулярной массой в углеродном каркасе имеют бензовисмолы, построенные с помощью реакций метатезиса с раскрытием кольца (схема 36 ).
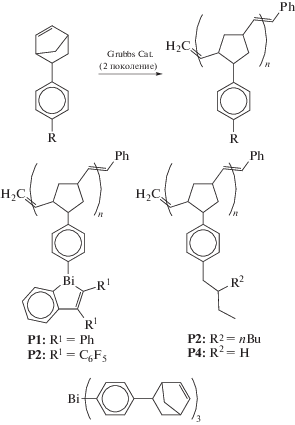
Схема 36 .
Дипиридиновисмол DPyBi получали по реакции дилитиевого производного 2,2'-дибром-4,4'-бипиридила с дииодфенилвисмутом (cхема 37). Циклическая вольтамперограмма данного соединения в ацетонитриле указывает на повышенное сродство к электрону по сравнению с бипиридилом без мостика. Это соединение и родственное производное сурьмы (DPySb) обладают слабой флуоресценцией при комнатной температуре и видимой фосфоресценцией при 77 K с максимумами излучения λmax = 453 нм и временем жизни τ = 1.03 мс для DPySb и λmax = 454 нм, τ = 0.26 мс для DPyBi соответственно. Твердофазная фосфоресценция также наблюдалась на этих дипиридиногетеролах при 77 К. Взаимодействие DPyBi с Cu2I2(PPh3)3 приводит к получению соответствующего медного комплекса [(PPh3)CuI(DPyBi)CuI(PPh3)]2, который демонстрирует красную фосфоресценцию в твердом состоянии при комнатной температуре [42].
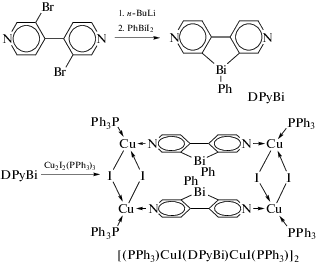
Схема 37 .
ПРОИЗВОДНЫЕ ВИСМУТА ОБЩЕЙ ФОРМУЛЫ RBiX2 И R2BiX
Производные трехвалентного висмута, содержащие одну или две органические группы у атома металла, получают несколькими способами. Наиболее простым и достаточно эффективным является метод отщепления от триорганилвисмута органических заместителей соединениями, содержащими активный атом водорода. Другие, не менее эффективные способы синтеза указанных соединений, основаны на реакциях замещения, присоединения и внедрения.
Реакции отщепления органических заместителей от триорганилвисмута. Опубликована серия работ, посвященных реакции дефенилирования трифенилвисмута карбоновыми кислотами, продуктами которых являются карбоксилаты арил- или диарилвисмута. Отличительна особенность карбоксилатов диарилвисмута − их полимерная структура, обусловленная бидентатными свойствами карбоксилатного лиганда. Однако подобные координационные полимеры представлены единичными примерами. Так, в [43] описан синтез 2-фенилкарборанилкарбоксилата дифенилвисмута по реакции замещения между трифенилвисмутом и 2‑фенилкарборанилкарбоновой кислотой в бензоле (cхема 38) и определены его структурные особенности методом РСА.
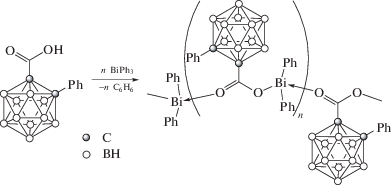
Схема 38 .
По данным РСА, 2-фенилкарборанилкарбоксилат дифенилвисмута представляет собой координационный полимер, закристаллизовавшийся в виде сольвата с бензолом. Атомы висмута имеют бисфеноидную координацию с апикально расположенными атомами кислорода 2-фенилкарборанилкарбоксилатных заместителей. В экваториальной плоскости находятся два фенильных лиганда и неподеленная электронная пара.
Авторами [44] изучено взаимодействие трифенилвисмута с 4-нитрофенилуксусной и 2-нитробензойной кислотами в толуоле при 90°С. Показано, что при эквимолярном соотношении реагентов образуются бис(4-нитрофенилацетат) фенилвисмута и бис(2-нитробензоат) фенилвисмута с выходами 49 и 46% соответственно. Минорными продуктами реакций являются 4-нитрофенилацетат дифенилвисмута и 2-нитробензоат дифенилвисмута (27 и 16% соответственно).
С целью исследования влияния растворителя и природы карбоновой кислоты на образование карбоксилатов висмута, трифенилвисмут обрабатывали салициловой, 5-бромсалициловой, 3‑метоксисалициловой, 5-нитрозалициловой, 3-метилантраниловой, 5-хлорантраниловой и N-ацетилантраниловой кислотами при мольном соотношении исходных реагентов 1 : 2 в различных условиях (схема 39 ) [45].
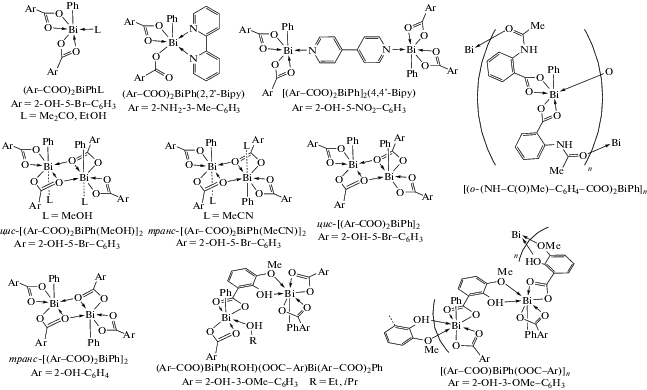
Схема 39 .
Соединения (Ar−COO)2BiPhL (L = Me2C=O, EtOH) мономерны и содержат молекулу координированного растворителя в экваториальном положении у атома висмута в пентагональной пирамиде. Такая же геометрия найдена в [(Ar-COO)2BiPh]2-(4,4'-Bipy), где две такие единицы связаны вместе через 4,4'-бипиридиновый лиганд. Соединения (Ar−COO)BiPh(ROH)(OOC−Ar)Bi(Ar−COO)2Ph образуют димеры, в которых наблюдаются подобные координационные полиэдры атомов Bi. Соединения [(o-(NH−C(O)Me)−C6H4−COO)2BiPh]n и [(Ar−COO)BiPh(OOC−Ar)]n полимерные. В (Ar−COO)2BiPh(2,2'-Bipy) один из карбоксилатных лигандов бидентатный, в то время как другой остается монодентным, отображая ту же структурную геометрию, что и для описанных ранее. Соединения цис-/транс-[(Ar−COO)2BiPh(L)]2 (L = = MeOH/MeCN), цис-[(Ar−COO)2BiPh]2, транс-[(Ar−COO)2BiPh]2 представляют собой димерные структуры, связанные между собой общими атомами кислорода карбоксилатных групп.
Три дикарбоксилата фенилвисмута получены дефенилированием трифенилвисмута o-метоксибензойной, м-метоксибензойной, 5-[(R/S)-2,3-дигидроксипропилкарбамоил]-2-пиридинкарбоновой кислотами (мольное соотношении 1 : 2) при кипячении смеси реагентов с обратным холодильником в метаноле или этаноле [46]. Прибавление к реакционной смеси эквимолярного количества 2,2′-бипиридила (Вipy) приводит к синтезу устойчивого комплекса [PhBi(O2CC6H4OMe-o)2-(Вipy)] · 0.5EtOH. Полученные комплексы охарактеризованы ЯМР-спектроскопией и протестированы на антилейшманиозную активность. Дополнительно оценена их токсичность для клеток млекопитающих. Комплексы висмута замещенных бензойных кислот показывают значительную антилейшманиозную активность против промастиготов L, majorV121 при очень низких концентрациях, в то время как соответствующие свободные карбоновые кислоты не проявляют действенной активности. Однако соединения висмута ингибируют рост клеток млекопитающих при всех изученных концентрациях (от 1.95 до 500 мкг/мл) после 48 ч инкубации.
Показано, что реакции трифенилвисмута с такими гетероциклическими карбоновыми кислотами как 3-гидроксипиколиновая, пиразин-2-карбоновая, хинолин-2-карбоновая (хинальдиновая), фуран-2-карбоновая и тиофен-2-карбоновая кислоты, приводят к образованию карбоксилатов дифенилвисмута и дикарбоксилатов фенилвисмута [47]. По данным РСА, координационное число (КЧ) висмута в полученных комплексах вследствие координации потенциальных координирующих центров (гетероатомов и карбонильных атомов кислорода) изменяется от 5 до 8.
Повышение КЧ атома висмута имеет место в бис(хлорацетате) фенилвисмута, где хлорацетатные лианды являются тридентатными хелатно-мостиковыми, связывающими через атомы кислорода соседние молекулы в полимерные цепочки [48].
Два комплекса висмута (MICA)2BiPh и (IGA)2BiPh получали из индолкарбоновых кислот (MICAH = 1-метил-1Н-индол-3-карбоновая кислота, IGAH = 2-(1H-индол-3-ил)-2-оксоуксусная кислота) и трифенилвисмута в кипящем этаноле (cхема 40) [49].
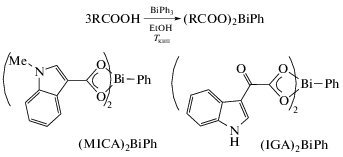
Схема 40 .
Комплексы охарактеризованы с помощью элементного анализа, ИК-, масс-спектроскопии, спектроскопии ЯМР (1Н, 13С). Комплекс (IGA)2BiPh охарактеризован рентгеновской кристаллографией как димер в твердом состоянии. In vitro антибактериальную активность индолкарбоновых кислот и их комплексов висмута оценивали в отношении Helicobacter pylori. Соединения проявляют высокую активность против лейшманиоза без какой-либо токсичности в отношении клеток млекопитающих при их эффективной концентрации.
В отсутствие растворителя из трифенилвисмута и 3-гидроксипиколиновой кислоты (3-НpicH) с последующей перекристаллизацией из диметилформамида был получен дикарбоксилат фенилвисмута (3-Нpic)2BiPh (схема 41 ) [50].
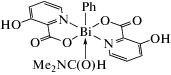
Схема 41 .
В молекулах комплекса атомы висмута гексакоординированы в искаженной пентагонально-пирамидальной геометрии с двумя атомами N, двумя атомами O хелатирующих 3‑Нpic-лигандов, атомом кислорода молекулы растворителя в экваториальной плоскости и фенильным заместителем в апикальном положении.
Полиядерные оксокластеры висмута получены из трифенилвисмута и орто-нитробензойной кислоты в различных условиях (схема 42 ) [51].
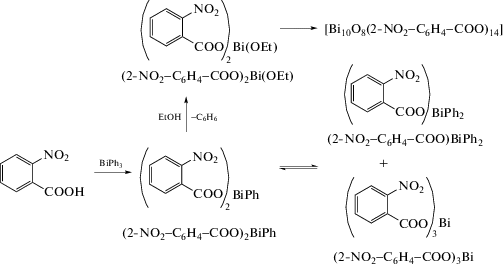
Схема 42 .
При мольном соотношении исходных реагентов 1 : 2 в этаноле получены (2-NO2–C6H4−COO)2Bi(OEt) ∙ EtOH и (2-NO2–C6H4− COO)BiPh2, которые первоначально кристаллизовались вместе, в то время как (2-NO2–C6H4− COO)3Bi ∙ H2O появился позже из отфильтрованного маточного раствора. Соединение (2-NO2–C6H4− COO)2Bi(OEt) ∙ EtOH является результатом этанолиза in situ продукта (2-NO2–C6H4−COO)2BiPh и далее подвергается гидролизу с получением кристаллов оксокластера [Bi10O8(2-NO2–C6H4−COO)14](EtOH)x. Рентгеноструктурные исследования монокристаллов четырех из пяти соединений (кроме (2-NO2–C6H4−COO)2BiPh) показывают, что все они являются полимерными в твердом состоянии, КЧ(Bi) равно 9, 8, 5 соответственно.
N,C,N-Внутримолекулярно координированный оксид висмута(III) (ArBiO)2, где Ar = 2,6-(Me2NCH2)2C6H3, реагирует с 1,1'-ферроцендикарбоновой кислотой с образованием соответствующего биядерного карбоксилата (Fc(COO)2BiAr)2 (схема 43 ) [52].
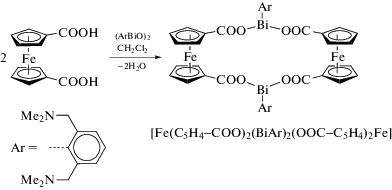
Схема 43 .
Соединение охарактеризовано с помощью ЯМР, рамановской, ИК-, УФ-видимой спектроскопии и РСА.
Фенантролиновый комплекс висмута [Bi(Рhen)- (C6H5COO)(C6H4COO)] (Рhen = 1,10-фенантролин) в виде коричневых кристалов был синтезирован из нитрата висмута, 2-меркаптобензойной кислоты,1,10-фенантролина в качестве вспомогательноголиганда, азотной кислоты и оксида неодима вводе при 160°С в течение 3 сут (схема 44 ) [53]. Комплекс охарактеризован РСА, ИК-спектроскопией, TГ-анализом.
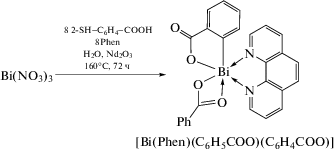
Схема 44 .
Очевидно, что появление бензоатной группы в комплексе обусловлено протекающей в гидротермальных условиях реакцией десульфирования 2-меркаптобензойной кислоты.
Два комплекса висмута (Napht−C(O)S)2BiPh (cхема 45) и (4-Br–C6H4−C(O)S)2BiPh, полученные из тионафтойной и п-бромтиобензойной кислот и трифенилвисмута в растворе кипящего этанола (1 ч), были охарактеризованы и оценены на предмет активности in vitro против лейшманиоза и общей токсичности в отношении клеток фибробластов человека [54].
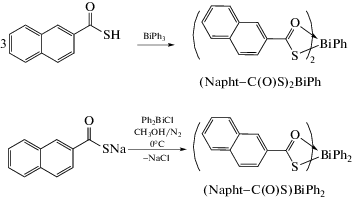
Схема 45 .
Необходимо отметить, что термолиз производных (R−С(O)S)BiPh2 приводил к образованию соединений PhBiX2 и трифенилвисмута по реакции перераспределения радикалов (cхема 46).
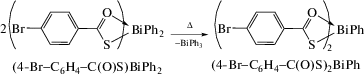
Схема 46 .
По данным РСА, в комплексе (4-Br–C6H4−C(O)S)2BiPh два тиокарбоксилатных лиганда координируются с атомом висмута, формируя искаженную октаэдрическую геометрию координационного узла, в которой фенильная группа и неподеленная пара ориентированы аксиально по отношению к плоскости, образованной двумя тиокарбоксилатными лигандами. Межмолекулярные взаимодействия Bi⋅⋅⋅S (3.54 Å) связывают эти мономерные звенья в единое целое. Показано, что тиокарбоксилатные производные висмута в биологическом плане оказались более активными, чем соответствующие кислоты. Отмечают наиболее высокую активность комплексов (Napht−C(O)S)BiPh2 и (4-Br-C6H4−C(O)S)2BiPh.
Несколько соединений висмута(III): (Ph-C(O)S)2BiPh, (м-NO2-C6H4−C(O)S)2BiPh и (3-SO3-C6H4−C(O)S)2BiPh синтезировано из тиобензойных кислот и трифенилвисмута (схема 47 ) [55]. По реакции замещения из хлорида дифенилвисмута синтезировано производное с двумя фенильными группами при атоме висмута (Ph−C(O)S)BiPh2, легко превращаемое по реакции перераспределения лигандов в монофенильный комплекс (Ph−C(O)S)2BiPh.
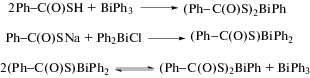
Схема 47 .
По данным РСА, комплекс (Ph−C(O)S)2BiPh образует дискретные тетрамерные единицы, скрепленные длинными межмолекулярными связями Bi⋅⋅⋅S (3.774 Å). Исследована активность комплексов (Ph−C(O)S)2BiPh и (Ph−C(O)S)BiPh2 против трех штаммов Helicobacter pylori. Показано, что высокий уровень бактерицидной активности не чувствителен к степени замещения у атома висмута.
Серия моноорганических дитиокарбоксилатных комплексов висмута (Ar−C(S)S)2BiR (R = Me, Ph, п-Тol; Ar = Ph, п-Тol) была синтезирована по реакциям замещения из триорганилвисмута, либо из дихлорида метилвисмута и дитиокарбоновых кислот в присутствии триэтиламина в качестве акцептора HCl (схема 48 ) [56].
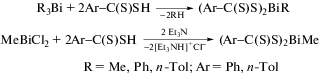
Схема 48 .
Соединения были охарактеризованы элементным анализом и спектроскопическими исследованиями. Молекулярное строение (п-Tol−C(S)S)2BiR (R = Me или Ph) в кристаллическом виде установлено с помощью РСА. Показано, что атом висмута в этих соединениях принимает квадратно-пирамидальную конфигурацию с группой R в апикальном положении. Термолиз (п-Tol−C(S)S)2BiR (R = Me или Ph) при кипячении с обратным холодильником в дифениловом эфире приводил к образованию нанокристаллов Bi2S3. Комплекс (п-Tol−C(S)S)2BiPh использовался для осаждения тонких пленок Bi2S3.
Обработка трифенилвисмута 4-метил-4Н-1,2,4-триазол-3-тиолом (4-МТТН) или 2-меркапто-1-метилимидазолом (2-MMIH) в смеси растворителей толуол−этанол при кипячении или микроволновом облучении приводит к окислению производных висмута и образованию в обоих случаях ярко-желтых веществ (4-MMT)4BiPh и (2-MMI)4BiPh, соответственно, время синтеза которых при облучении было наименьшим (7 мин) (схема 49 ) [57].
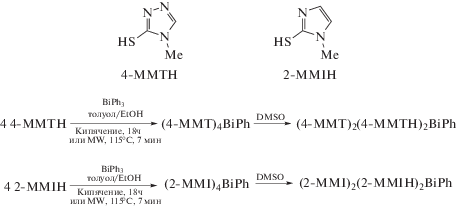
Схема 49 .
Однако перекристаллизация комплексов (4-MMT)4BiPh и (2-MMI)4BiPh из ДМСО приводила к образованию производных трехвалентного висмута [(4-MMT)2(4-MMTH)2BiPh]3 и [(2-MMI)2-(2-MMIH)2BiPh]4.
Продуктами реакций тетразол-, имидазол- и тиадиазол-гетероциклических тиолов: 1-метил-1Н-тетразол-5-тиол (1-MMTZH); 4-МТТН; 5-метил-1,3,4-тиадиазол-2-тиол (5-MMTDH); 1,3,4-тиадиазол-2-дитиол (2,5-DMTDH2) с трифенилвисмутом являются гетеролептические комплексы тиолатофенилвисмута общего вида (RS)2BiPh, которые также были получены из дихлорида фенилвисмута и натриевой соли тиола (схема 50 ) [58]. Полученные комплексы были охарактеризованы спектральными методами и РСА.
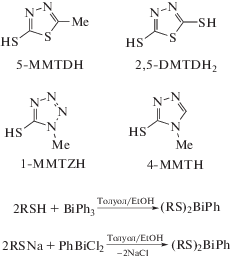
Схема 50 .
Проведена оценка бактерицидных свойств полученных соединений против Mycobacterium smegmatis (М. smegmatis), золотистого стафилококка (S. aureus), метициллинрезистентного золотистого стафилококка (MRSA), устойчивого к ванкомицину энтерококка (VRE), Enterococcus faecalis (E. faecalis) и Escherichia coli (E. coli), наибольшую эффективность среди которых показали комплексы, содержащие лиганды 1-MMTZ и 4-MTT: (1-MMTZ)2(1-MMTZH)2BiPh и (4-МТТ)2(4-МТТН)2BiPh. Все комплексы показали незначительную или нулевую токсичность в отношении клеток COS-7 млекопитающих при 20 мг/мл.
Тиолатовисмутовый комплекс (5-MMTD)2(4-MMTH)BiPh был синтезирован из тиадиазол- и триазол-гетероциклических тионов и структурно охарактеризован (cхема 51) [59].
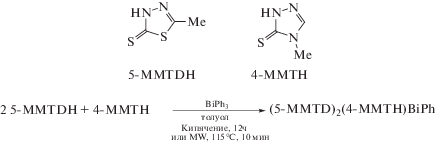
Схема 51 .
Показано, что комплекс проявляет антибактериальные свойств против золотистого стафилококка, ванкомицин-резистентного энтерококка, E. faecalis, E. coli и низкую токсичность по отношению к клеточным линиям COS-7 млекопитающих в дозе 20 мкг/мл.
Из 4-фенилтиазол-2-тиола (MBTH) и трифенилвисмута или BiPhCl2 и соответствующего тиолата натрия (NaMBT) был получен (MBT)2BiPh (cхема 52) с выходом до 89% в различных условиях (без растворителя, 100°С, 4 ч; кипячение раствора толуола, 6 ч; микроволновое облучение, толуол, 115°С, 15 мин; метиловый спирт, 12 ч, 24°С), охарактеризованный методом РСА [60].
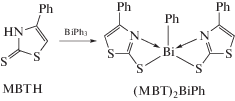
Схема 52 .
Показано, что комплекс (MBT)2BiPh, являющийся в кристалле димером, проявляет хорошую антилейшманиозную активность, обладает активными бактерицидными свойствами против микобактерий смегматис, MRSA, E. faecalis, устойчивый к VRE и E.coli и имеет низкую токсичность по отношению к клеткам COS-7 млекопитающих при 20 мкг/мл.
Ряд 5-замещенных фенилтиазолоксадиазолетионов вида (Х-PTOT)2BiPh, где X = Me, MeO, MeS, F, Cl, Br, CF3; PTOTH = 5-(2-фенилтиазол-4-ил)-1,3,4-оксадиазол-2-тиол) (cхема 53), был синтезирован из трифенилвисмута и соответствующих тиоамидов или из дихлорида фенилвисмута и натриевых солей тиоамидов [61].
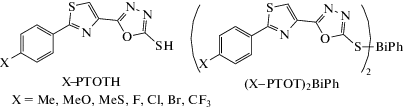
Схема 53 .
Комплексы (Cl–PTOT)2BiPh и (Br–PTOT)2BiPh после перекристаллизации из ДМСО были структурно охарактеризованы с помощью РСА как (X–PTOT)2BiPh ∙ 2DMSO (X = Cl, Br). Оценены антибактериальные свойства тионов и их Bi(III)-комплексов против Mycobacterium smegmatis, S. aureus, MRSA, VRE, E. faecalis и E. coli. Показано, что все комплексы висмута(III) высокоэффективны против всех бактерий, так как имеют очень низкие значения минимальной ингибирующей концентрации (МИК) (1.1–2.1 мкМ). Эти комплексы показали незначительную токсичность или отсутствие токсичности в отношении клеток COS-7 млекопитающих при 20 мкг/мл.
Комплексы висмута(III) [(Sac)BiPh2]n, [(Sac)2BiPh]n, [(Tsac)BiPh2]n, [(Tsac)2BiPh]n (SacH = = сахарин, TsacH = тиосахарин) были синтезированы и охарактеризованы в [62]. Отщепление одной фенильной группы от атома висмута наблюдалось при кипячении эквимолярных количеств трифенилвисмута и сахарина или тиосахарина в этаноле в течение 30 мин (схема 54 ).
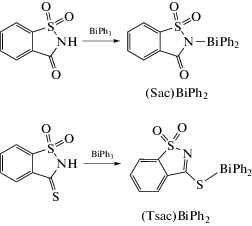
Схема 54 .
При мольном соотношении 1 : 2 и увеличении времени нагревания до 1 ч имело место образование производных PhBiX2 с выходом до 73%. Структуры [(Sac)BiPh2]n и [(Tsac)BiPh2]n были подтверждены методом рентгеновской кристаллографии. В [(Sac)BiPh2]n мостиковые лиганды Sac связывают группировки Ph2Bi с четырехкоординированным атомом висмута через атом азота (Bi−N 2.353(4) Å) и один из атомов кислорода SO2-группы (Bi⋅⋅⋅O 2.605(4) Å). Однако в структуре [(Tsac)BiPh2]n тиосахариновый лиганд σ-связан через экзоциклический атом серы, образуя тиолатный комплекс, подтверждая более тиофильный характер висмута(III). В кристалле комплекса [(Tsac)BiPh2]n также содержатся полимерные цепочки с формально четырехкоординированными атомами висмута. Была оценена активность комплексов против H. pylori. Активность зависит как от лиганда, так и от степени замещения лиганда. Сахаринатные комплексы [(Sac)BiPh2]n и [(Sac)2BiPh]n проявляют активность, сравнимую со стандартными для трикарбоксилатов висмута(III) (6.25 мкг/мл), в то время как активность тиолатных комплексов висмута резко возрастала с увеличением количества тиолатных групп. Сахарин, тиосахарин и трифенилвисмут являлись неактивными.
В [63] сообщалось о синтезе бис(2,5-диметилбензолсульфоната) фенилвисмута с выходом 94% из трифенилвисмута и 2,5-диметилбензлсульфоновой кислоты в толуоле. По данным РСА, атомы висмута с учетом стереохимически активной неподеленной электронной пары имеют искаженную октаэдрическую координацию, которую без “фантом”-лиганда можно рассматривать как квадратно-пирамидальную с атомами кислорода в экваториальных положениях и атомом углерода фенильной группы в аксиальном. Атом висмута выходит из средней экваториальной плоскости О4 на 0.19 Å в направлении, противоположном атому углерода. Транс-углы в экваториальной плоскости ОBiO равны 177.7(1)° и 164.1(1)°. Два угла СBiO (89.0(5)°, 88.8(5)°) близки к теоретическому значению, тогда как другие два угла СBiO (81.6(5)°, 82.5(5)°) значительно отклоняются от него. Длина связи Bi–C равна 2.247(5) Å. Валентные связи Bi–O (2.394(9), 2.390(9) Å) и координационные связи Bi⋅⋅⋅O (2.396(10), 2.403(10) Å) практически не отличаются между собой.
О получении и характеристиках двух координационных полимеров винилсульфонатов висмута(III) сообщалось в [64]. Синтез комплексов висмута осуществляли из трифенилвисмута и винилсульфоновой кислоты в этаноле или ацетонитриле (cхема 55).
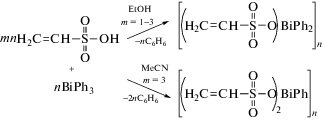
Схема 55 .
Строение соединений доказано спектральными методами анализа и РСА. В кристалле координационного полимера [(Vin-SO3)BiPh2]n (Vin = винил) атомы висмута с учетом стереохимически активной неподеленной электронной пары имеют координацию искаженной тригональной бипирамиды, в которой атомы кислорода занимают апикальные позиции с углом OBiO 164.7(3)°. Один фенильный заместитель соседней молекулы ориентирован на атом висмута (Bi⋅⋅⋅арен 3.42 Å) при почти перпендикулярном расположении атома висмута над центром фенильного кольца (схема 56 ).
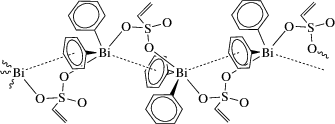
Схема 56 .
Кристалл координационного полимера [(Vin-SO3)2BiPh]n содержит два типа кристаллографически независимых молекул, которые не взаимодействуют друг с другом. Молекулы каждого типа образуют вдоль кристаллографической оси b полимерную цепочку, в которой атомы висмута связаны между собой через атомы кислорода сульфонатных лигандов. Координационные сферы атомов висмута лучше всего описать как квадратную пирамиду с фенильным заместителем в вершине, а с учетом стереохимически активной неподеленной пары электронов как псевдооктаэдр.
Три органосульфоната дифенилвисмута (R−SO3)BiPh2, где R = п-толил (п-Tol), мезитил (Mes) или S-(+)-10-камфорил (S-(+)-10-Camph), были синтезированы взаимодействием эквимолярных количеств трифенилвисмута и органосульфоновой кислоты в спирте с выходом целевого продукта до 99% (cхема 57) [65]. Перекристаллизация соединений из ацетона сопровождается реакцией перераспределения лигандов и образованием полимерного бис(органосульфонато)фенилвисмута [(R−SO3)2BiPh]x и трифенилвисмута.
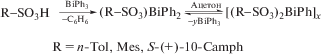
Схема 57 .
По данным РСА, комплексы (Mes−SO3)BiPh2 и (S-(+)-10-Camph−SO3)BiPh2 структурно очень похожи со структурами полимерных спиральных цепей, в которых атомы висмута, имеющие тригонально-бипирамидальное окружение, соединены с атомами кислорода сульфонатных групп с почти линейными углами OBiO. Два фенильных кольца с стереохимически активной парой электронов находятся в экваториальной плоскости, аксиальные положения занимают атомы кислорода. Присутствие одного сульфонатного лиганда в соединениях (R−SO3)BiPh2 приводило к резкому увеличению бактерицидной активности в отношении бактерии H. pylori относительно трифенилвисмута и сульфокислоты, которые были практически неактивными. В комплексах [(R−SO3)2BiPh]x (R = = п-Tol, Mes) атомы висмута связаны между собой двумя мостиковыми органосульфонатными лигандами через атомы кислорода. Координация атомов висмута октаэдрическая, одно положение занято неподеленной электронной парой; длины связей Bi−C и Bi−O составляют 2.226(7), 2.221(10) Å и 2.361(4)−2.384(7) Å соответственно. Восьмичленные кольца, состоящие из атомов висмута, кислорода и серы, обладают конформацией “кресло”, два противоположных атома кислорода выходят из плоскости остальных (компланарных в пределах 0.09 Å) в разные стороны на 0.93 Å.
Обработка трифенилвисмута 5-сульфосалициловой кислотой (H3Ssal) приводит к образованию гидрата сульфосалицилата фенилвисмута (HSsal)BiPh ∙ ∙ H2O и его этанольного аналога (HSsal)BiPh ∙ EtOH [66]. По данным РСА, оба комплекса в твердом состоянии являются полимерами с каркасами, построенными из димерных [(HSsal)Bi]2. Первый из данных гетеролептических комплексов демонстрирует замечательную растворимость в воде (10 мг/мл), в результате чего получается прозрачный раствор с pH 1.5. Напротив, второй комплекс (с этанолом) практически нерастворим в воде. Комплексы проявляют значительную активность в отношении бактерии H. pylori.
Трифлат дифенилвисмута был получен из трифлата димезитилтеллура и трифенилвисмута и встречным синтезом из трифенилвисмута и трифторметансульфоновой кислоты (схема 58 ) [67].
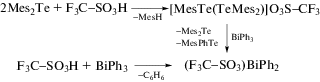
Схема 58 .
По данным РСА, трифлат дифенилвисмута представляет собой полимер, в котором фрагменты Ph2Bi связаны между собой через мостиковые атомы кислорода трифлатных групп (2.531(6), 2.473(5) Å).
Взаимодействие трифенилвисмута с 1,1,2,3,3-пентаметилтриметилен-фосфиновой кислотой {cyc-P(O)OH · 2H2O} в тетрагидрофуране при нагревании и комнатной температуре приводит к образованию 16-членного макроцикла [(cyc-PO2)8 -(BiPh)4] и полимера [(cyc-PO2)BiPh2]n соответственно (cyc-PO2 = 1,1,2,3,3-пентаметилтриметиленфосфинат) (схема 59 ) [68].
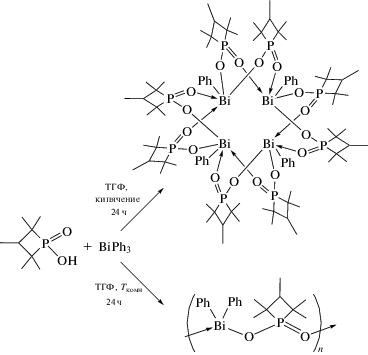
Схема 59 .
В обоих комплексах, охарактеризованных методом РСА, анизобидентатные фосфинатные лиганды мостиковые.
Взаимодействие фосфатного диэфира (tBuO)2PO(OH) с BiPh3 в соотношении 1 : 1 при комнатной температуре в этаноле дает координационный полимер [((tBuO)2PO2)BiPh2]n, в котором атомы висмута соединены изобидентатными лигандами [(tBuO)2PO2] (схема 60 ) [69].
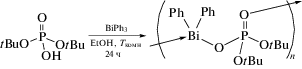
Схема 60 .
Термолиз соединения при 700°С дает чистую фазу BiPO4.
Новый координационный полимер [(Me2N− C(S)S)BiPhCl]n был синтезирован из диметилдитиокарбамата натрия и трифенилвисмута (мольное соотношение 2 : 1) в смеси растворителей (метанол−тетрагидрофуран (25°С, 24 ч) и охарактеризован ИК-, ЯМР 1Н-спектроскопией и РСА [70]. В кристалле квадратно-пирамидальные блоки с фенильным лигандом в апикальном положении связываются между собой мостиковыми атомами хлора, образуя одномерную спиральную цепь (схема 61 ).
Схема 61 .
Комплекс обладает высокой фотокаталитической активностью, показанной на примерах метиленового синего, родамина B и метилового фиолетового.
Иные методы синтеза производных висмута RBiX2 и R2BiX. Соединения трехвалентного висмута с одним или двумя органическими заместителями при атоме висмута можно получать несколькими способами, среди которых достаточно эффективными являются методы, основанные на реакциях органических производных активных металлов с тригалогенидами висмута. Так, была получена серия производных трехвалентного висмута tBu2BiX (X = Cl, Br, I, CN, N3, SCN) [71]. Хлорид tBu2BiCl получали реакцией трихлорида висмута с двумя эквивалентами tBuMgCl, тогда как соединения tBu2BiX (X = Br, I, CN, SCN) синтезированы из tBu3BiX2. Азид tBu2BiN3 был получен по реакции tBu2BiCl с NaN3. Кристалл tBu2Bi(CN) состоит из полимерных цепей, в которых группы tBu2Bi связаны между собой Bi−C≡N⋅⋅⋅Bi мостиками (расстояние N⋅⋅⋅Bi составляет 2.548 Å).
Описаны подходы к синтезу первых адамантильных комплексов висмута из адамантилмагнийбромида или адамантиллития и 1- и 2-адамантилцинкбромидов (схема 62 ) [72].
Схема 62 .
Молекулярное строение бромида бис(2-адамантил)висмута в кристаллическом состоянии подтверждено методом РСА.
По схемам классического элементоорганического синтеза получен ряд пентафторэтильных производных висмута EtFnBiX3 –n (EtF = CF2−CF3, X = F, Cl, Br, I), охарактеризованых РСА [8]. Их химические свойства, индуцированные сильным электроноакцепторным характером пентафторэтильных групп, показаны на примерах реакций с галогенводородными кислотами и солями некоторых элементов (схема 63 ).
Схема 63 .
Четыре новых стерически перегруженных терфенилзамещенных дигалогенида висмута типа [ArmBiX2]2, m = 1, 2 (Ar1 = 2,6-Mes2-C6H3, X = Br, I; Ar2 = 2,6-Mes2-4-tBu-C6H2, X = Cl, Br) синтезированы и структурно охарактеризованы (схема 64 ) [73]. В то время как соединения [Ar1BiBr2]2, [Ar2BiCl2]2, [Ar2BiBr2]2 димерны в твердом состоянии, диодид висмута [Ar1BiI2]n − одномерный координационный полимер.
Схема 64 .
При гидролизе соединения [Ar2BiBr2]2 имеет место образование гидроксида арилбромвисмута Ar2Bi(OH)Br, который был выделен вместо ожидаемого монооргановисмутдигидроксида. Устойчивость Ar2Bi(OH)Br к дальнейшему гидролизу можно объяснить внутримолекулярными π-взаимодействиями висмут⋅⋅⋅арен. Реакция Ar2Bi(OH)Br со стерически перегруженной фосфиновой кислотой Ar2PH(O)(OH) дает фосфинаторгановисмут (Ar2HP(O)O)BiAr2Br. Рентгеноструктурный анализ данного соединения выявил необычную двойную π-внутримолекулярную координацию висмут⋅⋅⋅арен (схема 65 ).
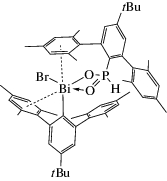
Схема 65 .
C целью исследования внутримолекулярных взаимодействий Bi···π-арен был синтезирован ряд соединений непереходных металлов, в состав которых входили объемные амидные лиганды [(R3Si)N(Ar*)] (Ar* = 2,6-(CHPh2)2-4-tBu-C6H2, R = Me, Ph), которые синтезировали по схеме 66 [74].
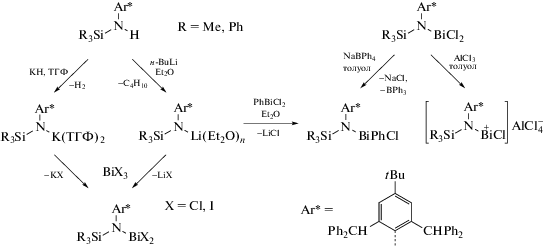
Схема 66 .
Наименьший контакт Bi···η6−π-арен наблюдается в катионном комплексе [(R3Si)N(Ar*)BiCl]+ -[AlCl4]− (2.85−2.98 Å). В остальных комплексах висмута расстояния Bi···C (η6−π-арен) приближаются к сумме ван-дер-ваальсовых радиусов висмута и углерода, указывающие на слабое взаимодействие между ними. Отметим, что подобный контакт наблюдался в 3,4,5-трифторбензоатном четырехъядерном комплексе висмута с толуолом Bi4(O)2(O2CC6H2F3-3,4,5)8 · 2(η6-C6H5Me), в котором расстояние Bi···C (η6-π-арен) достигало значения 3.02 Å [75].
Реакция Ph2BiCl с PhSLi или (2,6-Me2C6H3)SLi дает Ph2BiSPh и Ph2BiSC6H3Me2-2,6 соответственно [76]. Оба соединения охарактеризованы ИК-, Раман, ЯМР 1Н- и 13С-спектроскопией и РСА. Показано, что структура Ph2BiSPh является полимерной c межмолекулярными взаимодействиями Bi(1)⋅⋅⋅S(2) (3.309(1) Å) и длиной связи Bi−S 2.588(1) Å. При увеличении объема фенилтиолатного лиганда в Ph2BiSC6H3Me2-2,6 комплекс кристаллизуется в виде мономера.
Плохо растворимые в органических растворителях бесцветные кристаллы метоксида диметилвисмута [Me2BiOMe]∞ образуются при взаимодействии бензольного раствора триметилвисмута с кислородом воздуха (12 ч) [77]. По данным РСА, комплекс является координационным полимером, в котором MeO-лиганды соединяют фрагменты Me2Bi в цепи; длины связей Bi–O равны 2.359(6) и 2.344(6) Å, что больше суммы ковалентных радиусов Bi и O (2.18 Å [20]), а расстояния С−Bi (2.243(6), 2.243(6) Å) обычны для такого типа соединений [78].
В основе одного из методов синтеза некоторых висмуторганических соединений лежат реакции внедрения малых молекул. Так, показано, что в катионный висмутамид встраивается монооксид углерода по связи Bi–N в мягких условиях (схема 67 ) [79].
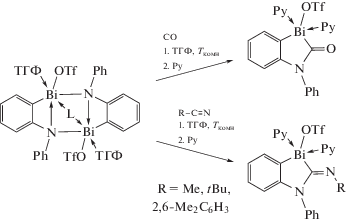
Схема 67 .
Комбинированный экспериментальный и теоретический подход позволил понять механизм введения СО, который можно распространить и на изонитрилы.
Найдено, что полученный из 1,8-бис((триметилсилил)амино)нафталина и трис(диметиламида)висмута амид висмута 1,8-C10H6(NSiMe3)2Bi–NMe2 реагирует с 2-бензоилпиридином, 3-пиридинкарбоксальдегидом, 2-метил-2-пропеннитрилом и диэтилацетилендикарбоксилатом с образованием продуктов присоединения по связи Bi–N (схема 68 ) [80]. Полученные соединения охарактеризованы РСА и спектральными методами анализа.
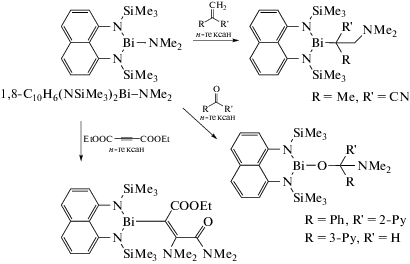
Схема 68 .
С помощью амида висмута 1,8-C10H6(NSiMe3)2Bi−NMe2 можно синтезировать производные трехвалентного висмута с такими углеводородными заместителями, как Me, C5Me5 и C≡CPh (схема 69 ) [81].
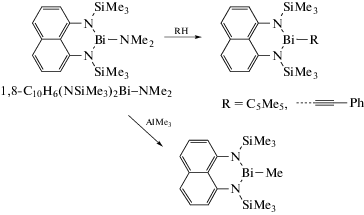
Схема 69 .
Строение соединений доказано спектроскопией ЯМР 1H, 13C и 29Si и РСА.
Показано, что для 1,2,4,3-триазаборол-3-ил-дифенилвисмута (LN3B)BiPh2 характерны реакции внедрения малых молекул арилизонитрилов (ArNC) и монооксида углерода по связи B−Bi (схема 70 ) [82].
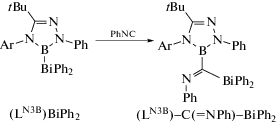
Схема 70 .
Исходное соединение (LN3B)BiPh2 получали из амидразона (схема 71 ), комплексы (LN3B)BiPh2 и (LN3B)−C(=NPh)−BiPh2 были охарактеризованы методом РСА.
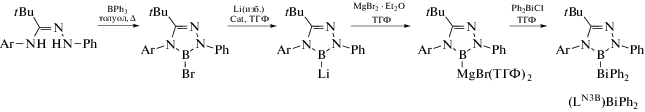
Схема 71 .
Изучена реакционная способность трибромида висмута по отношению к N-гетероциклическому карбену 1,3-бис(2,6-диизопропилфенил)-имидазол-2-илидену (Dipp-NHC) [83]. Показано, что добавление одного молярного эквивалента трибромида висмута к раствору IPr в диэтиловом эфире приводит к образованию аддукта (Dipp-NHC) ⋅ BiBr3, который мгновенно осаждается из реакционной смеси в виде ярко-желтых кристаллов c выходом 90% (cхема 72). Показано, что 1 : 1 аддукт (Dipp-NHC) ⋅ BiBr3 легко изомеризуется при нагревании до 75°C (12 ч) с образованием бесцветных кристаллов цвиттер-иона (схема 72 , внизу слева), а его реакция с бромидом алюминия к ионному комплексу [(Dipp-NHC)BiBr2]+[AlBr4]− (cхема 72, внизу справа).
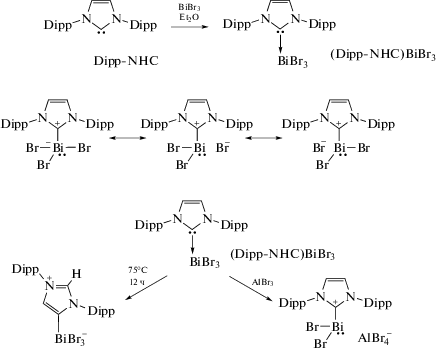
Схема 72 .
В [84] описаны синтез и структурные характеристики первых аддуктов N-гетероциклического карбена с хлоридом висмута (схема 73 ). По реакции N-гетероциклического карбена iPr−NHCMe с BiCl3 получено соединение (iPr−NHCMe)BiCl3 (схема 73 ), прибавление к раствору которого триметилсилилтрифторметансульфоната приводило к образованию аддукта (iPr−NHCMe)BiCl2(OTf)(THF), димерное строение которого в кристаллическом виде установлено с помощью РСА.
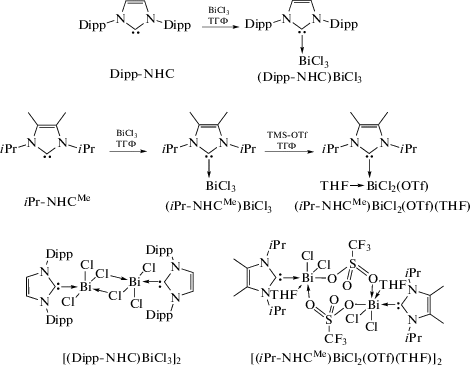
Схема 73 .
Описан синтез первых стабилизированных комплексами висмута циклических (алкил)(амино)карбенов (Et2CAAC)Bi(Ph)Cl2 и (CyCAAC)Bi(Ph)Cl2, которые получали из карбенов и дихлорида фенилвисмута (схема 74 ) [85].
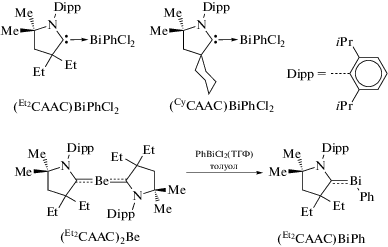
Схема 74 .
Комплексы висмута также могут быть получены депротонированием стабильных на воздухе солей [Et2CAAC−${\text{H]}}_{2}^{{2 + }}$[Cl2(Ph)Bi(μ-Cl2)Bi(Ph)Cl2]2− и [CyCAAC−${\text{H]}}_{2}^{{2 + }}$[Cl2(Ph)Bi(μ-Cl2)Bi(Ph)Cl2]2− с помощью бис(триметилсилил)амида калия K[N(SiMe3)2]. В другой работе этих же авторов сообщается о синтезе карбен-стабилизированного висмутиниденового комплекса (Et2CAAC)BiPh из комплекса бериллия (Et2CAAC)2Be, который используют в качестве восстанавливающего агента и реагента переноса лиганда (cхема 74) [86].
Аддукт (CDPPh)BiCl3 гексафенилкарбодифосфорана (CDPPh) с трихлоридом висмута получен в растворе ТГФ при комнатной температуре (cхема 75) [87]. Обработка (CDPPh)BiCl3 трифлатом TMS−OTf привела к образованию комплекса (CDPPh)BiCl(OTf)2.
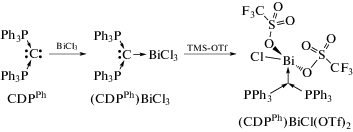
Схема 75 .
Все соединения охарактеризованыспектральными методамианализа и РСА.
Кинетическистабилизированныйаналог карбена, содержащий ион висмутения [(2,6-Mes2C6H3)2Bi]+[${\text{BAr}}_{4}^{{\text{F}}}$]− (Mes = 2,4,6-Me3C6H2, ArF = = 3,5-(CF3)2C6H3)) был получен из гидрида диарилвисмута и соли трифенилкарбения (cхема 76) [88].
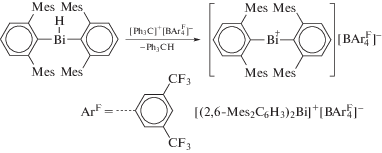
Схема 76 .
Для тяжелого бирадикала [Bi(µ-N(2,6-Mes2C6H3))]2 проведены реакции [2+2]-присоединения с использованием ацетилена или толана (cхема 77), в результате чего образуются гетероциклические соединения, строение которых доказано РСАF [89].
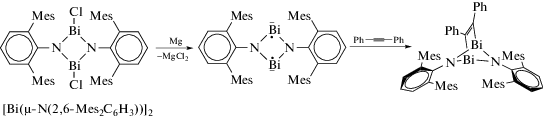
Схема 77 .
Весьма интересны реакции висмуторганических производных, сопровождающиеся образованием органических соединений неординарного строения. Так, в [90] была изучена реакционная способность аминовисмутана Mes*N(SiMe3)BiCl2 (Mes* = 2,4,6-три-трет-бу--тилфенил) с органическими производными металлов. Показано,что синтез Mes*N(SiMe3)BiCl2 с выходом 33% сопровождается образованием продукта сочетания двух молекул Mes*N(SiMe3)H с выходом 60% (cхема 78).
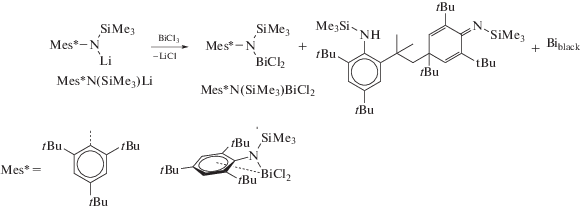
Схема 78 .
Реакция Mes*N(SiMe3)BiCl2 с GaCl3 привела к образованию только [Mes*N(SiMe3)BiCl]+[GaCl4]− (схема 79 ). Использование соли Ag[WCA] (WCA = = слабо координирующий анион) для связывания хлоридов позволило выделить продукт сочетания C–C. Также были синтезированы дииодид Mes*N(SiMe3)BiI2 и азидохлоридное соединение Mes*N(SiMe3)Bi(N3)Cl, однако синтез указанных производных не сопровождался образованием продуктов сочетания.
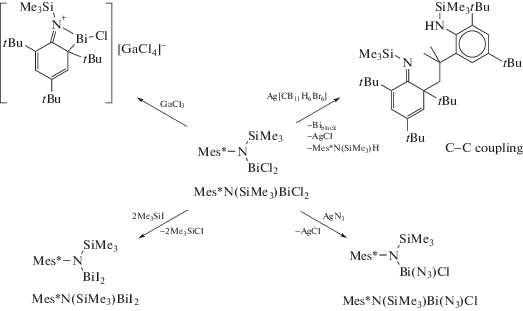
Схема 79 .
В [91] описаны синтез и свойства фосфинатов висмута(III). Так, при обработке трифенилвисмута перфторалкилфосфиновой кислотой имеет место отщепление одной или двух фенильных групп от атома висмута (схема 80 ).
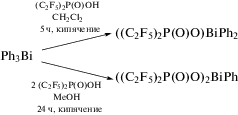
Схема 80 .
Приведены примеры успешного применения полученных фосфинатов висмута в реакциях образования углерод-углеродных связей (например, схема 81 ).
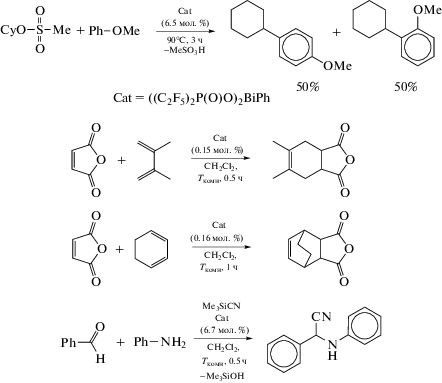
Схема 81 .
В [92] описаны реакции карбометаллирования смешением галогенида висмута, углеродного нуклеофила и ненасыщенного углеводорода, причем изменение типа галогена в соли висмута переключает региоселективность реакции (схема 82 ).
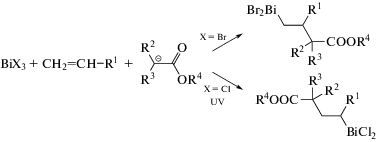
Схема 82 .
Необходимо отметить, что условия проведения реакций определяют количественный состав продуктов (схема 83 ).
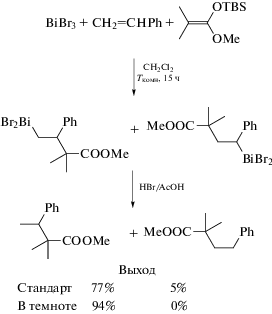
Схема 83 .
Карбовисмутинизация алкина была подтверждена РСА продукта реакции трибромида висмута с 3,5-ди(трет-бутил)фенилацетиленом и диметилкетентриметилсилилметилацеталем в хлористом метилене при комнатной температуре, когда единственным продуктом является дибромид моноалкенилвисмута,выделенныйиз реакционной смеси ввидебесцветныхкристаллов с количественнымвыходом(схема 84 ) [93].
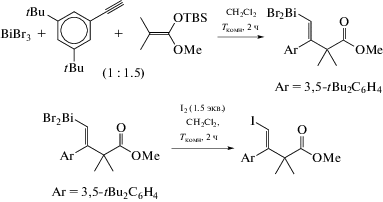
Схема 84 .
Рентгеноструктурный анализ данного дибромида моноалкенилвисмута выявил цис-конформацию висмута и ароматического заместителя при двойной связи; что подтверждает регио- и стереоселективный характер карбовисмутинирования. Кристалл состоит из тетрамеров, образующихся за счет бромидных мостиков. Геометрия атомов висмута − искаженная тригонально-бипирамидальная с атомами брома в аксиальных положениях. Алкенильная группа, атом бром и неподеленная электронная пара занимают экваториальные позиции. Показано, что алкенилвисмут легко реагирует с иодом, при этом образуется алкенилйодид с сохранением стереохимии.
В [94] описано превращение связей С−Н в более реакционноспособные связи С−М, поддающиеся дальнейшей функционализации, что имеет фундаментальное значение в синтетической химии. Показано, что преобразование нейтральных соединений висмута в их катионные аналоги может быть использовано в качестве стратегии для облегчения реакций активации связей C−H (схема 85 ).
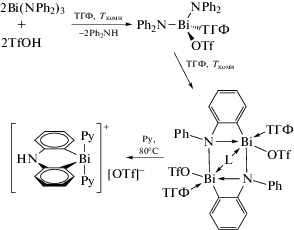
Схема 85 .
Металлоорганические продукты первого и второго этапов активации связей С−Н были выделены с высоким выходом. Данные РСА и DFT-расчеты показали необычные свойства основного состояния этих соединений (кольцевая деформация и умеренная гетероароматичность).
Синтез соединений висмута с полидентатными арильными лигандами. В последнее время интенсивно развивается химия арильных соединений висмута с полидентатными лигандами при центральном атоме металла. Наиболее простыми представителями данного класса являются β-дикетонатные производные висмута, которые можно синтезировать из трифенилвисмута и β-дикетона. Так, были получены первые примеры F-содержащих дикетонатных комплексов арилвисмута − бис(гексафторацетилацетонат) фенилвисмута(III) (Нfac)2BiPh и его аддукты (Нfac)2BiPh(L) (HfacH = 1,1,1,5,5,5-гексафтор-2,4-пентандион; L = H2O, Me2CO, THF, DMAA (N,N-диметилацетамид), ДМСО, PhCN, а также смешанный комплекс гексафторацетилацетонатотрифторацетат фенилвисмута, [(Нfac)BiPh(CF3COO)]2 (схема 86 ), которые охарактеризованы РСА [95].
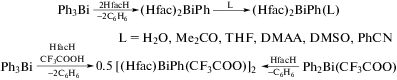
Схема 86 .
Комплекс (Нfac)2BiPh выделяют из реакции трифенилвисмута с 1,1,1,5,5,5-гексафтор-2,4-пентандионом (мольное соотношение 1 : 2) в сухом гексане. Соединение [(Нfac)BiPh(O2CCF3)]2 было синтезировано из эквимолярных количеств Ph3Bi, HfacH и CF3COOH, по второму методу использовали трифторацетат дифенилвисмута Ph2BiO2CCF3 и β-дикетон (Hhfac). Попытки вырастить монокристаллы (Нfac)2BiPh из растворов некоординирующих растворителей оказались безуспешными. Однако в присутствии координирующих растворителей (Нfac)2BiPh образует желтые кристаллы соответствующих аддуктов (Нfac)2BiPh(L), выделяющихся из гексанового раствора (Нfac)2BiPh в присутствии малых количеств H2O, Me2CO, THF, DMAA, DMSO и PhCN. Все выделенные комплексы чувствительны к воздуху, умеренно растворимы в метаноле, ацетоне, дихлорметане и хлороформе, в меньшей степени – в диэтиловом эфире и углеводородах. Полученные соединения охарактеризованы ИК-, ЯМР-спектроскопией и РСА. В ИК-спектрах, как и ожидалось, наблюдаются полосы, отвечающие валентным колебаниям групп C=O в области 1634–1640 см−1, что значительно отличается от аналогичной полосы, наблюдаемой в ИК-спектре свободного HfacH (1689 см−1), и указывают на хелатирующий характер лиганда. Из данных РСА следует, что конфигурация атома металла в аддуктах (Нfac)2BiPh(L) представляет собой пятиугольную пирамиду. Двухъядерный комплекс [(Нfac)BiPh(CF3COO)]2 состоит из двух искаженных пятиугольных пирамид, соединенных в димеры с помощью мостиковых карбоксилатных групп.
Несколько большим числом в литературе представлены соединения висмута, содержащие бидентатные C,N-лиганды у атома висмута, которые, как правило, синтезируют из галогенидов висмута и производных активных металлов. Например, бромид (2-(iPr2NCH2)C6H4)2BiBr был получен из реактива Гриньяра и трихлорида висмута (схема 87 ) [96].
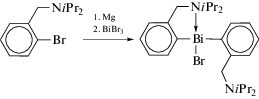
Схема 87 .
Соединение (2-(iPr2NCH2)C6H4)2BiBr было охарактеризовано методами многоядерной ЯМР-спектроскопии, масс-спектрометрии и РСА. Длина связи Bi−Br (2.7294(10) Å) короче, чем у других родственных производных R2BiBr (для R = = 2-(Me2NCH2)C6H4 2.8452(7), 2-(Et2NCH2)C6H4 2.7517(10), 2.7484(10), 2.8084(11) Å) [78]. Один из атомов азота не координирован с центральным атомом металла, однако расстояние между вторым атомом азота и атомом висмута Bi⋅⋅⋅N (2.737(6) Å) несколько больше суммы их ковалентных радиусов (2.19 Å, [20]), но существенно меньше суммы ван-дер-ваальсовых радиусов указанных элементов (3.94 Å, [20]), что указывает на взаимодействие между ними. Валентный угол BrBiN (164.80(13)°) меньше идеального значения 180° и сопоставим c найденными в соединениях R2BiBr.
С помощью комбинации выше указанных методов были получены хлориды диарилвисмута [13]. Так, реакция RLi или RMgBr (R = 2-(Et2NCH2)C6H4) с трихлоридом висмута в молярном соотношении 2 : 1 дает R2BiCl и R2BiBr соответственно (схема 88 ); по реакции перераспределения радикалов может быть получен дихлорид арилвисмута RBiCl2, в котором атомы хлора легко могут быть заменены на атомы брома или иода действием водных растворов бромистого или иодистого калия.
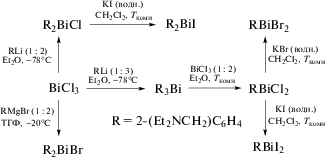
Схема 88 .
В полученных моногалогенидах один азот координируется с атомом висмута (2.557(8)− 2.645(6) Å), тогда как второй практически не координирован с центральным атомом металла (2.992(12)–3.170(8) Å). Общее ядро (C,N)2BiX (X = = Cl, Br, I) имеет искаженную квадратно-пирамидальную форму.
Аналогичносинтезировали бромиды R(C6F5)BiBr, R(Mes)BiBr иR(Ph)BiBr(R = 2-(Me2NCH2)C6H4) из эквимолярныхколичествRBiBr2 и C6F5MgBr, MesMgBr или PhMgBr илииз PhBiBr2 и RLi в молярномсоотношении 1 : 1 (схема 89 ) [12].
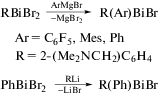
Схема 89 .
По сходным схемам были получены и структурно охарактеризованы бис(2-((диметиламино)- метил)фенил)азидовисмут (2-(Me2NCH2)C6H4)2BiN3 и диазид (2-(диметиламинометил)фенил)висмута (2-(Me2NCH2)C6H4)Bi(N3)2 [14]. В качестве промежуточного продукта использовали триарилвисмут, из которого по реакции перераспределения радикалов получали соответствующие хлориды арил- и диарилвисмута. Последние действием избытка иодида натрия в тетрагидрофуране превращали в иодиды, обработка которых азидом серебра приводила к целевым продуктам (схема 90 ).
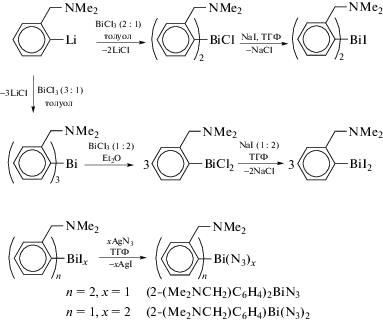
Схема 90 .
Полученные соединения представляют собой редкие примеры азидов висмута. В то время как в кристалле моноазида (2-(Me2NCH2)C6H4)2BiN3 присутствовали мономерные молекулы с координацией атомов азота аминогрупп на центральный атом (2.555(2), 3.131(3) Å), то диазид (2-(Me2NCH2)C6H4)Bi(N3)2, в котором координация атома азота аминогруппы с центральным атомом металла весьма существенна (2.568(2) Å), представлен в виде димера с двумя типами соединения азидогрупп. Кроме того, слабые ван-дер-ваальсовы взаимодействия между этими центросимметричными димерами приводят к цепочечной структуре в кристалле.
Триоргановисмутины R(C6F5)2Bi и R[2,4,6-(C6F5)3C6H2]2Bi, где R = 2-(Me2NCH2)C6H4, были синтезированы из RBiBr2 и C6F5MgBr или 2,4,6-(C6F5)3C6H2Li соответственно в молярном соотношении 1 : 2 (схема 91 ) [12].
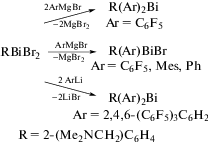
Схема 91 .
Аналогичным образом были получены бромиды R(C6F5)BiBr, R(Mes)BiBr и R(Ph)BiBr из RBiBr2 и C6F5MgBr, MesMgBr, PhMgBr или из PhBiBr2 и RLi в эквимолярном соотношении (схема 91 ). Молекулярные структуры данных соединений определены методом РСА. В хиральных бромидах R(C6F5)BiBr, R(Mes)BiBr и R(Ph)BiBr (R = 2-(Me2NCH2)C6H4) в твердом состоянии наблюдается сильная внутримолекулярная координация N → Bi. Расстояния N–Bi в данных соединениях примерно одинаковы, что указывает на отсутствие влияния второго органического заместителя (C6F5, Ph, Mes) на длину координационной связи N⋅⋅⋅Bi.
Хлориды (2-{E(CH2CH2)2NCH2}C6H4)2BiCl, где E = O, MeN, и дихлориды (2-{E(CH2CH2)2NCH2}C6H4)BiCl2, где E = O, MeN, синтезированы по реакции соответствующего орто-литиевого производного с трихлоридом висмута в соответствующих молярных соотношениях (схема 92 ) [23].
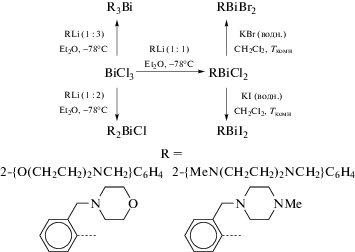
Схема 92 .
Дигалогениды (2-{E(CH2CH2)2NCH2}C6H4)BiX2, где X = Br, E = O, MeN; X = I, E = O, MeN, а также (2-(Me2NCH2)C6H4)BiBr2 получены реакциями галогенидного обмена между RBiCl2 и избытком водного раствора KX. Во всех соединениях атомы азота координированы с атомами висмута. В монохлоридах один атом азота сильно координирован с атомом висмута (2.660(11) Å в (2-{O(CH2CH2)2NCH2}C6H4)2BiCl и 2.744(14) Å в (2-{MeN(CH2CH2)2NCH2}C6H4)2BiCl), тогда как второй участвует в слабом внутримолекулярном взаимодействии N → Bi (3.095(11) Å в (2-{O(CH2CH2)2NCH2}C6H4)2BiCl и 3.061(14) Å в (2-{MeN(CH2CH2)2NCH2}C6H4)2BiCl). В целом ядро (C,N)2BiCl тетрагонально-пирамидальное. Кристаллы дигалогенидов (2-{O(CH2CH2)2NCH2}C6H4)2BiCl2 и (2-(Me2NCH2)C6H4)BiBr2 содержат дискретные димерные единицы. Атом азота аминогрупп в (C,N)BiX2 (X = Cl, Br) ядра координирован на атом металла (2.548(9) Å в (2-{O(CH2CH2)2NCH2}C6H4)2BiCl2 и 2.485(13) Å в (2-(Me2NCH2)C6H4)BiBr2). В кристаллах данных двух соединений и (2-{O(CH2CH2)2NCH2}C6H4)2BiCl образуются супрамолекулярные архитектуры, основанные на межмолекулярных взаимодействиях Bi⋅⋅⋅Br, Cl⋅⋅⋅H и Br⋅⋅⋅H. Шестичленные морфолиновые и пиперазиновые кольца принимают конформацию стула, которая препятствует внутримолекулярной координации атомов кислорода или азота на атом висмута.
Четыре устойчивых к воздуху гипервалентных соединения органовисмута R2BiCl с (C,O)- или (C,S)-хелатирующими лигандами, где R = = 2‑(MeECH2)С6Н4 (E = O или S), а также (2‑(MeOCH2)C6H4)2Bi(OTf) и [{2-(MeSCH2)-C6H4}2Bi]+[OTf]– получены из трихлорида висмута и литийарила (cхема 93) [97].
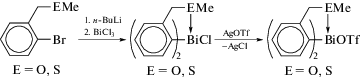
Схема 93 .
В отличие от первых трех соединений, ионный комплекс [{2-(MeSCH2)C6H4}2Bi]+[OTf]− состоит из висмутсодержащих катионов и трифлатных анионов. Соединение [2-(MeOCH2)C6H4]2Bi(OTf) показало хорошую каталитическую эффективность и возможность повторного использования в реакциях аллилирования различных альдегидов тетрааллилоловом в метаноле (или ТГФ, MeCN, EtOH, Tкомн, 1 ч) для получения соответствующих гомоаллильных спиртов с выходом до 96% (схема 94 ).
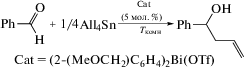
Схема 94 .
Взаимодействием хлоридов органовисмута(III) R2BiCl и RBiCl2, где R = 2-(Me2NCH2)C6H4, с псевдогалогенидами щелочных металлов в молярном соотношении 1 : 1 и 1 : 2 соответственно, получены соединения гипервалентного висмута R2BiX (X = NCO, SeCN) и RBiX2 (X = NCO, NCS и SeCN) (схема 95 ) [98].
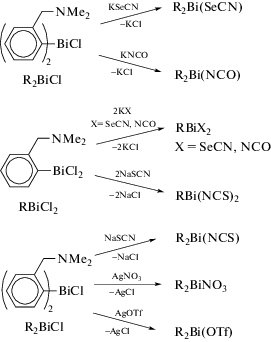
Схема 95 .
Определена молекулярная структура соединений R2Bi(NCO), R2Bi(SeCN). Во всех комплексах атомы азота аминогрупп участвуют во внутримолекулярной координации с металлом, что приводит к искаженной квадратной координационной геометрии атома висмута.
Реакции эквимолярных количеств хлорида R2-BiCl, где R = 2-(Me2NCH2)C6H4, с солями натрия или серебра (NaSCN, AgOTf или AgNO3) приводят к замещению атома хлора на другой электроотрицательный заместитель и образованию производных висмута R2Bi(NCS), R2Bi(OTf) и R2BiNO3 соответственно (схема 95 ). Структуры данных соединений были определены методом РСА [99]. Оба атома азота аминогрупп участвуют во внутримолекулярной N⋅⋅⋅Bi координации разной силы. Для изотиоцианата R2Bi(NCS) это приводит к искаженному квадратно-пирамидальному ядру (C,N)2BiN. В случае трифлата R2Bi(OTf) и нитрата R2BiNO3 оксоанионы сильно координированы к асимметрично атому Bi через атомы кислорода (Bi⋅⋅⋅O 2.337(12)− 3.317(15) Å в R2Bi(OTf) и 2.476(5)−3.088(5) Å в R2BiNO3). Таким образом, для соединений R2Bi(OTf) и R2BiNO3 наблюдается пентагонально-пирамидальная координация атома висмута.
Хлорид мезитил(2-диметиламинометилфенил) висмута R(Mes)BiCl, где R = 2-(Me2NCH2)C6H4, получен из дихлорида мезитилвисмута (схема 96 ) [100].
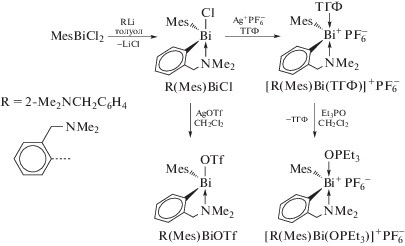
Схема 96 .
Комплекс R(Mes)BiCl при обработкетрифлатом серебра превращается в R(Mes)BiOTf. Последовательные реакции R(Mes)BiCl с AgPF6 и Et3PO приводят к синтезу [R(Mes)Bi(OPEt3)]+${\text{PF}}_{6}^{ - }$, в котором связь Bi−N (2.501(5) Å) более длинная, чем в R(Mes)BiOTf (2.446(2) Å).
Неоднозначность реакций литийорганических соединений с трихлоридом висмута показана в [101]. Так, взаимодействие эквимолярных количеств BiCl3 с (Dipp−NacNac)Li (Dipp-NacNac = Dipp−N−C(Me)=CH−C(Me)=N−Dipp, Dipp = 2,6-iPr2C6H3) в различных условиях приводили к синтезу разнообразныхсоединений висмута:[(Dipp−NacNac)BiCl2]2, [(Dipp−NacNac)Bi,Cl(μ-Cl)Bi(nBu)Cl(μ-Cl)]2, [LBiCl(μ-Cl)]2 (L =N(Ar)=C(Me)CH=C(NHAr)CH2) и L'Bi2Cl4 (L' = N(Ar)=C(Me)CC(Me)=N(Ar)) (cхема 97).
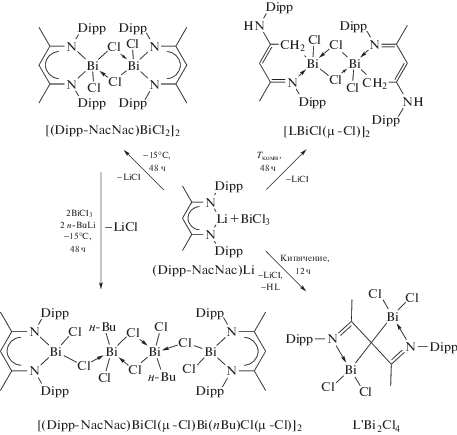
Схема 97 .
Соединения [(Dipp−NacNac)BiCl2]2 и [LBiCl(μ-Cl)]2 являются изомерами, итермическая конверсия от[(Dipp−NacNac)BiCl2]2 в [LBiCl(μ-Cl)]2 была реализована.В этойреакционной системе при небольшомизбытке н-BuLi и BiCl3 выделяли [(Dipp−NacNac)BiCl(μ-Cl)Bi(н-Bu)Cl(μ-Cl)]2 как побочныйпродуктпосле выделения [(Dipp−NacNac)BiCl2]2. Строение комплексов подтверждено данными спектроскопии ЯМР 1H и 13C и рентгеновской кристаллографии.
Дихлорид арилвисмута LBiCl2 (L = o-(CH=N-2,6-iPr2C6H3)C6H4]) реагирует с дифенилдихалькогенидом PhEEPh (E = S, Se или Te) с образованием соответствующих комплексов LBi(EPh)2 (E = = S, Se, Te), из которых в различных условиях можно синтезировать производные висмута с одной или двумя группами EPh (cхема 98) [102].
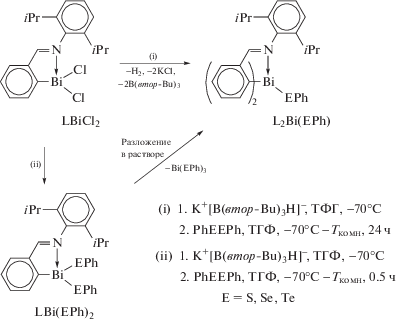
Схема 98 .
Устойчивость соединений L2Bi(EPh) обусловлена жесткой координацией обоих атомов-доноров азота лиганда L с атомом висмута.
По сходной схеме из эквимолярных количеств LLi, где L = 2,6-(MeN(CH2CH2)2NCH2)2C6H3, и BiCl3 былполучен дихлорид арилвисмута LBiCl2, содержащий(N,C,N)-лиганд(схема 99 ). LBiBr2 и RBiI2 былиполучены реакциями обмена галогена изLBiCl2 [103].
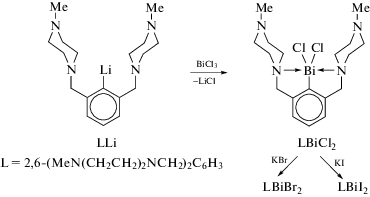
Схема 99 .
Данные дигалогениды арилвисмута(III) LBiHal2 были охарактеризованы как в растворе, так и в твердом состоянии. Молекулярное строение соединений в кристаллическом виде установлено методом РСА. Все они имеют Т-образное ядро CBiHal2, стабилизированное двумя сильными внутримолекулярными взаимодействиями N → Bi в транс-положениях друг к другу. Общее (N,C,N)BiHal2-ядро имеет искаженную квадратно-пирамидальную координационную геометрию с арильным лигандом в вершине. ЯМР-спектроскопические исследования подтверждают наличие внутренней координации азот−висмут в растворе.
Дихлорид арилвисмута вида L*BiCl2, содержащий тридентатный N,C,N-лиганд L* (схема 100 ), использовали в качестве прекурсора для синтеза мономерного соединения висмута L*Bi [104].
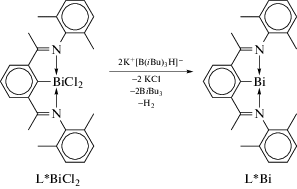
Схема 100 .
Реакция L*BiCl2 с двумя эквивалентами K[B(iBu)3H] в ТГФ протекает с изменением цвета реакционной смеси на темно-синий, при этом заметно выделение газа, что указывало на образование нестабильного гидрида LBiH2, который сразу теряет водород. Соединение L*Bi выделяли кристаллизацией из насыщенного гексанового раствора в виде темно-синего микрокристаллического порошка с выходом 35%. Соединения охарактеризованы элементным анализом и спектрами ЯМР 1Н и 13С в дейтерированном бензоле, в которых присутствовали сигналы, соответствующие лиганду L*.
С целью сравнения координационной химии Bi3+ и лантанидов Ln3+, имеющих аналогичные размеры ионов, исследованы некоторые реакции хлорида диарилвисмута с (N,C,N)-лиган-дами (2,6-(Me2NCH2)2C6H3)2BiCl (L2BiCl) (схема 101 ) [6].
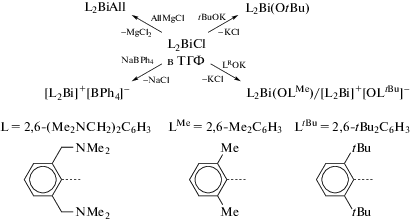
Схема 101 .
Комплекс L2BiCl (L = 2,6-(Me2NCH2)2C6H3) реагирует с tBuOK и LMeOK с образованием алкоксида L2Bi(OtBu) и арилоксида L2Bi(OLMe) соответственно, но аналогичная реакция с калиевой солью более объемного фенола LtBuOK приводила к образованию ионного комплекса [L2Bi]+[OLtBu], в котором арилоксидный лиганд действует как анион внешней сферы. Заместитель Cl удаляется из L2BiCl с помощью NaBPh4, при этом образуется другой ионный комплекс [L2Bi]+[BPh4]−.
В работе [105] описаны синтез и молекулярные структуры производных висмута c O,C,O-лигандом (4-tBu-2,6-[(EtO)2P=O]2C6H2)BiCl2 и (4-tBu-2,6-[(EtO)2P=O]2C6H2)BiCl (cхема 102).
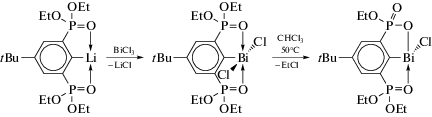
Схема 102 .
Соединение (4-tBu-2,6-[(EtO)2P=O]2C6H2)BiCl2 кристаллизуется в триклинной пространственной группе с двумя парами кристаллографически независимых молекул на элементарную ячейку. Каждый атом висмута имеет искаженную октаэдрическую конфигурацию CCl2O2Bi с атомами хлора и кислорода в транс-положении. Внутримолекулярные расстояния Bi⋅⋅⋅O находятся в диапазоне от 2.378(5) до 2.414(5) Å. Производное фосфависмола (4-tBu-2,6-[(EtO)2P=O]2C6H2)BiCl образует димер по типу “голова к хвосту” через межмолекулярные связи Bi⋅⋅⋅O (2.426(2), 2.278(3) Å) (cхема 103).
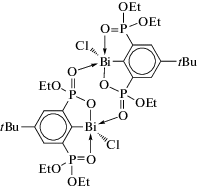
Схема 103 .
Расчеты DFT показывают высокий s-характер неподеленных электронных пар на атомах висмута в (4-tBu-2,6-[(EtO)2P=O]2C6H2)BiCl2 и (4-tBu-2,6-[(EtO)2P=O]2C6H2)BiCl.
Взаимодействие эквимолярных количеств ариллития LLi, L = 2-(Me2NCH2)-6-(MeOCH2)-C6H3, с трихлоридом висмута привело к образованию соответствующего дихлорида арилвисмута LBiCl2 c хорошим выходом (cхема 104) [106].
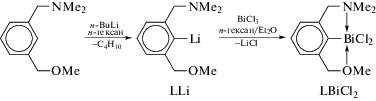
Схема 104 .
Новый хелатирующий лиганд L, 2-(Me2NCH2)-6-(MeOCH2)-C6H3, получали из м-толуолнитрила. Последующее литирование этого лиганда бутиллитием в гексане и прибавление к реакционной смеси трихлорида висмута завершилось синтезом целевого продукта, строение которого доказано методом РСА.
В нескольких публикациях описан синтез и особенности строения производных висмута с C,E,C-лигандами (E = N, O, S). Так, несколько соединений циклических хлоридов и трифенилгермилпропионатов висмута(III), [(C6H4CH2)2X]BiCl и [(C6H4CH2)2X]BiOC(O)CH2CH2GePh3 (X = S или NR с атомом азота или серы в качестве дополнительного внутримолекулярного координирующего центра) были синтезированы из дилитиевого производного и хлорида висмута (cхема 105) [107].
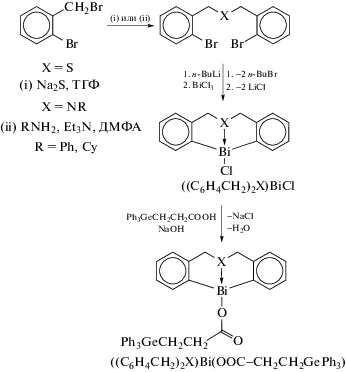
Схема 105 .
Результаты РСА показывают, что длины связи Bi–S или Bi–N в тиабисмоцине или азабисмоцине в восьмичленных циклах зависят от природы замещенных групп при атомах Bi. Замена атома хлора в азависмоцине и тиависмоцине на трифенилгермилпропионовую группу (Ph3GeCH2CH2COO) приводит к удлинению связей Bi–N и Bi–S. Обнаружено, что соединения проявляют более высокую антипролиферативную активность в отношении клеток карциномы желудка, чем у цисплатина. Более того, наблюдается усиление антипролиферативной активности, когда атом хлора бисмоциновых соединений заменяется на трифенилгермилпропионовый заместитель.
Несколько других соединений диоргановисмута(III), например [(C6H4CH2)2S]BiX на основе гетероциклического каркаса типа бабочки − тетрагидродибензо[c,f][1,5]-тиависмоцина (схема 106 ) − также были получены и структурно охарактеризованы [108].
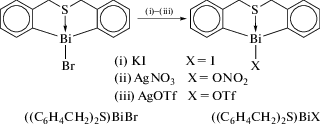
Схема 106 .
Реакция между дилитиевым производным бис(2-бромбензил)сульфида с трибромидом висмута в мольном соотношении 1 : 1 привелa к образованию ((C6H4CH2)2S)BiBr. Дальнейшие обменные реакции [(C6H4CH2)2S]BiBr с KI, AgNO3 и AgOTf, соответственно, дали соединения гипервалентного висмута [(C6H4CH2)2S]BiX (X = I, ONO2 и OTf). Во всех соединениях атом серы внутримолекулярно координирован с висмутом, межмолекулярные взаимодействия X⋅⋅⋅НС, Bi⋅⋅⋅Ar и Bi⋅⋅⋅O приводят к образованию полимерных цепей в кристаллах.
В другой работе этих авторов сообщается о синтезе бромидов диоргановисмута(III) [(C6H4CH2)2NR]BiBr (R = C6H5CH2, C6H5CH2CH2 и MeOCH2CH2), содержащих гетероциклический каркас дибензо[1,5]азависмоцина, из соответствующего дибромида (2-Br-C6H4CH2)2NR последовательными реакциями, включая орто-литирование и обработку дилитиевого производного бромидом висмута в молярном соотношении 1 : 1 (Схема 107 ) [109].
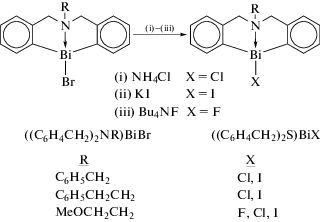
Схема 107 .
Дальнейшие обменные реакции между бромидами и соответствующими галогенидами металлов или фторидом аммония (схема 107 ) приводили к образованию [(C6H4CH2)2NR]BiX, где R = = C6H5CH2, X = Cl, I; R = C6H5CH2CH2, X = Cl, I; и R = MeOCH2CH2, X = F, Cl и I. Все десять соединений были охарактеризованы методами ЯМР и РСА. Сильные трансаннулярные взаимодействия N → Bi наблюдались во всех исследованных галогенидах диоргановисмута(III). Молекулы связаны в димеры сильными взаимодействиями Bi⋯π-арен в [(C6H4CH2)2N(CH2CH2OMe)]BiBr, [(C6H4CH2)2N(CH2C6H5)]BiCl и [(C6H4CH2)2N-(CH2CH2OMe)]BiI (около 3.50 Å) и взаимодействиями Bi⋯X в [(C6H4CH2)2N(CH2C6H5)]BiBr, [(C6H4CH2)2N(CH2C6H5)]BiI и [(C6H4CH2)2N-(CH2CH2C6H5)]BiI.
Два прекурсора асимметричного тридентатного C,E,C-хелатирующего лиганда 1-Br-2-[(2'-BrC6H4CH2E)CH2]C10H6 (E = O, S) были получены с большим выходом (схема 108 ) [110].
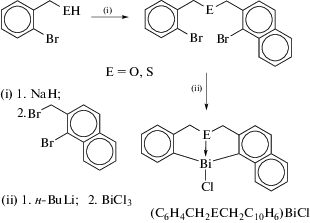
Схема 108 .
После их литирования бутиллитием и обработки треххлористым висмутом получены два хлорида гипервалентного висмута с асимметричным C,E,C-хелатным лигандом (C6H4CH2OCH2C10H6)BiCl и (C6H4CH2SCH2C10H6)BiCl (E = O, S). Рентгеноструктурный анализ соединений выявил, что донорные атомы (O, S) сильно координированы с атомами висмута.
Отметим, что некоторые из соединений висмута с С,E,C-лигандами являются эффективными катализаторами различных реакций органического синтеза. Например, стабильный на воздухе трифлатный комплекс органовисмута {[(C6H4CH2)2O]Bi(H2O)}+[OTf]− с дибен-зо[1,5]ок-сависмоциновым каркасом проявляет высокую каталитическую активность по отношению к открытию кольца в реакциях эпоксидов в водных средах с ароматическими аминами при комнатной температуре (схема 109 ) [111].
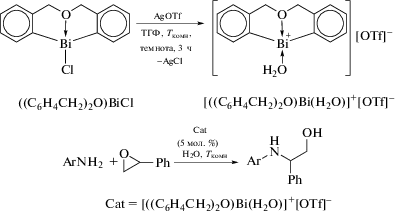
Схема 109 .
Этот катализатор демонстрирует хорошую стабильность, пригодность к регенерации и повторному использованию. Каталитическая система обеспечивает простой и эффективный способ синтеза β-аминоспиртов с выходом до 93%.
В другой работе сообщается о синтезе двухъядерных органовисмутовых комплексов {[(C6H4CH2)2NR]Bi}2E (E = O, S; R = tBu, Cy, Ph) с двумя дибензо[1,5]азависмоциновыми каркасами, сшитыми через атом серы или кислорода путем обработки хлоридов органовисмута гидроксидом натрия или Na2S ⋅ 9H2O (схема 110 ) [112].

Схема 110 .
Комплексы {[(C6H4CH2)2NR]Bi}2E показывают высокую каталитическую эффективность в синтезе циклических карбонатов из 2-(хлорметил)оксирана и СО2, наибольшую активность из которых проявляет комплекс {[(C6H4CH2)2NtBu)Bi]2S (схема 111 ).
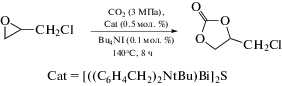
Схема 111 .
По сравнению с их предшественниками − хлоридом [(C6H4CH2)2NR]BiCl, метоксидом [(C6H4CH2)2NtBu]BiOMe и метантиолатом [(C6H4CH2)2NtBu]BiSMe, которые являются одноядерными органовисмутовыми комплексами, двухъядерные комплексы органовисмута показывают более высокую каталитическую активность. Однако комплексы с кислородным мостиком [((C6H4CH2)2NR)Bi]2O не стабильны на воздухе и теряют свою каталитическую эффективность из-за гидролиза или адсорбции СО2 (с образованием карбонатов органовисмута в последнем случае). Тем не менее двуядерные комплексы органовисмута с серным мостиком {[(C6H4CH2)2NR]Bi}2S очень устойчивы на воздухе и могут применяться при синтезе циклических карбонатов (в присутствии Bu4NI) через различные виды эпоксидов, демонстрируя удовлетворительную эффективность и селективность.
Ионные комплексы, содержащие С,S,C-лиганды, проявляют высокую каталитическую активность в реакциях Манниха для получения α,β-ненасыщенных кетонов [113]. Прекурсором для получения катализатора является хлорид органовисмута [(C6H4CH2)2S]BiCl, который синтезируют из лиганда в форме бабочки с серой, н-BuLi и треххлористого висмута в диэтиловом эфире. Комплекс [(C6H4CH2)2S]BiCl содержит тридентатный лиганд, в котором атом серы имеет две пары электронов: одна координируется с центром висмута, а другая свободна. Из этого соединения были синтезированы комплексы с противоанионом общего вида {[(C6H4CH2)2S]Bi(H2O)}+X− (X− = ${\text{ClO}}_{4}^{ - }$, ${\text{BF}}_{4}^{ - }$, ${\text{OS}}{{{\text{O}}}_{2}}{{{\text{C}}}_{4}}{\text{F}}_{9}^{ - }$, ${\text{OS}}{{{\text{O}}}_{2}}{{{\text{C}}}_{8}}{\text{F}}_{{17}}^{ - }$), которые показывали высокую электроноакцепторную способность и проявляли свойства эффективных катализаторов в реакции Манниха (схема 112 ).
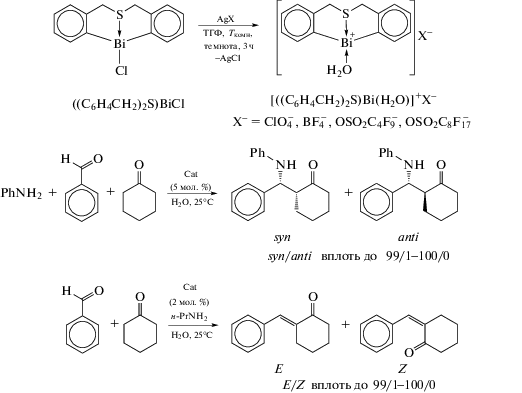
Схема 112 .
При этом наблюдается высокая диастереоселективность, продукты транс-конформации выделяются из реакционной смеси с выходом до 99%.
N,C,N-хелатные комплексы висмута с объемными замещенными бис(дифенил(арилимино) фосфорано)метановыми заместителями H2C(Ph2PN- Dipp)2 могут быть получены из трихлорида висмута и соответствующих соединений лития (схема 113 ) [114].
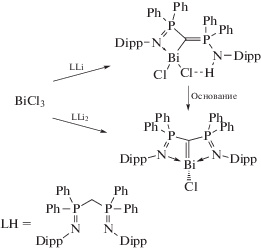
Схема 113 .
Комплексы охарактеризованы с помощью РСА и ЯМР-исследований. Обнаружено, что дианионный комплекс имеет редкий структурный мотив формальной двойной связи углерод-висмут(III).
Во многих работах, посвященных синтезу арильных производных трехвалентного висмута, в качестве исходного органического соединения металла используют оксиды арилвисмута, содержащие в своем составе тридентатные арильные лиганды. Так, взаимодействие RBiCl2, (R = 2,6-MeN(CH2CH2)2NCH2]2C6H3) с КОН приводит к образованию оксида цикло-R2Bi2O2 (схема 114 ) [115]. Циклический оксид способен улавливать газообразный CO2 с образованием “RBiCO3”. Реакция дихлорида RBiCl2 с этиленгликолем, пинаколом или пирокатехином (CatH2) в присутствии КОН приводит к образованию 2-органо-1,3,2-диоксависмоланов RBi(OCH2)2, RBi(OCMe2)2 и к 2‑органо-1,3,2-диоксависмолу RBi(Cat) соответственно.
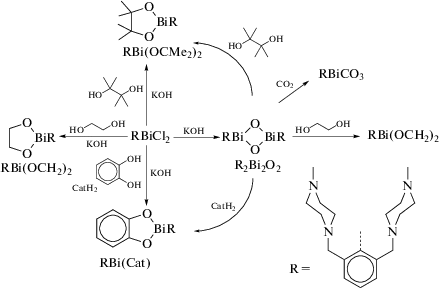
Схема 114 .
Строение полученных соединений исследовано методами ЯМР-спектроскопиии и РСА. Выявлено, что органическая группа R действует как хелатный N,C,N-лиганд. Непланарность пятичленных хелатных циклов BiC3N объясняется внутримолекулярными взаимодействиями N → Bi. Молекулы соединений R2Bi2O2, RBi(OCH2)2 и RBi(Cat), независимо от природы оксолиганда, имеют искаженную квадратно-пирамидальную форму.
Реакция висмуторганического оксида (LBiO)2 (L = [2,6-бис(диметиламино)метил]фенил) с борорганическими кислотами (1 : 4 мольн.) дает гетеробороксины LBi[(OBR)2O], R = Ph, 4-CF3C6H4 и Fc (схема 115 ) [116].
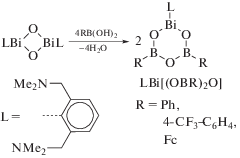
Схема 115 .
Соединения охарактеризованы методами элементного анализа и ЯМР-спектроскопии. Их структура описана как в растворе (исследования ЯМР), так и в твердом состоянии. По данным РСА, все комплексы содержат центральный фрагмент BiB2O3, неароматический характер которого подтвердили DFT-расчетами.
Синтез и строение висмагетеробороксинов общей формулы LBi[(OBR)2O] с N,C,N-хелатирующим лигандом L = 2,6-(Me2NCH2)2C6H3 (схема 116 ) описаны в [117]. Целевые соединения получали из оксида (LBiO)2 и соответствующей борорганической кислоты (1 : 4 мольн.).
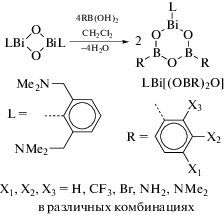
Схема 116 .
Гетеробороксин LBi[(OBR)2O], содержащий донорную группу (R = 4-пиридил) в борной кислоте, был синтезирован по аналогичной схеме (схема 117 ).
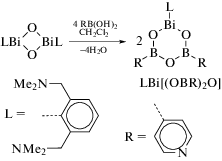
Схема 117 .
Полученные соединения были охарактеризованы методами многоядерной ЯМР-спектроскопии и РСА.
Взаимодействием N,C,N-внутримолекулярнокоординированного оксида висмута(III) (LBiO)2, где L = 2,6-(Me2NCH2)2C6H3), с (HO)SiPh2OSiPh2(OH) при молярном соотношении 1 : 2 получен цикло-LBi[(OSiPh2)2O], содержащий шестичленный цикл BiSi2O3 (схема 118 ) [118].
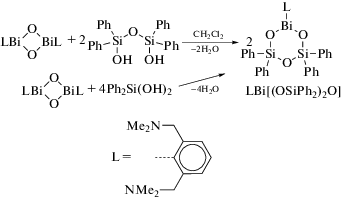
Схема 118 .
Альтернативно, цикло-LBi[(OSiPh2)2O] может быть получен из дигидроксида дифенилолова Ph2Si(OH)2 и (LBiO)2 при молярном соотношении 4 : 1. Соединение (LBiO)2 реагирует с циклосилоксаном (Me2SiO)3 с образованием шестичленного висмутсилоксана цикло-LBi(OSiMe2)2O (схема 119 ).
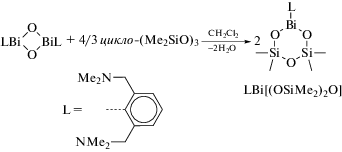
Схема 119 .
Соединения были охарактеризованы с помощью элементного анализа, спектроскопии ЯМР 1Н, 13C, 29Si и РСА.
Для гетеробороксинового комплекса висмута(III) LBi[(OBDipp)2O], полученного из оксида (LBiO)2, R = 2,6-(Me2NCH2)2C6H3, и замещенной борной кислоты (cхема 120), были получены кристаллы с сольватной молекулой бензола. Обнаруженная непланарность бензольного кольца в кристалле сольвата хелатного комплекса не была поддержана DFT-D квантово-химическими расчетами [119]. Наблюдаемая изогнутая структура бензола на самом деле является наложением (тепловое среднее) ансамбля термонаселенных бензольных структур в комплексе.
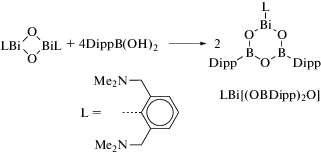
Схема 120 .
Беспрецедентный перенос арильной группы от бора на атом висмута наблюдается в реакции гетеробороксинов общей формулы LBi[(OBR)2O] (L = 2,6-(Me2NCH2)2C6H3; R = Ph, 4-CF3C6H4, 4‑BrC6H4) с соответствующей борной кислотой RB(OH)2 (cхема 121) [120].
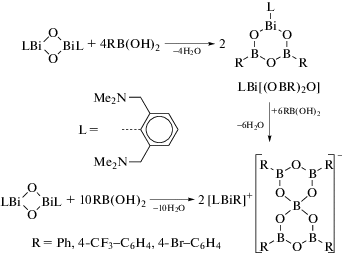
Схема 121 .
Были получены ионные пары [LBiR]+-[R4B5O6]− (R = Ph, 4-CF3C6H4, 4-BrC6H4), строение фенил-содержащего комплекса доказано методом РСА.
Взаимодействие димерного оксида висмута(III) (LBiO)2 (R = (Me2NCH2)2C6H3) с уксусной кислотой приводило к образованию ацетата LBi(OAc)2 [121]. При использовании трифторметансульфоновой кислоты был получен гидроксид LBi(OH)(OTf) (cхема 122).
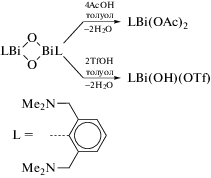
Схема 122 .
Комплексы LBi(OAc)2 и LBi(OH)(OTf) охарактеризованы с помощью масс-спектрометрии, ЯМР 1Н-, 13С-спектроскопии и РСА. В кристалле LBi(OH)(OTf) присутствуют слабосвязанные димерные звенья LBi(μ-OH)2BiL, а трифлат-анионы связаны с мостиковыми фрагментами OH посредством водородных связей, наряду с взаимодействиями Bi⋅⋅⋅O, приводящими к бесконечной цепочке супрамолекулярной структуры LBi(OH)- (OTf).
Реакции (LBiO)2, где R = (Me2NCH2)2C6H3, с фосфорорганическими кислотами при молярном соотношении 1 : 4 давали фосфонаты органовисмута LBi[OP(tBu)(O)(OH)]2 (схема 123 ) [122]. При молярном соотношении 1 : 2 имеет место образование [LBi(O(O)P(tBu)O)]3. Реакция [LBi(O(O)P(tBu)O)]3 с EtP(O)(OH)2 приводит к смешанному фосфонату LBi[OP(Et)(O)(OH)][OP(tBu)(O)(OH)].
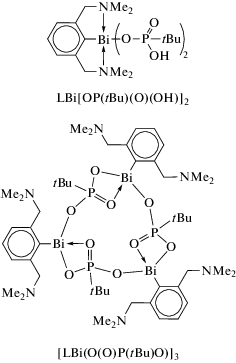
Схема 123 .
Все соединения были охарактеризованы элементным анализом, масс-спектрометрией, 1Н-, 13С-, 31P-спектроскопии и ИК-спектроскопией. Вторичный фосфонат LBi[OP(tBu)(O)(OH)]2 состоит из слабосвязанных димерных единиц через водородные мостики типа PO–H···O=P. С другой стороны, комплекс [LBi(O(O)P(tBu)O)]3 является тримером с центральным 12-членным циклом, сформированым тремя блоками LBi(O(O)P(tBu)O) через межмолекулярные контакты Bi···O=P.
Этот же оксид (LBiO)2 реагирует с оксидами мышьяка As2O5 и As2O3 с образованием молекулярных соединений (LBi)3(AsO4)2 и (LBi)2(As2O5) (схема 124 ) [123].
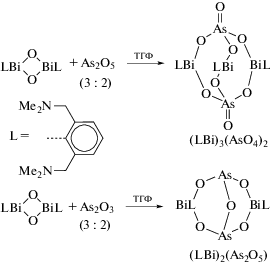
Схема 124 .
Полученные комплексы охарактеризованы с помощью масс-спектрометрии, спектроскопии 1H и 13C ЯМР, а в случае (LBi)2(As2O5) − РСА.
В [124] сообщается о синтезе N→Bi внутримолекулярно координированного селенита висмута [LBi(O(O)SeO)]3 (схема 125 ).
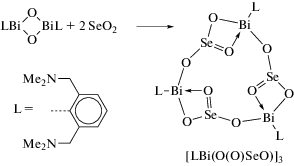
Схема 125 .
Висмуторганический селенит [LBi(O(O)SeO)]3 является редким примером смешанного оксида селена и висмута, который охарактеризован спектроскопией ЯМР 1H, 13C, 77Se, ИК-спектроскопией и РСА.
Реже в синтезе производных трехвалентного висмута с тридентатными лигандами используют сульфиды арилвисмута, которые получают из дихлоридов арилвисмута и сульфидов натрия или лития. Так, взаимодействием дихлорида арилвисмута(III) LBiCl2, содержащим O,C,O-хелатирующий лиганд L = 2,6-(tBuOCH2)2C6H3, с сульфидом натрия в смеси толуол−вода синтезирован сульфид висмута (LBiS)2 (схема 126 ), стабильный при −30°C, но разлагающийся при комнатной температуре [125].
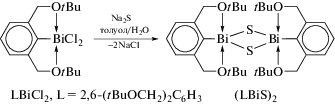
Схема 126 .
Сульфид (LBiS)2 охарактеризован элементным анализом, масс-спектрометрией, ЯМР 1Н- и 13С-спектроскопией и РСА.
Сульфид органовисмута(III) (LBiS)2 (L = 2,6-(Me2NCH2)2C6H3), полученный из дихлорида арилвисмута и сульфида лития, также является димерным в твердом состоянии (схема 127 ) [126].
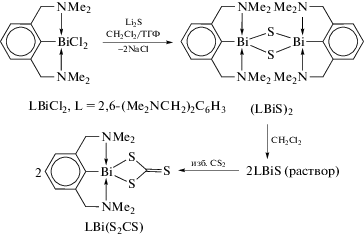
Схема 127 .
Тем не менее наличие в растворе мономерной структуры с концевыми связями Bi−S было доказано [2+2]-реакцией циклоприсоединения с CS2, приводящей к образованию молекулярного тритиокарбоната LBi(S2CS). Оба соединения были охарактеризованы в твердом состоянии методами дифракции рентгеновских лучей на монокристаллах и ИК-спектроскопии. Дисульфид углерода может быть удален из тритиокарбоната LBi(S2CS) при его нагревании до 160°С с восстанавлением до исходного сульфида. В растворе тритиокарбонат LBi(S2CS) находится в равновесии с исходным сульфидом, но это равновесие может быть сдвинуто влево путем добавления избытка сероуглерода.
Сульфид арилвисмута (LBiS)2 также может быть использован в элементоорганическом синтезе. Так, (LBiS)2, содержащий NCN-хелатирующий лиганд L = 2,6-(Me2NCH2)2C6H3, реагирует с одним молярным эквивалентом элементарной серы с образованием циклического бис(пентасульфида) LBi(μ-S5)2BiL с центральным двенад-цатичленным циклом Bi2S10 (схема 128 ) [127].
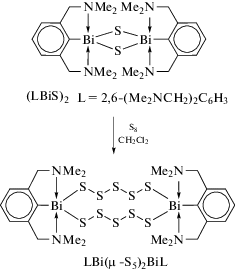
Схема 128 .
Соединение LBi(μ-S5)2BiL получено в виде стабильных оранжевых кристаллов и охарактеризовано с помощью РСА, ИК- и Рамановской спектроскопии.
Реакционная способность комплексов висмута с полидентатными лигандами мало изучена, за исключением реакций замещения, которые более всего представлены реакциями органических га-логенидов висмута с серебряными солями раз-личных кислот. Например, из 12-хлоро-6-циклогексил-5,6,7,12-дибензо[1,5]азависмоцина и трифлата серебра был получен соответствующий трифлат гетероциклического соединения висмута [(C6H4CH2)2NCy]Bi(OTf) (схема 129 ) и определена его кристаллическая структура [128].
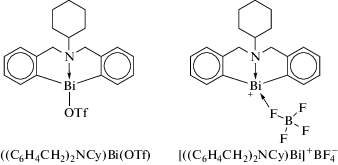
Схема 129 .
Центральная висмутсодержащая часть комплекса обладает искаженной псевдотригонально-бипирамидальной структурой. Атомы углерода и неподеленная электронная пара атома Bi располагаются в экваториальной плоскости, в то время как атомы азота и кислорода расположены в апикальных положениях. Расстояния Bi−C составляют 2.216(9) и 2.219(9) Å. Угол CBiC составляет 96.3(3)°, а угол NBiO равен 151.7(2)° (а не 180°). Расстояние Bi−N составляет 2.430(6) Å, циклогексильная группа разупорядочена по двум положениям.
По этой же схеме синтезирован стабильный на воздухе тетрафторборат гетероциклического арилвисмута [(C6H4CH2)2NCy]Bi}+BF4− (схема 129 ), который проявляет каталитическую эффективность в реакциях аллилирования различных альдегидов тетрааллилоловом в водном метаноле (схема 130 ), давая соответствующие гомоаллильные спирты с превосходной селективностью [129]. Такая активность комплекса {[(C6H4CH2)2NCy]Bi}+B${\text{F}}_{4}^{ - }$ близка к каталитической активности ранее описанного [2-(MeOCH2)C6H4]2Bi(OTf) (схема 94 ) [97].
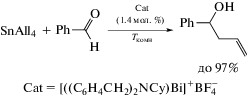
Схема 130 .
Аналогично получен воздухостойкий перфтороктансульфонат органовисмута {[(C6H4CH2)2S]-Bi(H2O)}+[OSO2C8F17]−, обладающий высокой каталитической активностью и возможностью повторного использования в синтезе (E)-α,β-ненасыщенных кетонов благодаря высокоселективной перекрестной конденсации кетонов и альдегидов в воде [130]. Стабильный на воздухе катионный висмуторганический комплекс, полученный из перхлората серебра и хлорида диарилвисмута, использовали в качестве высокоэффективного катализатора прямой диастереоселективной реакции Манниха в воде (схема 112 ) [131].
Органовисмутовый комплекс 5H-дибензо[1,5]оксависмоцин-12(7H)-ил нитрат [(C6H4CH2)2-O]BiONO2 синтезировали прибавлением раствора хлорида арилвисмута в ТГФ к раствору нитрата серебра в воде [132]. Обнаружено, что этот комплекс проявляет противоопухолевую активность и имеет большой потенциал в лечении рака.
Реакции бромидов органилвисмута [(C6H4CH2)2NR]BiBr (R = C6H5CH2, C6H5CH2CH2) и соответствующих солей серебра привели к образованию соединений висмута общей формулы [(C6H4CH2)2NR]BiX (схема 131 ) [133].
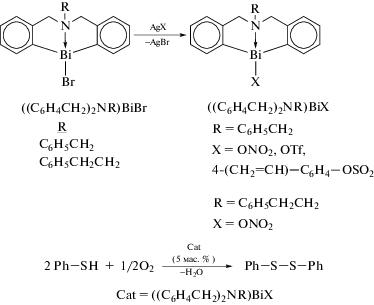
Схема 131 .
Полученные соединения катализируют реакции окисления тиофенола в дифенилдисульфид с использованием воздуха в качестве окислителя в циклогексане при температуре ниже 100°С, что обеспечивает высокие скорости реакции (100% конверсии через 5 ч).
Аналогично полученный стабильный на воздухе перфтороктилсульфонат органовисмута(III) [(C6H4CH2)2NCy]BiOSO2C8F17 проявляет высокую каталитическую эффективность к реакции Манниха с ароматическими альдегидами и ароматическими аминами в воде (схема 132 ) [134] (как и его серасодержащие аналоги {[(C6H4CH2)2S]Bi(H2O)}+X− (X− = ${\text{ClO}}_{4}^{ - }$, ${\text{BF}}_{4}^{ - }$, ${\text{OS}}{{{\text{O}}}_{2}}{{{\text{C}}}_{4}}{\text{F}}_{9}^{ - }$, ${\text{OS}}{{{\text{O}}}_{2}}{{{\text{C}}}_{8}}{\text{F}}_{{17}}^{ - }$), представленные на схеме 113 ).
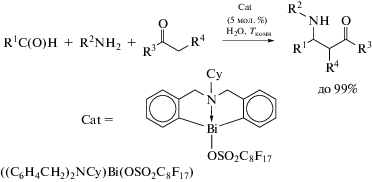
Схема 132 .
Этот катализатор также показывает хорошую рециркуляцию и возможность повторного использования в синтезе β-аминокетонов.
Из 12-хлор-6-фенил-дибензо[1,5]азависмоцина и перхлората серебра в тетрагидрофуране получен комплекс висмута 6-фенил-дибензо[1,5]азaвисмоцин-12(5H)-ил перхлорат с выходом 93% [135]. По данным РСА, центральный атом имеет тригонально-бипирамидальное окружение с атомами кислорода и азота в аксиальных позициях и двумя атомами углерода и неподеленной электронной парой в экваториальных положениях. Длины связей Bi–C составляют 2.250(13), 2.204(12) Å, углы CBiC и NBiO равны 92.5(5)° и 154.0(3)° соответственно. Расстояние Bi⋅⋅⋅N (2.388(10) Å) короче, чем в прекурсоре C6H5N(CH2C6H4)2BiCl (2.607(5) Å).
Ряд гетероциклических карбоксилатов органовисмута(III) [(C6H4)2SO2]BiOC(O)R (схема 133 ) был синтезирован для определения влияния структуры карбоксилатного лиганда на липофильность и противогрибковую активность в отношении дрожжей Saccharomyces cerevisiae [136].
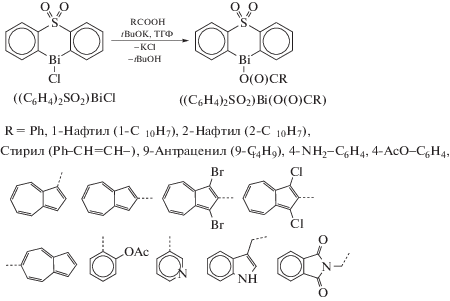
Схема 133 .
Взаимодействием дихлорида арилвисмута LBiCl2 (L = 2,6-(Me2NCH2)2C6H3) с Na2CO3 или Ag2SO4 (мольное соотношение 1 : 1) получены карбонат RBiCO3 и сульфат арилвисмута RBiSO4 соответственно (схема 134 ) [137]. Динитрат арилвисмута RBi(NO3)2 синтезировали из дихлорида арилвисмута и нитрата серебра при мольном соотношении исходных реагентов 1 : 2.
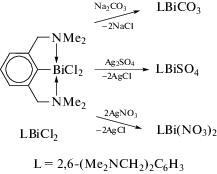
Схема 134 .
Молекулярные структуры RBiCO3 · 0.5CH2Cl2, RBiSO4, RBi(NO3)2 · H2O были установлены методом РСА. Карбонат и сульфат имеют полимерную структуру на основе мостиковых оксоанионов, тогда как динитраты являются димерами с мостиковыми и концевыми нитрат-анионами.
Из дихлорида арилвисмута с арильным NCO-лигандом L = 2-(Me2NCH2)-6-(tBuOCH2)C6H3 и солей серебра были синтезированы два ионных комплекса висмута вида [LBiCl]+X− (схема 135 ) [138].
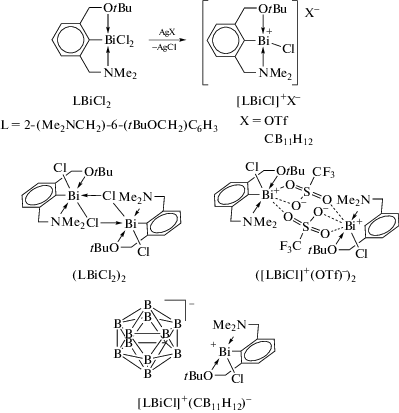
Схема 135 .
Если кристаллы комплексов LBiCl2 и [LBiCl]+-(OTf)− представляют собой в твердом состоянии димеры, то ионный комплекс [LBiCl]+(CB11H12)− состоит из мономерных структурных единиц. Все изученные соединения охарактеризованы с помощью спектроскопии ЯМР 1Н и 13С, ESI масс-спектрометрии и методом дифракции рентгеновских лучей на монокристаллах.
Нитрат диарилвисмута с потенциальными координирующими центрами в арильных лигандах [(C6H4CH2)2NCy]Bi(NO3) получен из хлорида диарилвисмута и нитрата серебра в воде [139]. В кристалле атомы висмута имеют тригонально-бипирамидальное окружение с атомами N (Bi⋅⋅⋅N 2.495(3) Å) и O в апикальных позициях и двумя арильными лигaндами и неподеленной электронной парой в экваториальной плоскости. Нитратная группа является несимметричным бидентатным лигандом (Bi−O 2.416(3) и 3.0451(4) Å).
Обработка N,C,N-хелатного дихлорида висмута LBiCl2 (L = 2,6-(R-N=CH)2C6H3, R = tBu, 2,6-Me2C6H3) одним мольным эквивалентом Ag[CB11H12] приводит к образованию ионных пар [LBiCl]+[CB11H12]− [140]. Аналогичная реакция C,N-хелатного аналога L'BiCl2 (L' = 2-(Dipp− N=CH)-4,6-(tBu)2C6H2) дает соединение [L'BiCl]+-[CB11H12]− (cхема 136).
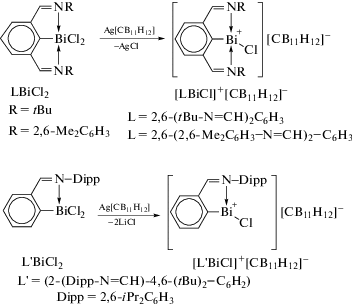
Схема 136 .
При обработке комплекса [LBiCl]+[CB11H12]− (L = 2,6-(tBu−N=CH)2C6H3) другим эквивалентом Ag[CB11H12] был выделен аддукт исходного материала с Ag[CB11H12], а именно [(2,6-(tBu−N=CH)2C6H3)BiCl]+[Ag(CB11H12)2]−. Кристаллы данного ионного соединения разлагаются при дневном свете, судя по спектрам 1Н ЯМР, до исходных соединений.
Висмуторганический карбонат {[(C6H4CH2)2O]-Bi}2CO3, в котором плоская группа CO3 присоединена к двум дибензо[1,5]оксабисмоциновым каркасам (схема 137 ), синтезирован из хлорида диарилвисмута и карбоната натрия в смеси растворителей вода−дихлорметан [141]. Длины двух ковалентных связей висмут–кислород составляют 2.217(3) и 2.223(3) Å.
Схема 137 .
Расстояния Bi⋅⋅⋅O c CH2OCH2-группой (2.587(4) и 2.618(3) Å) указывают на сильную координацию O → Bi в комплексе.
Аналогично получен другой карбонат диарилвисмута [(2-Et2NCH2C6H4)2Bi]2CO3 (схема 138 ), в котором мостиковая карбонатная группа связывает две группы (2-Et2NCH2C6H4)2Bi. Выход атомов висмута и ипсо-атомов углерода из плоскости карбонатной группы составляет 0.323(1) и 0.330(9) Å соответственно. Арильные лиганды находятся в транс-положении относительно квазиплоских групп (CBi)2CO3 [142]. Атом металла сильно координируется на атом N одной аминогруппы (Bi⋅⋅⋅N 2.739(6) Å), в то время как атом N другой аминогруппы слабо связан с атомом металла (Bi⋅⋅⋅N 3.659(7) Å). С учетом этих внутримолекулярных взаимодействий можно считать, что общая координационная геометрия у висмута становится искаженной квадратно-пирамидальной.
Реакция между N,C,N-хелатным арилвисмутом(I) [LBi]n, приготовленным in situ из LBiCl2 [L = 2,6-C6H3(CH2NMe2)2] и K[B(втор-Bu)3H], и диорганодисульфидами ArSSAr приводит к образованию висмуторганических соединений LBi(SAr)2 (Ar = 2-пиридил, 4-метилтиазол-2-ил, тиофен-2-ил, 4-трет-бутил-1-изопропил-1H-имидазол-2-ил, 1-фенил-1H-тетразол-5-ил- 2-аминофенил) (схема 138 ) [143].
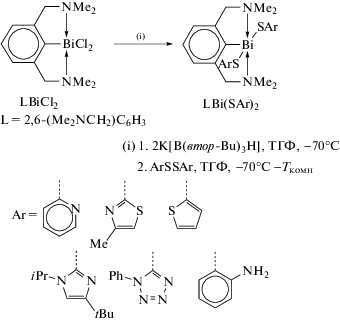
Схема 138 .
Соединения охарактеризованы спектроскопией ЯМР 1H и 13C, а в случае 2-пиридильного, 4-метил-тиазол-2-ильного и 1-фенил-1H-тетразол-5-ильного производных − с помощью РСА. Комплекс на основе о-аминотиофенола неустойчив в растворе и разлагается до соединения LBi[S(NH)C6H4], содержащего пятичленное кольцо BiSNC2, и 2-аминотиофенола (схема 139 ).
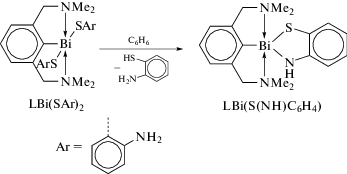
Схема 139 .
Попытки селективного расщепления связи Bi−N в этом кольце соляной или уксусной кислотами приводили только к выделению LBiCl2 или диацетата LBi(OAc)2 и 2-аминотиофенола.
N,C,N-пинцерный комплекс дихлорида арилвисмута LBiCl2, где L = 2,6-(Me2NCH2)2C6H3, реагирует с двумя эквивалентами калиевых солей фенолов (2,6-Me2C6H3O)K и (2,6-iPr2C6H3O)K с образованием ожидаемых диарилоксидов висмута LBi(OArR)2, где ArR = 2,6-R2C6H3, R = Me, iPr (схема 140 ) [144].
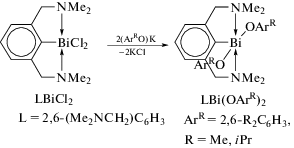
Схема 140 .
Однако аналогичная реакция с двумя эквивалентами (ArtBuO)K, где ArtBu = 2,6-tBu2C6H3O, приводит к образованию фенола ArtBuOH и темно-оранжевого ароксида арилвисмута LBi(C6H2tBu2O), являющимся продуктом активации пара-СН-связи (схема 141 ).
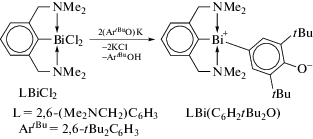
Схема 141 .
Комплекс LBi(C6H2tBu2O), где L = 2,6-(Me2NCH2)2C6H3, при действии эквимолярного количества тетрафторбората триэтиламмония в тетрагидрофуране превращается в ионный комплекс [LBi(C6H2tBu2OH)]+[BPh4]− (схема 142 ).
Весьма интересны реакции комплексов висмута, содержащие тридентатные лиганды, с малыми молекулами. Так, были исследованы реакции пинцерного комплекса висмута LBi(C6H2tBu2O), где L = 2,6-(Me2NCH2)2C6H3, с СО2 и COS в ацетонитриле (схема 142 ) [145]. Показано, что красная окраска раствора LBi(C6H2tBu2O) в течение 1 ч меняется на желтую, присущую растворам оксиарилкарбоксильных комплексов LBi[O(E)C− C6H2tBu2O], где E = O, S соответственно. В этих реакциях введения СО2 и COS по связи Bi−C наблюдаются генерация новых дианионов, которые имеют хиноидный характер, сходный с оксиарилдианионным лигандом в LBi(C6H2tBu2O).
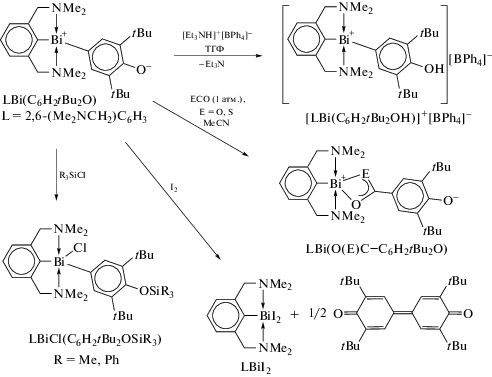
Схема 142 .
Силилгалогениды и псевдогалогениды R3SiX (X = Cl, CN, N3; R = Me, Ph) реагируют с LBi(C6H2tBu2O), где L = 2,6-(Me2NCH2)2C6H3, присоединяя X с образованием комплексов LBiX(C6H2tBu2OSiR3) (схема 142 ), в которых имело место увеличение КЧ центрального атома металла до пяти. Они реагируют с дополнительным количеством R3SiX с образованием комплексов LBiX2, L = 2,6-(Me2NCH2)2C6H3 и 2,6-tBu2C6H3− OSiR3. Реакция LBi(C6H2tBu2O) [L = 2,6-(Me2NCH2)2C6H3] с иодом протекает по схеме окислительного сочетания с образованием дииодида Ar'BiI2 и 3,3',5,5'-тетра-трет-бутил-4,4′-дифенохинона (схема 142 ).
Красный ацетонитрильный раствор оксиарильного комплекса LBi(C6H2tBu2O), где L = = 2,6-(Me2NCH2)2C6H3, реагирует с NO при 1 атм. с образованием темно-зеленого раствора, содержащего несколько идентифицированных с помощью спектроскопии ЯМР 1Н продуктов (схема 143 ). Реакции, проводимые при низкой температуре (−35°C) и со стехиометрическими количествами газа NO, дали сложные смеси продуктов [146].
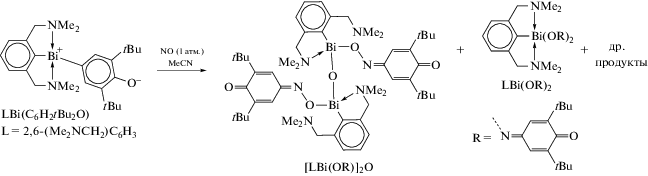
Схема 143 .
При доведении температуры реакционной смеси до комнатной, удаления растворителя и перекристаллизации остатка из сырого ацетонитрила были выделены желтые кристаллы [LBi(OR)]2O (схема 143 ). Из сухого ацетонитрила были выделены желтые кристаллы LBi(OR)2 (R = −N=C6H2tBu2=O).
АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ВИСМУТА(V)
Сообщалось о галогенировании арильных соединений трехвалентного висмута бромом, хлористым сульфурилом и дифторидом ксенона. Так, из три-п-толилвисмута и брома в растворе четыреххлористого углерода получен дибромид три-п-толилвисмута [147]. Тригалогениды диарилвисмута получить указанным способом невозможно, однако в случае присутствия при атоме висмута N,C-хелатного лиганда L (L = (2-Dipp−N=CH)C6H4, Dipp = 2,6-ди-изопропилфенил) и обработке исходного хлорида LBi(Ph)Cl хлористым сульфурилом имеет место стабилизация комплекса LBi(Ph)Cl3, который был выделен и структурно охарактеризован (cхема 144) [11].
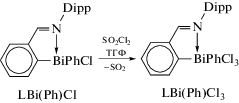
Схема 144 .
Отметим, что аналогичным образом было синтезировано и производное LBiPh2Cl2.
Об эффективном фторировании дифторидом ксенона дифениларилвисмута [2-(Me2NCH2)C6H4]-BiPh2, содержащего в орто-положении арильного лиганда диметиламинометильный заместитель (схема 145 ), сообщалось в [148].
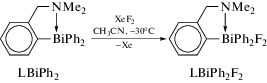
Схема 145 .
Соединения [2-(Me2NCH2)C6H4]2BiN3 и [2-(Me2NCH2)C6H4]Bi(N3)2 представляют собой редкие примеры азидов висмута [14]. В то время как в кристалле моноазида [2-(Me2NCH2)C6H4]2BiN3 присутствовали мономерные молекулы, то диазид [2-(Me2NCH2)C6H4]Bi(N3)2 представлен в виде димера с двумя типами соединения азидогрупп. Кроме того, слабые ван-дер-ваальсовы взаимодействия между этими центросимметричными димерами приводят к цепочечной структуре в кристалле.
Весьма обширным классом органических соединений висмута являются дикарбоксилаты триорганилвисмута, которые представлены прежде всего арильными производными. Известно, что дикарбоксилаты триарилвисмута успешно получают по реакции окислительного присоединения из триарилвисмута и карбоновой кислоты в диэтиловом эфире в присутствии гидропероксидов [149–156]. Синтез дикарбоксилатов триарилвисмута проводили, как правило, с использованием гидропероксида третичного бутила в диэтиловом эфире (схема 146 ).
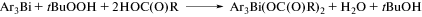
Схема 146 .
Однако в случае триарилвисмута,содержащего в своем составепотенциальныекоординирующие центры,напримертрис(2-метокси-5-бромфенил)висмута,целесообразнеебыло использовать в реакции в качестве окислителя пероксид водорода [31]. Целевые продукты выделяли из реакционной смеси с выходом не менее 70%.
Серия дикарбоксилатов трифенилвисмута общей формулы (RCOO)2BiPh3 (схема 147 ) получена по этой же схеме с целью определения их антилейшманиозной активности [157, 158].
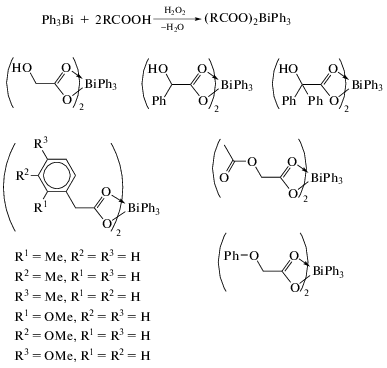
Схема 147 .
Показано, что они менее активны по сравнению с аналогичными производными сурьмы.
По той же схеме были синтезированы одиннадцать дикарбоксилатов трифенилвисмута на основе функционализированных производных бензойной или салициловой кислот (схема 148 ), которые показали хорошую активность против лейшманиоза [159].
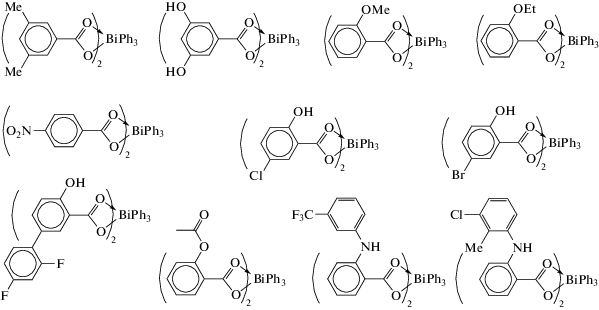
Схема 148 .
Подобная большая серия дикарбоксилатов три-толилвисмута (RCOO)2BiTol3 на основе функционализированных производных бензойной или салициловой кислот с орто-, мета- или пара-толильными лигандами была синтезирована аналогично [160]. Из них 15 были оценены на токсичность по отношению к промастиготам лейшмании и клеткам фибробластов человека, с десятью затем проводилась последующая оценка против амастигот паразитов. Наилучшая активность и селективность наблюдается у соединений висмута, содержащими о- и м-толильные лиганды.
Замена диэтилового эфира на изопропиловый спирт не изменяет схемы реакции. В этом случае из трифенилвисмута, карбоновой кислоты и пероксида водорода в растворе изопропанола были получены дикарбоксилаты трифенилвисмута (RCOO)2BiPh3, где R = 5-Br-2-OH−C6H3, 2-OH−C6H4, 2,6-(OH)2-C6H3, 3-Me-2-NH2−C6H3, Ph, Me, с более высокими выходами, чем в известных способах синтеза целевых продуктов [161].
В основе другого метода получения дикарбоксилатов триарилвисмута лежит реакция замещения атомов галогена в дигалогенидах триарилвисмута. Именно по этой схеме были синтезированы дикарбоксилаты трифенилвисмута Ph3Bi[OC(O)R]2 (R = C6H3F2-3,5, C6H4CF3-4, C4H3S), которые получали смешением растворов дихлорида трифенилвисмута и натриевой соли кислоты в метаноле [162].
Из натриевых или калиевых солей пара- и мета-пиридинкарбоновых кислот и дихлорида трифенилвисмута (схема 149 ) в растворе спирта были синтезированы соответствующие дикарбоксилаты трифенилвисмута [163].
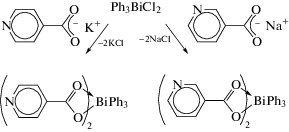
Схема 149 .
Реакции полученных дикарбоксилатов трифенилвисмута с трифлатом серебра приводили к образованию координационных полимеров, в которых атомы серебра сшивают молекулы дикарбоксилатов в цепочку за счет координации атомов переходного металла с атомами азота пиридинкарбоксилатных лигандов.
Дикарбоксилат трифенилвисмута (4-FC6H4COO)2BiPh3 был выбран в качестве объекта для изучения фотохимической активности из-за его химической стабильности, малой токсичности и простоты синтеза в реакциях фотодеградации трех распространенных красителей [164]: метиленового синего, родамина B и метилового фиолетового. К раствору кислоты и метоксида натрия в метаноле прибавляли раствор дихлорида трифенилвисмута в толуоле, реакционную смесь перемешивали 2 ч при 25°С. Выход 66%. Показано, что комплекс обладает хорошей фотокаталитической способностью в деградации органических красителей под действием ультрафиолета или видимого света. Результат этого исследования может помочь в разработке новых фотокаталитических материалов.
В некоторых случаях эффективный синтез дикарбоксилатов триарилвисмута можно осуществить из триарилвисмута и карбоновой кислоты в присутствии триэтиламина (схема 150 ) [165].
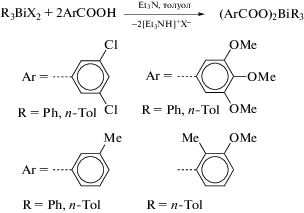
Схема 150 .
Соединение [(2-Me)(3-OMe)C6H3C(O)O]2Bi(п-Tol)3 оказалось весьма эффективным против лейшманиоза.
Реакция дихлорида трифенилвисмута с лапахолом (Lp) в присутствии триэтиламина приводила к образованию биядерного соединения висмута (LpPh3Bi)2O, который был охарактеризован РСА [166]. Двухъядерный комплекс содержит два шестикоординированных атома висмута, соединенных через атом кислорода (Lp)2(Ph3Bi)2O. Соединение ингибирует рост линии клеток хронического миелогенного лейкоза, причем комплекс примерно в 5 раз более активен, чем свободный лапахол.
Известен случай, когда дихлорид трифенилвисмута реагирует с карбоновой кислотой с образованием дикарбоксилата трифенилвисмута, например с салициловой кислотой (схема 151 ) [167].
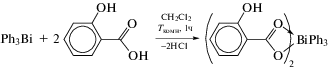
Схема 151 .
Известно, что в дикарбоксилатах триарилвисмута реализуется внутримолекулярное взаимодействие атома висмута с карбонильными атомами кислорода − потенциальными координирующими центрами карбоксилатных лигандов, что позволяет отнести эти производные к комплексам высококоординированного висмута [152–172]. Прочность внутримолекулярных контактов Bi⋅⋅⋅O(=C), основой которых являются донорно-акцепторные взаимодействия, во многом определяется природой заместителей в арильных кольцах при атоме металла (влияют на акцепторные способности металла) и в органическом радикале остатка карбоновой кислоты (усиливают или ослабляют донорные свойства карбонильного кислорода). Показано, что внутримолекулярные расстояния Bi⋅⋅⋅O(=C) различаются между собой в бóльшей степени: меньшему расстоянию Bi⋅⋅⋅O(=C) соответствует бoльшая длина связи Bi–O, что свидетельствует о перераспределении электронной плотности при возникновении прочного донорно-акцепторного взаимодействия. Меньшим расстояниям Bi⋅⋅⋅O(=C) в соединениях (RCOO)2BiAr3 соответствует бoльшая длина связи Bi–O, что свидетельствует о перераспределении электронной плотности при возникновении прочного донорно-акцепторного взаимодействия. Самые слабые внутримолекулярные взаимодействия наблюдаются в молекулах тех дикарбоксилатов триарилвисмута, в которых донорные способности карбонильного атома кислорода ослаблены из-за смещения электронной плотности, обусловленного наличием электроотрицательных заместителей в органическом радикале кислоты (−I-эффект). В молекулах дикарбоксилатов триарилвисмута с одинаковыми арильными заместителями при атомах висмута невалентные взаимодействия усиливаются с повышением донорных свойств карбонильного атома кислорода за счет +М-эффекта радикала. В молекулах дикарбоксилатов укорочение расстояний Bi⋅⋅⋅O(=C) коррелирует с увеличением одного из экваториальных углов СBiC (до 152.9°) со стороны внутримолекулярных контактов.
Дикарбоксилаты триарилвисмута могут быть использованы для получения других классов соединений, например в синтезе дисульфонатов триарилвисмута, когда диацетат трифенилвисмута при действии трифторметансульфоновой кислоты превращался в соответствующий дисульфонат (cхема 152), весьма эффективный в реакциях гликозилирования при комнатной температуре. Этот промотирующий агент продемонстрировал преимущества перед большинством современных тиогликозидных активаторов, а именно высокую растворимость и стабильность к действию воздуха и света [173].
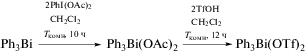
Схема 152 .
Ряд бис(аренсульфонатов) триарилвисмута синтезирован из трифенил-, трис(мета-толил)- и трис(2-метокси,5-бромфенил)висмута и аренсульфоновой кислоты в эфире. В качестве окислителя использовали пероксид водорода, поскольку в присутствии трет-бутилгидропероксида не наблюдалось образование целевого продукта. При соотношении исходных реагентов 1 : 2 : 1 (мольн.) из реакционной смеси выделяли дисульфонаты триарилвисмута Ph3Bi(OSO2C6H3Me2-3,4)2 [172], (м-Tol)3Bi(OSO2C6H3Me2-3,4)2 [171] и [(2-MeO)(5-Br)C6H3]3Bi(OSO2Ph)2 [174] с выходом до 85%. Из данных РСА следует, что атомы висмута в молекулах дисульфонатов триарилвисмута имеют тригонально-бипирамидальную координацию с аренсульфонатными заместителями в аксиальных положениях. Относительно экваториального фрагмента С3Bi аренcульфонатные группы в первых двух дисульфонатах имеют цис-ориентацию. Со стороны максимального экваториального угла СBiC (140.77(11)° и 133.69(17)°) наблюдаются внутримолекулярные контакты между центральным атомом Bi и атомами O аренсульфонатных групп (3.189(4), 3.122(3) и 3.244(6), 3.406(6) Å соответственно). Для третьего соединения также обнаруживаются внутримолекулярные контакты между атомом металла и атомами кислорода сульфонатных групп, однако расстояния Bi⋅⋅⋅O в двух кристаллографически независимых молекулах (3.265(4)−3.296(4) Å) несколько больше, чем в первых двух соединениях. Отметим, что подобная тенденция лигандов к проявлению бидентатных свойств характерна для дикарбоксилатов триарилвисмута [78]. Кроме того, в молекулах третьего дисульфоната присутствует внутримолекулярная координация атомов кислорода метоксигрупп на атом висмута (Bi⋅⋅⋅OMe 3.062(9)−3.215(9) Å).
Реакция трис(2-метокси,5-бромфенил)висмута с бензолсульфоновой кислотой (1 : 2 мольн.), проходящая в растворе диэтилового эфира в присутствии кислорода воздуха, сопровождалась образованием бис(бензолсульфоната) трис(5-бром-2-метоксифенил)висмута [174], который через 48 ч был выделен из реакционной смеси с выходом 7%. Очевидно, что в отсутствие пероксида роль окислителя триарилвисмута выполнял кислород воздуха.
Из дихлорида трифенилвисмута и трифлата серебра может быть получен бис(трифторметансульфонат) трифенилвисмута, реакции которого с донорными лигандами, такими как оксид трифенилфосфина, аминопиридин и бипиридил, приводят к образованию ионных комплексов с катионами пятикоординированного висмута (схема 153 ) [175].
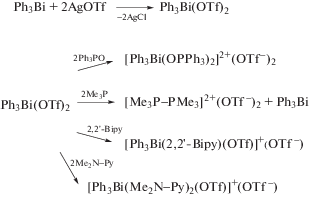
Схема 153 .
Этот синтетический метод имеет потенциал для широкого развития химии координационных соединений висмута.
Взаимодействием дибромида три-п-толилвисмута с перхлоратом серебра и его гидратом получены диперхлорат три-п-толилвисмута и μ-оксо-бис((перхлорато)три-п-толилвисмут) [176]. В молекулярной структуре первого атомы висмута имеют искаженную тригонально-бипирамидальную координацию с апикально расположенными атомами кислорода перхлоратных групп (связи Bi–C 2.180(5)–2.201(5), Bi−O 2.324(4)–2.355(4) Å; аксиальные углы OBiO 170.1(1)°, 174.5(1)°). Структура второго соединения содержит биядерные молекулы [п-Tol3Bi(ClO4)]2O (связи Bi−O 2.371(15), 1.9107(7) Å, аксиальный угол OBiO 180.0°).
Первый перфторалкилфосфинат трифенилвисмута, [(C2F5)2PO2]2BiPh3, был синтезирован из Ph3BiCl2 и [(C2F5)2PO2]Ag (схема 154 ). Данный фосфинат был успешно использован в качестве катализатора в реакции Дильса−Альдера [91].

Схема 154 .
Авторы [148] разработали эффективный метод генеририрования катионов трифенилфторвисмутония [Ph3BiF]+ и трифенил(диацетонитрило)- висмутония [Ph3Bi(NCMe)2]2+ из легкодоступного Ph3BiF2 (схема 155 ).
Схема 155 .
При действии кислот на пентафенилвисмут образуются соли тетрафенилвисмутония. Так, титрование пентафенилвисмута эфирным раствором хлористого водорода сопровождается исчезновением фиолетовой окраски, характерной для пентафенилвисмута, и образованием лабильных бесцветных кристаллов хлорида тетрафенилвисмута, разлагающихся при комнатной температуре до трифенилвисмута и хлорбензола [177]. Авторы [178] методом РСА установили его строение и нашли, что в тригонально-бипирамидальном окружении центрального атома хлор занимает аксиальное положение. Атом висмута выходит из экваториальной плоскости в направлении аксиально расположенного атома углерода. Длина связи Bi–Cl (2.9116(19) Å) превышает сумму ковалентных радиусов атомов висмута и хлора (2.50 Å), но существенно меньше суммы их ван-дер-ваальсовых радиусов (3.82 Å) [20].
Аналогичной структурой обладает и кинетически неустойчивый бромид тетрафенилвисмута, полученный из пентафенилвисмута и раствора бромистого водорода в ацетоне [179].
Взаимодействием пентафенилвисмута с эквимолярными количествами серной, 2,4-динитробензолсульфоновой и азотной кислот синтезированы гидросульфат тетрафенилвисмута (HОSO3)BiPh4, 2,4-динитробензолсульфонат тетрафенилвисмута (2,4-(NO2)2C6H3SO2O)BiPh4 и гидрат нитрата тетрафенилвисмута Ph4BiNO3 ∙ 1/3H2O [180]. Кристаллические структуры соединений висмута определены методом РСА. Если в первых двух атомы висмута пентакоординированы (окружение С4BiО), то в последнем присутствуют молекула нитратотетрафенилвисмута и два типа катионов тетрафенилвисмутония, один из которых координирован с нитрат-анионом и молекулой воды.
С целью установления природы заместителей в ароксильной группе на значения валентных углов и длин связей при атоме висмута в ароксидах тетрафенилсурьмы был синтезирован ряд указанных производных по реакции пентафенилвисмута (толуол, 0.5−5 мин, 20°С) с фенолами, содержащими электроноакцепторные заместители (схема 156 ) [181].
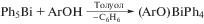
Схема 156 .
Цвет раствора в результате взаимодействия реагентов изменялся на желтый или желто-коричневый; целевые продукты выделяли кристаллизацией из смеси бензол−октан. Ароксиды тетрафенилвисмута представляют собой устойчивые на воздухе кристаллические вещества желтого или желто-коричневого цвета, растворимые в алифатических и ароматических углеводородах. Выходы полученных ароксидов тетрафенилвисмута достигали 86%.
В 1999 году была открыта реакция перераспределения лигандов для фенильных соединений пятивалентного висмута на примере взаимодействия пентафенилвисмута с бис(2,5-диметилбензолсульфонатом) трифенилвисмута и бис(2,4-диметилбензолсульфонатом) трифенилвисмута [182]. Ароксиды тетрафенилвисмута были синтезированы также аналогичным способом из пентафенилвисмута и диароксида трифенилвисмута в бензоле (схема 157 ) [181].
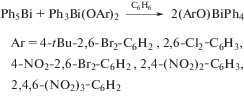
Схема 157 .
Молекулы ароксидов тетрафенилвисмута имеют характерную для большинства производных пентакоординированного висмута тригонально-бипирамидальную конфигурацию, причем наиболее электроотрицательный заместитель − ароксильный лиганд − занимает одно из аксиальных положений. Аксиальные углы СBiO близки к идеальному значению, атом висмута выходит из экваториальной плоскости в сторону атома углерода аксиально расположенного фенильного лиганда, что вызывает искажение валентных углов между аксиальными и экваториальными заместителями. Расстояния Bi−O (2.451−2.925 Å) больше суммы ковалентных радиусов атомов (2.31 Å [20]), причем наибольшее значение длины связи Bi−O наблюдалось в пикрате тетрафенилвисмута [183].
Во всех исследуемых структурах выявлена общая закономерность в расположении экваториальных фенильных групп. Так, два фенильных кольца в каждой структуре повернуты вокруг экваториальных связей Bi−C на значительные торсионные углы, тогда как плоскость третьего практически перпендикулярна аксиальной связи Bi−O. Ароксигруппа располагается над этим экваториальным фенилом, что обусловливает взаимодействие их π-систем (так называемый π−π-стекинг-эффект). В ароксидах тетрафенилвисмута характерная для π−π-стекинг-взаимодействия геометрия искажена – межцентровые расстояния равны 3.666− 4.021 Å, а межплоскостные углы составляют 14.2°−32.4°, что близко по значению к идеальным для этого типа взаимодействий [184, 185].
В препаративной химии органических соединений пятивалентного висмута с помощью реакций замещения синтезируют ряд производных платины и золота. Так, продуктом взаимодействия хлорида тетрафенилвисмута Ph4BiCl с гексабромоплатинатом калия (2 : 1 мольн.) в воде после перекристаллизации из диметилсульфоксида является S-диметилсульфоксидотрибромплатинат О-диметилсульфоксидотетрафенилвисмута [Ph4Bi ∙ DMSO-O]+[PtBr3 ∙ DMSO-S]−. Перекристаллизация из ацетонитрила комплекса, полученного из хлорида тетрафенилвисмута и гексахлороплатината калия, дает гексахлорплатинат тетрафенилвисмута $[{\text{P}}{{{\text{h}}}_{4}}{\text{Bi}}]_{2}^{ + }$[PtCl6]2− [178].
Взаимодействием бромида тетрафенилвисмута с дихлоро- и дибромодицианоауратом калия в воде с последующим удалением воды и перекристаллизацией твердого остатка из ацетонитрила синтезированы и структурно охарактеризованы комплексы золота [Ph4Bi]+[Au(CN)2Cl2]− и [Ph4Bi]+-[Au(CN)2Br2]− [186].
Эквимолярные количества сульфосалицилата тетрафенилвисмута и иодида висмута в ацетоне реагируют с образованием красно-оранжевых кристаллов ионного комплекса $[{\text{P}}{{{\text{h}}}_{4}}{\text{Bi}}]_{4}^{ + }$[Bi4I16]4− ⋅ ⋅ 2(Me2C=O) [187]. Из данных РСА следует, что в комплексе два независимых катиона тетрафенилвисмутония имеют несколько различную геометрию. В одном из них координация атома висмута искаженная тетраэдрическая (длины связей Bi−C лежат в интервале 2.184−2.207 Å, а валентные углы CBiC – 106.0°−113.7°). В координационной сфере другого катиона находится молекула ацетона (расстояние Bi⋅⋅⋅O составляет 3.094 Å), что приводит к появлению в тетраэдрической структуре вклада тригонально-бипирамидальной составляющей: заметное отклонение валентных углов CBiC от идеального для тетраэдра значения (102.1°−120.8°). Четырехъядерный центросимметричный анион [Bi4I16]4− (схема 158 ) в комплексе состоит из двух пар объединенных по общим ребрам октаэдров BiI6. Атомы Bi(2) и Bi(2') имеют в своем окружении по три концевых и мостиковых атомов иода (расстояния Bi−I составляют 2.909−2.947 и 3.284−3.337 Å соответственно), атомы Bi(1) и Bi(1') – по два концевых и четыре мостиковых атомов иода (соответствующие связи равны 2.898, 2.904 и 3.027−3.312 Å).
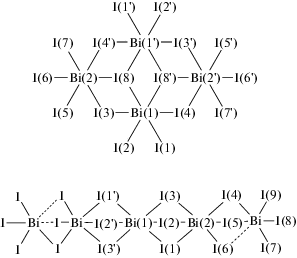
Схема 158 .
При увеличении количества иодида висмута (мольное соотношение аренсульфоната тетрафенилвисмута и иодида висмута 1 : 2) наблюдается образование комплекса с линейным пятиядерным трехзарядным анионом [Ph4Bi]+3[Bi5I18]3− [188]. В центросимметричном анионе [Bi5I18]3− (схема 158 ) октаэдрически координированные атомы Bi попарно объединены тройными иодными мостиками. Концевой атом Bi(3) соединен с соседним атомом Bi(2) менее прочными связями Bi(3)−I(4−6), чем Bi(2)−I(4−6) (3.423−3.582 и 2.940−2.954 Å соответственно). Концевые связи Bi(3)−I(7,8,9) 2.842−2.860 Å – самые короткие в анионе [Bi5I18]3−.
Отметим эффективный метод синтеза фторидов трифениларилвисмута, заключающийся в обработке дифторида трифенилвисмута фенилборной кислотой в присутствии эфирата трифторида бора в хлористом метилене с последующей обработкой реакционной смеси избытком фторида цезия, который впервые описали японские авторы в 2003 году [189] и продолжили авторы [190], получившие по аналогичной схеме фториды трифениларилвисмутония и изучившие транспортные свойства катионов общей формулы [Ph3BiAr]+, где Ar = фенил, нафтил, антрил или пиренил. Показано, что эти катионы эффективно переносят гидроксид-, фторид- и хлорид-анионы через фосфолипидный бислой.
ЗАКЛЮЧЕНИЕ
Органические соединения висмута привлекают все большее внимание исследователей по всему миру. Обусловлено это обнаруженной в последнее время каталитической активностью ряда органических соединений висмута по отношению к различным группам реакций важного значения в органической и элементоорганической химии, а также их большим потенциалом применения в качестве реагентов в тонком органическом синтезе. С точки зрения биохимии и медицины данный класс соединений висмута также имеет большой потенциал применения в качестве противораковых, противогрибковых и противобактериальных препаратов. Кроме этого, органические соединения висмута(III,V) способны образовывать моно-, би- и полиядерные структуры разнообразного строения, стабильные гетеролигандные соединения как молекулярного, так и ионного типов, что несомненно важно для развития фундаментальных исследований висмуторганических соединений. В ближайшее время следует ожидать еще более активного развития катализа висмуторганическими соединениями, областей их биохимического и медицинского использования.
Список литературы
Разуваев Г.А., Осанова Н.А., Шарутин В.В. // Докл. АН СССР. 1975. Т. 225. № 3. С. 581.
Шарутин В.В., Мосунова Т.В. // Вестник ЮУрГУ. Сер. Химия. 2020. Т. 12. № 3. С. 7.
Kindra D.R., Peterson J.K., Ziller J.W., Evans W.J. // Organometallics. 2015. V. 34. P. 395.
Schulz S., Kuczkowski A., Blaser D. et al. // Organometallics. 2013. V. 32. P. 5445.
Lichtenberg C., Pan F., Spaniol T.P. et al. // Angew. Chem., Int. Ed. 2012. V. 51. P. 13011.
Casely I.J., Ziller J.W., Mincher B.J., Evans W.J. // Inorg. Chem. 2011. V. 50. P. 1513.
Auer A.A., Mansfeld D., Nolde C. et al. // Organometallics. 2009. V. 28. P. 5405.
Solyntjes S., Bader J., Neumann B. et al. // Chem. Eur. J. 2017. V. 23. P. 1557.
Ishii T., Suzuki K., Nakamura T., Yamashita M. // J. Am. Chem. Soc. 2016. V. 138. P. 12787.
Tomaschautzky J., Neumann B., Stammler H.-G. et al. // Dalton Trans. 2017. V. 46. P. 1645.
Urbanova I., Jambor R., Ruzicka A. et al. // Dalton Trans. 2014. V. 43. P. 505.
Olaru M., Nema M.G., Soran A. et al. // Dalton Trans. 2016. V. 45. P. 9419.
Soran A., Breunig H.J., Lippolis V. et al. // J. Organomet. Chem. 2010. V. 695. P. 850.
Schulz A., Villinger A. // Organometallics. 2011. V. 30. P. 284.
Chalmers B.A., Meigh C.B.E., Nejman P.S. et al. // Inorg. Chem. 2016. V. 55. P. 7117.
Plajer A.J., Colebatch A.L., Rizzuto F.J. et al. // Angew. Chem., Int. Ed. 2018. V. 57. P. 6648.
Wade C.R., Saber M.R., Gabbai F.P. // Heteroat. Chem. 2011. V. 22. P. 500.
Tschersich C., Hoof S., Frank N. et al. // Inorg. Chem. 2016. V. 55. P. 1837.
Materne K., Braun-Cula B., Herwig C. et al. // Chem. Eur. J. 2017. V. 23. P. 11797.
Бацанов С.С. // Журн. неорган. химии. 1991. Т. 36. № 12. С. 3015.
Obata T., Matsumura M., Kawahata M. et al. // J. Organomet. Chem. 2016. V. 807. P. 17.
Kawahata M., Yasuike S., Kinebuchi I. et al. // Acta Crystallogr. E. 2011. V. 67. P. m25.
Breunig H.J., Nema M.G., Silvestru C. et al. // Z. Anorg. Allg. Chem. 2010. V. 636. P. 2378.
Alcantara E., Sharma P., Perez D. et al. // Synth. React. Inorg., Met.-Org., Nano-Met. Chem. 2012. V. 42. P. 1139.
Vranova I., Jambor R., Ruzicka A. et al. // Organometallics. 2015. V. 34. P. 534.
Rao M.L.N., Dhanorkar R.J. // RSC Advances. 2016. V. 6. P. 1012.
Hebert M., Petiot P., Benoit E. et al. // J. Org. Chem. 2016. V. 81. P. 5401.
Petiot P., Gagnon A. // Eur. J. Org. Chem. 2013. P. 5282.
Ahmad T., Dansereau J., Hebert M. et al. // Tetrahedron Lett. 2016. V. 57. P. 4284.
Шарутин В.В., Сенчурин В.С., Шарутина О.К., Чагарова О.В. // Журн. общ. химии. 2011. Т. 81. № 10. С. 1649.
Шарутин В.В., Шарутина О.К., Ермакова В.А., Смагина Я.Р. // Журн. неорган. химии. 2017. Т. 62. № 8. С. 1049.
Benjamin S.L., Karagiannidis L., Levason W. et al. // Organometallics. 2011. V. 30. P. 895.
Hirayama T., Mukaimine A., Nishigaki K. et al. // Dalton Trans. 2017. V. 46. P. 15991.
Ohshita J., Matsui S., Yamamoto R. et al. // Organometallics. 2010. V. 29. P. 3239.
Onishi K., Douke M., Nakamura T. et al. // J. Inorg. Biochem. 2012. V. 117. P. 77.
Preda A.M., Schneider W.B., Rainer M. et al. // Dalton Trans. 2017. V. 46. P. 8269.
Preda A.M., Schneider W.B., Schaarschmidt D. et al. // Dalton Trans. 2017. V.46. P. 13492.
Chen J., Murafuji T., Tsunashima R. // Organometallics. 2011. V. 30. P. 4532.
Parke S.M., Narreto M.A.B., Hupf E. et al. // Inorg. Chem. 2018. V. 57. P. 7536.
Worrell B.T., Ellery S.P., Fokin V.V. // Angew. Chem., Int. Ed. 2013. V. 52. P. 13037.
Parke S.M., Hupf E., Matharu G.K. et al. // Angew. Chem., Int. Ed. 2018. V. 57. P. 14841.
Ohshita J., Yamaji K., Ooyama Y. et al. // Organometallics. 2019. V. 38. P. 1516.
Брегадзе В.И., Глазун С.А., Ефремов А.Н., Шарутин В.В. // Вестник ЮУрГУ. Сер. Химия. 2020. Т. 12. № 1. С. 5.
Егорова И.В., Жидков В.В., Гринишак И.П. // Журн. общ. химии. 2015. Т. 85. № 7. С. 1172.
Kumar I., Bhattacharya P., Whitmire K.H. // J. Organomet. Chem. 2015. V. 794. P. 153.
Andrews P.C., Frank R., Junk P.C. et al. // J. Inorg. Biochem. 2011. V. 105. P. 454.
Anjaneyulu O., Maddileti D., Swamy K.C.K. // Dalton Trans. 2012. V. 41. P. 1004.
Егорова И.В., Шарутин В.В., Иваненко Т.К. и др. // Коорд. химия. 2006. Т. 32. № 5. С. 336.
Pathak A., Blair V.L., Ferrero R.L. et al. // J. Inorg. Biochem. 2017. V. 177. P. 266.
Stavila V., Whitmire K.H. // Acta Crystallogr. E. 2010. V. 66. P. m1547.
Andrews P.C., Deacon G.B., Junk P.C. et al. // Organometallics. 2009. V. 28. P. 3999.
Jambor R., Ružicková Z., Erben M., Dostál L. // Inorg. Chem. Commun. 2017. V. 76. P. 36.
Sun Y.-Q., Zhong J.-C., Liu L.-H. et al. // J. Mol. Struct. 2016. V. 1124. P. 138.
Andrews P.C., Junk P.C., Kedzierski L., Peiris R.M. et al. // Aust. J. Chem. 2013. V. 66. P. 1297.
Andrews P.C., Ferrero R.L., Junk P.C. et al. // Aust. J. Chem. 2012. V. 65. P. 883.
Chaudhari K.R., Yadav N., Wadawale A. et al. // Inorg. Chim. Acta. 2010. V. 363. P. 375.
Luqman A., Blair V.L., Bond A.M., Andrews P.C. // Angew. Chem., Int. Ed. 2013. V. 52. P. 7247.
Luqman A., Blair V.L., Brammananth R. et al. // Chem. Eur. J. 2014. V. 20. P. 14362.
Luqman A., Blair V.L., Brammananth R. et al. // Eur. J. Inorg. Chem. 2016. P. 2738.
Luqman A., Blair V.L., Brammananth R. et al. // Eur. J. Inorg. Chem. 2015. P. 725.
Luqman A., Blair V.L., Brammananth R. et al. // Eur. J. Inorg. Chem. 2015. P. 4935.
Andrews P.C., Ferrero R.L., Forsyth C.M. et al. // Organometallics. 2011. V. 30. P. 6283.
Шарутин В.В., Шарутина О.К. // Журн. неорган. химии. 2014. Т. 59. № 10. С. 1356.
Wrobel L., Ruffer T., Korb M. et al. // Chem. Eur. J. 2018. V. 24. P. 16630.
Andrews P.C., Busse M., Deacon G.B. et al. // Dalton Trans. 2010. V. 39. P. 9633.
Andrews P.C., Deacon G.B., Ferrero R.L. et al. // Dalton Trans. 2009. P. 6377.
Beckmann J., Bolsinger J., Duthie A. et al. // Inorg. Chem. 2012. V. 51. P. 12395.
Metre R.K., Kundu S., Narayanan R.S., Chandrasekhar V. // Phosphorus, Sulfur, Silicon Relat. Elem. 2015. V. 190. P. 2134.
Chandrasekhar V., Metre R.K., Narayanan R.S. // Dalton Trans. 2013. V. 42. P. 8709.
Cui L.-S., Meng J.-R., Gan Y.-L. et al. // Inorg. Nano-Metal Chem. 2017. V. 47. P. 1537.
Ritter C., Ringler B., Dankert F. et al. // Dalton Trans. 2019. V. 48. P. 5253.
Armstrong D., Taullaj F., Singh K. et al. // Dalton Trans. 2017. V. 46. P. 6212.
Breunig H.J., Haddad N., Lork E. et al. // Organometallics. 2009. V. 28. P. 1202.
Schwamm R.J., Fitchett C.M., Coles M.P. // Chem. Asian J. 2019. V. 14. P. 1204.
Шарутин В.В., Егорова И.В., Шарутина О.К. и др. // Коорд. химия. 2003. Т. 29. № 12. С. 902.
Briand G.G., Decken A., Hunter N.M. et al. // Polyhedron. 2012. V. 31. P. 796.
Breunig H.J., Lork E., Nema M.-G. // Z. Naturforsch. B. 2009. V. 64. P. 1213.
Cambridge Crystallographic Data Centre, 2019. http://www.ccdc.cam.ac.uk/data_request/cif.
Ramler J., Poater J., Hirsch F. et al. // Chem. Sci. 2019. V. 10. P. 4169.
Nekoueishahraki B., Sarish S.P., Roesky H.W. et al. // Angew. Chem., Int. Ed. 2009. V. 48. P. 4517. https://doi.org/10.1002/anie.200901215
Nekoueishahraki B., Samuel P.P., Roesky H.W. et al. // Organometallics. 2012. V. 31. P. 6697.
Lu W., Hu H., Li Y. et al. // J. Am. Chem. Soc. 2016. V. 138. P. 6650.
Waters J.B., Chen Q., Everitt T.A., Goicoechea J.M. // Dalton Trans. 2017. V. 46. P. 12053.
Aprile A., Corbo R., Tan K.V. et al. // Dalton Trans. 2014. V. 43. P. 764.
Wang G., Freeman L.A., Dickie D.A. et al. // Inorg. Chem. 2018. V. 57. P. 11687.
Wang G., Freeman L.A., Dickie D.A. et al. // Chem. Eur. J. 2019. V. 21. P. 4335.
Munzer J.E., Kneusels N.-J.H., Weinert B. et al. // Dalton Trans. 2019. V. 48. P. 11076.
Olaru M., Duvinage D., Lork E. et al. // Angew. Chem., Int. Ed. 2018. V. 57. P. 10080.
Bresien J., Hinz A., Schulz A., Villinger A. // Dalton Trans. 2018. V. 47. P. 4433.
Bresien J., Schulz A., Thomas M., Villinger A. // Eur. J. Inorg. Chem. 2019. P. 1279.
Solyntjes S., Neumann B., Stammler H.-G. et al. // Chem. Eur. J. 2017. V. 23. P. 1568.
Nishimoto Y., Takeuchi M., Yasuda M., Baba A. // Chem. Eur. J. 2013. V. 19. P. 14411.
Nishimoto Y., Takeuchi M., Yasuda M., Baba A. // Angew. Chem., Int. Ed. 2012. V. 51. P. 1051.
Ritschel B., Poater J., Dengel H. et al. // Angew. Chem., Int. Ed. 2018. V. 57. P. 3825.
Stavila V., Dikarev E.V. // J. Organomet. Chem. 2009. V. 694. P. 2956.
Chirca I., Silvestru C., Breunig H.J., Rat C.I. // Inorg. Chim. Acta. 2018. V. 475. P. 155.
Tan N., Chen Y., Zhou Y. et al. // ChemPlusChem. 2013. V. 78. P. 1363.
Toma A., Rat C.I., Silvestru A. et al. // J. Organomet. Chem. 2013. V. 745. P. 71.
Nema M.G., Breunig H.J., Soran A., Silvestru C. // J. Organomet. Chem. 2012. V. 705. P. 23.
Kannan R., Kumar S., Andrews A.P. et al. // Inorg. Chem. 2017. V. 56. P. 9391.
Li Y., Zhu H., Tan G. et al. // Eur. J. Inorg. Chem. 2011. P. 5265.
Simon P., Jambor R., Ruzicka A., Dostal L. // Organometallics. 2013. V. 32. P. 239.
Soran A., Breunig H.J., Lippolis V. et al. // Dalton Trans. 2009. V. 7. P. 77.
Simon P., Proft F., Jambor R. et al. // Angew. Chem., Int. Ed. 2010. V. 49. P. 5468.
Peveling K., Schurmann M., Herres-Pawlis S. et al. // Organometallics. 2011. V. 30. P. 5181.
Vrana J., Jambor R., Ruzicka A. et al. // Collect. Czech. Chem. Commun. 2010. V. 75. P. 1041.
Zhang X.-W., Xia J., Yan H.-W. et al. // J. Organomet. Chem. 2009. V. 694. P. 3019.
Toma A., Rat C.I., Silvestru A. et al. // J. Organomet. Chem. 2016. V. 806. P. 5.
Toma A.M., Pop A., Silvestru A. et al. // Dalton Trans. 2017. V. 46. P. 3953.
Tan N., Chen Y., Yin S.-F. et al. // Dalton Trans. 2013. V. 42. P. 9476.
Tan N., Yin S., Li Y. et al. // J. Organomet. Chem. 2011. V. 696. P. 1579.
Qiu R., Meng Z., Yin S. et al. // ChemPlusChem. 2012. V. 77. P. 404.
Qiu R., Yin S., Song X. et al. // Dalton Trans. 2011. V. 40. P. 9482.
Sindlinger C.P., Stasch A., Wesemann L. // Organometallics. 2014. V. 33. P. 322.
Strimb G., Pollnitz A., Rat C.I., Silvestru C. // Dalton Trans. 2015. V. 44. P. 9927.
Mairychova B., Svoboda T., Stepnicka P. et al. // Inorg. Chem. 2013. V. 52. P. 1424.
Korenkova M., Mairychova B., Ruzicka A. et al. // Dalton Trans. 2014. V. 43. P. 7096.
Fridrichova A., Mairychova B., Padelkova Z. et al. // Dalton Trans. 2013. V. 42. P. 16403.
Fanfrlik J., Sedlak R., Pecina A. et al. // Dalton Trans. 2016. V. 45. P. 462.
Dostal L., Jambor R., Ruzicka A. et al. // Inorg. Chem. 2015. V. 54. P. 6010.
Fridrichova A., Svoboda T., Jambor R. et al. // Organometallics. 2009. V. 28. P. 5522.
Svoboda T., Jambor R., Ruzicka A. et al. // Eur. J. Inorg. Chem. 2010. P. 1663.
Svoboda T., Jambor R., Ruzicka A. et al. // Organometallics. 2012. V. 31. P. 1725.
Mairychova B., Svoboda T., Erben M. et al. // Organometallics. 2013. V. 32. P. 157.
Chovancova M., Jambor R., Ruzicka A. et al. // Organometallics. 2009. V. 28. P. 1934.
Dostal L., Jambor R., Ruzicka A. et al. // Organometallics. 2010. V. 29. P. 4486.
Dostal L., Jambor R., Erben M., Ruzicka A. // Z. Anorg. Allg. Chem. 2012. Bd. 638. S. 614.
Tan N., Zhang X. // Acta Crystallogr. E. 2011. V. 67. P. m252.
Zhang X., Qiu R., Tan N. et al. // Tetrahedron Lett. 2010. V. 51. P. 153.
Qiu R., Qiu Y., Yin S. et al. // Adv. Synth. Catal. 2010. V. 352. P. 153.
Qiu R., Yin S., Zhang X. et al. // Chem. Commun. 2009. P. 4759.
Liu Y.-P., Lei J., Tang L.-W. et al. // Eur. J. Med. Chem. 2017. V. 139. P. 826.
Toma A.M., Rat C.I., Pavel O.D. et al. // Cat. Sci. Tech. 2017. V. 7. P. 5343.
Zhang X., Yin S., Qiu R. et al. // J. Organomet. Chem. 2009. V. 694. P. 3559.
Zhang X.-W., Fan T. // Acta Crystallogr. E. 2011. V. 67. P. m875.
Murafuji T., Kitagawa K., Yoshimatsu D. et al. // Eur. J. Med. Chem. 2013. V. 63. P. 531.
Breunig H.J., Nema M.G., Silvestru C. et al. // Dalton Trans. 2010. V. 39. P. 11277.
Dostal L., Jambor R., Ruzicka A. et al. // Dalton Trans. 2011. V. 40. P. 8922.
Tan N., Wu S., Huiqiong Y. et al. // Z. Kristallogr. New Cryst. Struct. 2019. V. 234. P. 509.
Vranova I., Erben M., Jambor R. et al. // Z. Anorg. Allg. Chem. 2016. V. 642. P. 1212.
Tan N., Dang L., Lan D. et al. // Z. Kristallogr.-New Cryst. Struct. 2018. V. 233. P. 875.
Soran A.P., Nema M.G., Breunig H.J., Silvestru C. // Acta Crystallogr. E. 2011. V. 67. P. m153.
Simon P., Jambor R., Ruzicka A., Dostal L. // J. Organomet. Chem. 2013. V. 740. P. 98.
Casely I.J., Ziller J.W., Fang M. et al. // J. Am. Chem. Soc. 2011. V. 133. P. 5244.
Kindra D.R., Casely I.J., Fieser M.E. et al. // J. Am. Chem. Soc. 2013. V. 135. P. 7777.
Kindra D.R., Casely I.J., Ziller J.W., Evans W.J. // Chem. Eur. J. 2014. V. 20. P. 15242.
Егорова И.В., Жидков В.В., Гринишак И.П., Резванова А.А. // Журн. общ. химии. 2014. Т. 84. № 7. С. 1179.
Solyntjes S., Neumann B., Stammler H.-G. et al. // Eur. J. Inorg. Chem. 2016. P. 3999.
Verkhovykh V.A., Kalistratova O.S., Grishina A.I. et al. // Вест. Южно-Урал. гос. ун-та. Сер. Химия. 2015. Т. 7. № 3. С. 61.
Gushchin F.V., Kalistratova O.S., Maleeva A.I. et al. // Вест. Южно-Урал. гос. ун-та. Сер. Химия. 2016. Т. 8. № 1. С. 51.
Гущин А.В., Шашкин Д.В., Прыткова Л.К. и др. // Журн. общ. химии. 2011. Т. 81. № 3. С. 397.
Шарутин В.В., Сенчурин В.С., Шарутина О.К. // Журн. неорган. химии. 2011. Т. 56. № 10. С. 1644.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2014. Т. 59. № 1. С. 42.
Шарутин В.В., Шарутина О.К. // Журн. неорган. химии. 2014. Т. 59. № 6. С. 734.
Гусаковская А.А., Калистратова О.С., Андреев П.В. и др. // Кристаллография. 2018. Т. 63. № 2. С. 203.
Шарутин В.В., Шарутина О.К., Ефремов А.Н. // Журн. неорган. химии. 2019. Т. 64. № 2. С. 159.
Duffin R.N., Blair V.L., Kedzierski L., Andrews P.C. // Dalton Trans. 2018. V. 47. P. 971.
Duffin R.N., Blair V.L., Kedzierski L., Andrews P.C. // J. Inorg. Biochem. 2018. V. 189. P. 151.
Ong Y.C., Blair V.L., Kedzierski L., Andrews P.C. // Dalton Trans. 2014. V. 43. P. 12904.
Ong Y.C., Blair V.L., Kedzierski L. et al. // Dalton Trans. 2015. V. 44. P. 18215.
Kumar I., Bhattacharya P., Whitmire K.H. // Organometallics. 2014. V. 33. P. 2906.
Cui L., Bi C., Fan Y. et al. // Inorg. Chim. Acta. 2015. V. 437. P. 41.
Kiran A.B., Mocanu T., Pollnitz A. et al. // Dalton Trans. 2018. V. 47. P. 2531.
Zhang X.-Y., Wu R.-X., Bi C.-F. et al. // Inorg. Chim. Acta. 2018. V. 483. P. 129.
Feham K., Benkadari A., Chouaih A. et al. // Cryst. Struct. Theor. Appl. 2013. V. 2. P. 28.
Андреев П.В., Сомов Н.В., Калистратова О.С. и др. // Кристаллография. 2015. Т. 60. № 4. С. 571.
Шарутин В.В., Егорова И.В., Казаков М.А., Шарутина О.К. // Журн. неорган. химии. 2009. Т. 54. № 7. С. 1156.
Andreev P.V., Somov N.V., Kalistratova O.S. et al. // Acta Crystallogr. E. 2013. V. 69. P. m333.
Шарутин В.В., Сенчурин В.С., Шарутина О.К. и др. // Журн. общ. химии. 2010. Т. 80. № 10. С. 1630.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. и др. // Бутлеровские сообщения. 2012. Т. 29. С. 51.
Шарутин В.В., Шарутина О.К. // Журн. общ. химии. 2016. Т. 86. № 5. С. 811.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2016. Т. 61. № 3. С. 334.
Goswami M., Ellern A., Pohl N.L.B. // Angew. Chem., Int. Ed. 2013. V. 52. P. 8441.
Шарутин В.В., Шарутина О.К. // Журн. неорган. химии. 2016. Т. 61. № 8. С. 1023.
Robertson A.P.M., Burford N., McDonald R., Ferguson M.J. // Angew. Chem., Int. Ed. 2014. V. 53. P. 3480.
Егорова И.В., Жидков В.В., Гринишак И.П. и др. // Журн. неорган. химии. 2018. Т. 63. № 7. С. 816.
Wittig G., Clauß K. // Lieb. Ann. 1952. Bd. 578. № 1. S. 136.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. неорган. химии. 2020. Т. 65. № 11. С. 1516.
Сенчурин В.С., Шарутин В.В., Шарутина О.К. // Журн. неорган. химии. 2020. Т. 65. № 3. С. 320.
Шарутин В.В., Шарутина О.К., Сенчурин В.С. // Журн. структур. химии. 2020. Т. 61. № 5. С. 776.
Шарутин В.В., Егорова И.В., Шарутина О.К. и др. // Коорд. химия. 2008. Т. 34. № 2. С. 89.
Шарутин В.В., Шарутина О.К., Егорова И.В. и др. // Изв. АН. Сер. хим. 1999. № 12. С. 2350.
Егорова И.В. Дис. ... докт. хим. наук. Н. Новгород, 2008. 298 с.
Glowka M.L., Martynowski D., Kozlowska K. // J. Mol. Struct. 1999. V. 474. P. 81.
Tsuzuki S., Honda K., Uchimaru T. et al. // J. Am. Chem. Soc. 2002. V. 124. № 1. P. 104.
Сенчурин В.С. // Вест. Южно-Урал. гос. ун-та. Сер. Химия. 2019. Т. 11. № 3. С. 50.
Шарутин В.В., Егорова И.В., Клепиков Н.Н. и др. // Журн. неорган. химии. 2009. Т. 54. № 1. С. 53.
Шарутин В.В., Егорова И.В., Клепиков Н.Н. и др. // Журн. неорган. химии. 2009. Т. 54. № 11. С. 1847.
Ooi T., Goto R., Maruoka K. // J. Am. Chem. Soc. 2003. V. 125. P. 10494.
Park G., Brock D.J., Pellois J.-P., Gabbai F.P. // Chem. Cell Press. 2019. V. 5. P. 2215.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия