

Координационная химия, 2021, T. 47, № 2, стр. 121-130
Тетра-, пента- и гексакоординированные стереоизомеры бислигандных комплексов Zn(II) и Cd(II) на основе (N,O,S(Se))-тридентатных азометинов. Квантово-химическое исследование
Н. Н. Харабаев *
НИИ физической и органической химии Южного федерального университета
Ростов-на-Дону, Россия
* E-mail: kharabayev@sfedu.ru
Поступила в редакцию 09.07.2020
После доработки 05.08.2020
Принята к публикации 07.08.2020
Аннотация
Методом теории функционала плотности и неэмпирическим методом Хартри–Фока рассчитаны молекулярные структуры и относительные энергии тетра-, пента- и гексакоординированных стереоизомеров бислигандных комплексов Zn(II) и Cd(II) на основе (N,O,Y (Y = S, Se))-тридентатных азометинов (координационные узлы конкурирующих стереоизомеров, соответственно, MN2O2, MN2O2Y, MN2O2Y2). Моделирование механизма формирования тетра-, пента- и гексакоординированных стереоизомеров с учетом возможной последующей стереоизомеризации позволило установить для комплексов цинка предпочтительность тетракоординации (в виде псевдотетраэдра), а для комплексов кадмия пента- или гексакоординации в зависимости от особенностей строения лигандов.
Бис-лигандные тетракоординированные комплексы 3d-переходных металлов на основе ароматических азометинов хелатного типа подробно изучены ранее [1–3] и установлено, что их строение, спектральные, магнитные и другие физические свойства определяются (главным образом) составом и конфигурацией координационного узла MN2X2 (X = NR, O, S, Se). Формирование одной из возможных псевдотетраэдрической, цис- или транс-планарной конфигурации координационного узла MN2X2 зависит от типа центрального иона, природы лигатирующих атомов и структурных особенностей лигандов, что экспериментально установлено [1–3] и теоретически (на основе квантово-химических исследований) интерпретировано [4, 5]. В случае бис-лигандных комплексов ML2 на основе азометинов, включающих дополнительные координационно-активные Y-донорные центры (II, III), ситуация существенно усложняется, так как возможность образования дополнительных координационных связей Y → M переводит бидентатные лиганды (N,O)-хелатного типа I в потенциально (N,O,Y)-тридентатные лиганды II, III.
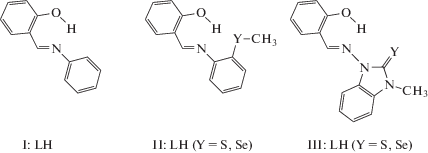
Для потенциально тридентатных азометинов II, III комплексы ML2 на их основе поливариантны по составу и конфигурации координационного узла (MN2O2, MN2O2Y, MN2O2Y2 (Y = S, Se)), что проявляется в рядах конкурирующих межу собой тетра-, пента- и гексакоординированных стереоизомеров IIа, IIb, IIc и IIIа, IIIb, IIIc.
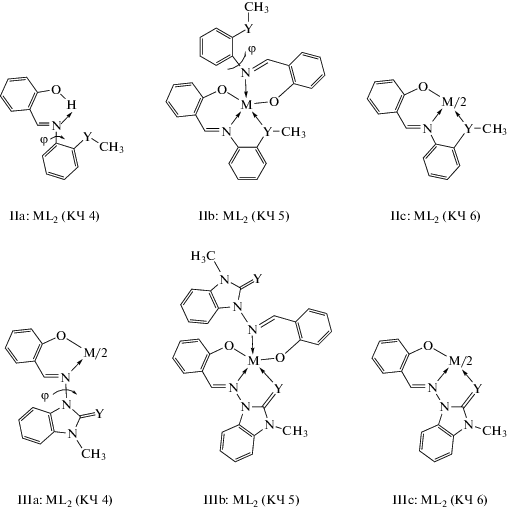
M = Co(II), Ni(II), Zn(II), Cd(II); Y = S, Se
Роль электронной конфигурации центрального атома металла в конкуренции тетра-, пента- и гексакоординированных стереоизомеров IIа, IIb, IIc и IIIа, IIIb, IIIc комплексов ML2 на основе азометинов II и III, соответственно, исследована ранее [6] на примере комплексов кобальта (d7(Co2+)) и никеля (d8(Ni2+)). Для определения роли и влияния размера центрального иона на конкуренцию тетра-, пента- и гексакоординации при формировании комплексов ML2 квантово-химическое исследование относительной устойчивости стереоизомеров IIа, IIb, IIc и IIIа, IIIb, IIIc продолжено в настоящей работе на примере комплексов d10-металлов: цинка(II) и кадмия(II). По аналогии с [6] изучено также влияние особенностей строения лигандов типа II (содержащих фенилтио(селено)эфирные фрагменты с атомом Y в тиольной форме) и лигандов типа III (содержащих тио(селено)бензимидазольные фрагменты с атомом Y в тионной форме) на конкуренцию тетра-, пента- и гексакоординации в комплексах ZnL2 и CdL2.
Следует отметить, что, согласно результатам РСА, для бис-лигандных комплексов d10-металлов (Zn2+, Cd2) на основе азометинов хелатного типа I характерна псевдотетраэдрическая конфигурация [1], а при наличии у лигандов дополнительных донорных центров для комплексов d10-металлов свойственно расширение состава координационного узла (от тетра- до пента- и гексакоординации) как в комплексах Zn(II) [7–13], так и в комплексах Cd(II) [8, 12, 14–16].
МЕТОДИКА РАСЧЕТОВ
Квантово-химические расчеты молекулярной структуры комплексов Zn(II) и Cd(II) (как и аналогичных комплексов Co(II) и Ni(II) в предыдущем исследовании [6]) проведены по программе Gaussian09 [17] методом теории функционала плотности (DFT) [18] с использованием гибридного функционала B3LYP [19, 20]. Вместе с тем, учитывая известное влияние на результаты DFT-исследований типа выбранного функционала плотности [21, 22], DFT/B3LYP-расчеты в настоящей работе продублированы расчетами стереоизомеров комплексов Zn(II) и Cd(II) неэмпирическим методом Хартри–Фока для молекулярных систем с закрытой оболочкой (RHF). В DFT- и RHF-расчетах использован базис 6-311++G(d,p) для комплексов Zn(II) и базис SDD для комплексов Cd(II). Локализация и анализ стационарных точек на поверхности потенциальной энергии проведены путем полной оптимизации геометрии молекул в сопровождении с расчетом колебательных спектров для основных состояний стереоизомеров комплексов Zn(II) и Cd(II) и переходных состояний в реакции стереоизомеризации в комплексах Zn(II). При проведении расчетов учтено, что основным у тетра-, пента- и гексакоординированных стереоизомеров комплексов Zn(II) и Cd(II) является низкоспиновое электронное состояние. Графические изображения молекулярных структур построены по программе ChemCraft [23].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Согласно результатам квантово-химических расчетов, проведенных DFT-методом и продублированных RHF-методом, более выгодными, судя по относительной энергии тетра-, пента- и гексакоординированных стереоизомеров бис-лигандных комплексов Zn(II) на основе потенциально тридентатных азометинов II и III (Y = S, Se), являются тетракоординированные изомеры (табл. 1) в виде псевдотетраэдров (табл. 2). Отметим согласие на качественном уровне результатов RHF- и DFT-расчетов (табл. 1).
Таблица 1.
Относительная энергия без учета (ΔE, ккал/моль) и с учетом нулевых колебаний (ΔEZPE, ккал/моль) стереоизомеров IIa, IIb, IIc и IIIа, IIIb, IIIс комплексов ZnL2 (Y = S, Se)
| Стереоизомеры IIа, IIb, IIс (Y) | Стереоизомеры IIIа, IIIb, IIIс (Y) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ZnL2 (Y) Y = S, Se |
DFT/B3LYP | RHF | ZnL2 (Y) Y = S, Se |
DFT/B3LYP | RHF | ||||
| ккал/моль | ккал/моль | ||||||||
| ΔE | ΔEZPE | ΔE | ΔEZPE | ΔE | ΔEZPE | ΔE | ΔEZPE | ||
| IIа (S) IIb (S) IIс (S) |
0.0 1.3 1.2 |
0.0 1.1 0.9 |
0.0 1.0 1.7 |
0.0 0.7 1.6 |
IIIа (S) IIIb (S) IIIс (S) |
0.0 2.2 7.1 |
0.0 1.7 6.3 |
0.0 2.2 11.1 |
0.0 1.8 10.5 |
| IIа (Se) IIb (Se) IIс (Se) |
0.0 1.8 2.1 |
0.0 1.5 1.8 |
0.0 0.6 3.6 |
0.0 0.3 3.5 |
IIIа (Se) IIIb (Se) IIIс (Se) |
0.0 1.8 7.3 |
0.0 1.5 6.8 |
0.0 2.2 12.2 |
0.0 1.8 11.5 |
Таблица 2.
Рассчитанные (DFT-метод) геометрические параметры координационных узлов ZnN2O2, ZnN2O2Y, ZnN2O2Y2 стереоизомеров IIа, IIb, IIс и IIIа, IIIb, IIIс комплексов ZnL2 на основе азометинов II и III (Y = S, Se)
| Стереоизомеры IIа, IIb, IIс комплексов ZnL2 (Y = S, Se) | ||||||
|---|---|---|---|---|---|---|
| ZnL2 (Y, КЧ) Y = S, Se |
Zn–N, Å | Zn–O, Å | ∠NZnO, град |
∠NZnN, град |
∠OZnO, град |
Zn–Y, Å |
| IIа (S, КЧ 4) | 2.031 | 1.947 | 95.0 | 124.8 | 120.7 | |
| IIb (S, КЧ 5) | 2.078 (2.035) |
1.987 (1.958) |
91.1 (95.7) |
121.3 | 103.1 | 3.019 |
| IIс (S, КЧ 6) | 2.128 | 2.030 | 88.1 | 166.4 | 94.8 | 2.757 |
| IIа (Se, КЧ 4) | 2.032 | 1.950 | 94.9 | 128.6 | 121.6 | |
| IIb (Se, КЧ 5) | 2.078 (2.037) |
1.993 (1.961) |
90.8 (95.5) |
124.1 | 102.1 | 3.100 |
| IIс (Se, КЧ 6) | 2.134 | 2.031 | 88.0 | 167.5 | 93.9 | 2.871 |
| Стереоизомеры IIIа, IIIb, IIIс комплексов ZnL2 (Y = S, Se) | ||||||
| IIIа (S, КЧ 4) | 2.042 | 1.936 | 93.6 | 117.1 | 115.4 | |
| IIIb (S, КЧ 5) | 2.181 (2.102) |
1.988 (1.976) |
84.4 (90.1) |
101.9 | 100.0 | 2.613 |
| IIIс (S, КЧ 6) | 2.165 | 2.044 | 83.6 | 169.5 | 91.3 | 2.684 |
| IIIа (Se, КЧ 4) | 2.039 | 1.937 | 93.5 | 118.0 | 115.2 | |
| IIIb (Se, КЧ 5) | 2.204 (2.109) |
1.990 (1.984) |
83.9 (89.6) |
100.9 | 99.3 | 2.685 |
| IIIс (Se, КЧ 6) | 2.184 | 2.049 | 82.8 | 169.1 | 89.9 | 2.786 |
В табл. 2 собраны рассчитанные (DFT-метод) геометрические параметры координационных узлов тетра-, пента- и гексакоординированных стереоизомеров IIа, IIb, IIс и IIIа, IIIb, IIIс комплексов ZnL2 (Y = S, Se) на основе азометинов типа II и III (числовые значения в скобках относятся к лигандам, проявившим бидентатность при формировании пентакоординированных стереоизомеров).
Для тетракоординированных стереоизомеров IIа и IIIа комплексов ZnL2 (Y = S, Se), как наиболее выгодных по полной энергии (табл. 1), проведена оценка возможности их формирования с использованием предложенной ранее [24, 25] постадийной модели механизма реакции образования бис-лигандных комплексов ML2. Первая стадия в этой модели: M++ + (L)– → (ML)+, т.е. связывание ионом металла первого лиганда, вторая стадия: (ML)+ + + (L)– → ML2, т.е. связывание ионом (ML)+ второго лиганда и формирование исходного стереоизомера комплекса ML2, третья стадия: стереоизомеризация от исходного до энергетически наиболее выгодного изомера комплекса ML2, оценка величины барьера которой характеризует возможность формирования конечной структуры. Если же исходный стереоизомер комплекса ML2, как продукт реакции (ML)+ + (L)– → ML2, является (в то же время) энергетически наиболее выгодным, то именно для этого изомера прогнозируется предпочтительность.
Для первой стадии модельной реакции – стадии формирования катионов (ZnL)+ на основе азометинов II, проведены квантово-химические расчеты молекулярной структуры катионов (ZnL)+. На основании этих расчетов установлено, что дополнительный Y-донорный центр первого лиганда (Y = S, Se) участвует в образовании координационной связи Y→Zn и первый лиганд при формировании катионов (ZnL)+ проявляет тридентатность (рис. 1). Это использовано при построении модели для второй стадии – стадии связывания катионом (ZnL)+ аниона второго лиганда (L)– и формирования исходного (для возможной последующей стереоизомеризации) изомера комплекса ZnL2 (рис. 1).
Рис. 1.
Циклические фрагменты рассчитанных (DFT-метод) структур катиона (ZnL)+, аниона (L)– и пентакоординированного изомера типа IIb комплекса ZnL2 (Y = S).
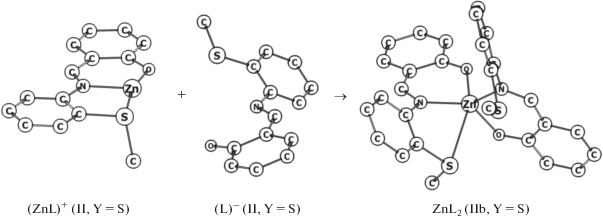
Для второй стадии модельной реакции (рис. 1) при стартовом расстоянии, равном 5 Å, между атомом цинка катиона (ZnL)+ и атомом азота аниона второго лиганда (L)– локализована (DFT-метод) пентакоординированная структура IIb комплексов ZnL2 (Y = S, Se), в которой тридентатность первого лиганда сочетается с бидентатностью второго.
Поскольку пентакоординированный изомер IIb комплексов ZnL2 (Y = S, Se), построенный на второй стадии модельной реакции (рис. 1), не является наиболее выгодным по полной энергии (табл. 1), то он может рассматриваться в качестве исходного изомера для последующего межконфигурационного перехода IIb → IIа в сторону энергетически более выгодного тетракоординированного изомера IIа (рис. 2).
Рис. 2.
Циклические фрагменты рассчитанных (DFT-метод) молекулярных структур тетра- и пентакоординированных изомеров IIа (КЧ 4) и IIb (КЧ 5) комплекса ZnL2 (Y = S).
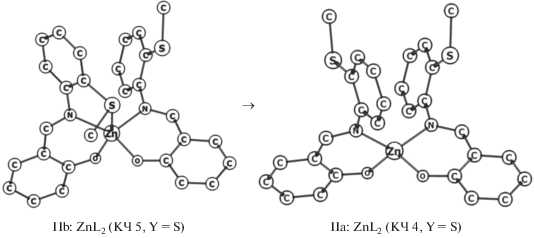
Механизм реакции стереоизомеризации IIb → IIа в комплексах ZnL2 сводится к разрыву координационной связи Y→Zn (Y = S, Se) за счет поворота вокруг связи C–N фенильного фрагмента первого лиганда (рис. 2). При этом стереоизомер IIа может быть образован из исходного IIb за счет поворота фенильного фрагмента как по, так и против часовой стрелки. Переходное состояние для реакции стереоизомеризации IIb → IIа (как показывают квантово-химические расчеты) достигается при повороте по часовой стрелке на 124° и на 121° для S- и Se-содержащих комплексов ZnL2 соответственно, а величина барьера реакции IIb → IIа равна 6.9 ккал/моль (Y = S) и 7.3 ккал/моль (Y = Se). При повороте вокруг связи C–N фенильного фрагмента первого лиганда против часовой стрелки переходное состояние достигается для S- и Se-содержащих комплексов ZnL2 при повороте на 51° и 55° соответственно, при этом значение барьера реакции IIb → IIа равно 2.8 и 2.9 ккал/моль. Такие значения барьера реакции стереоизомеризации IIb → IIа от исходного IIb до энергети-чески наиболее выгодного тетракоординированного изомера IIа (псевдотетраэдра (табл. 2)) позволяют заключить о его доступности при формировании комплексов ZnL2 на основе азометинов II (Y = S, Se).
Для энергетически наиболее выгодного тетракоординированного изомера IIIа (табл. 1) исследование его доступности при формировании комплексов ZnL2 на основе азометинов III (Y = S, Se) проведено в рамках постадийной модели механизма реакции образования бис-лигандных комплексов ML2 [24] и привело к таким же заключениям, что и сделанные выше для комплексов ZnL2 на основе азометинов II (Y = S, Se). Во-первых, для стадии формирования катионов (ZnL)+ на основе азометинов III (Y = S, Se) квантово-химические расчеты позволили также установить участие Y-донорного центра первого лиганда в образовании дополнительной координационной связи Y→Zn. Во-вторых, в модели связывания катионом (ZnL)+ аниона второго лиганда (L)–, также локализована (DFT-метод) пентакоординированная структура IIIb комплексов ZnL2 (рис. 3). В-третьих, не являясь энергетически наиболее выгодным (табл. 1), пентакоординированный изомер IIIb также может рассматриваться как исходный для последующего межконфигурационного перехода в сторону более выгодного тетракоординированного изомера IIIа (рис. 3).
Рис. 3.
Циклические фрагменты рассчитанных (DFT-метод) молекулярных структур тетра- и пентакоординированных изомеров IIIа (КЧ 4) и IIIb (КЧ 5) комплекса ZnL2 (Y = S).
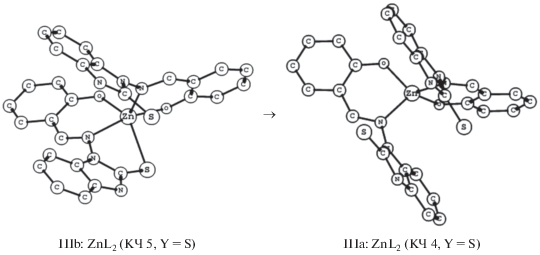
Межконфигурационный переход от исходного пентакоординированного IIIb до наиболее выгодного тетракоординированного изомера IIIа комплексов ZnL2 на основе азометинов III, так же как и в случае комплексов ZnL2 на основе азометинов II, сводится к разрыву координационной связи Y→Zn (Y = S, Se) за счет поворота участвующего в дополнительной координации бензимидазольного фрагмента вокруг связи N–N (рис. 3). При повороте этого фрагмента против часовой стрелки значение барьера реакции IIIb → IIIа для комплексов ZnL2 (Y = S) и ZnL2 (Y = Se) равно 2.0 и 2.5 ккал/моль соответственно [24]. Это позволяет сделать вывод о возможности формирования наиболее выгодного тетракоординированного изомера IIIа (табл. 1), как и для рассмотренного выше тетракоординированного изомера IIа комплексов ZnL2 на основе азометинов II (Y = S, Se).
Таким образом, согласно квантово-химическому исследованию тетра-, пента- и гексакоординированных стереоизомеров комплексов ZnL2 на основе потенциально тридентатных азометинов III (Y = S, Se) с учетом возможных межконфигурационных переходов, более предпочтительным является тетракоординированный стереоизомер IIIа.
Согласно результатам квантово-химических расчетов молекулярной структуры и относительной энергии тетра-, пента- и гексакоординированных стереоизомеров бис-лигандных комплексов Cd(II) на основе тридентатных азометинов II и III (табл. 3, 4), наиболее выгодными являются, с одной стороны, гексакоординированный изомер IIс комплексов CdL2 на основе азометинов II, с другой – пентакоординированный изомер IIIb комплексов CdL2 на основе азометинов III. Для комплексов CdL2 также можно отметить согласие на качественном уровне результатов RHF- и DFT-расчетов (табл. 3).
Таблица 3.
Относительная энергия без учета (ΔE, ккал/моль) и с учетом нулевых колебаний (ΔEZPE, ккал/моль) стереоизомеров IIa, IIb, IIc и IIIа, IIIb, IIIс комплексов CdL2 (Y = S, Se)
| Стереоизомеры IIа, IIb, IIс (Y) | Стереоизомеры IIIа, IIIb, IIIс (Y) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CdL2 (Y) Y = S, Se |
DFT/B3LYP | RHF | CdL2 (Y) Y = S, Se |
DFT/B3LYP | RHF | ||||
| ккал/моль | ккал/моль | ||||||||
| ΔE | ΔEZPE | ΔE | ΔEZPE | ΔE | ΔEZPE | ΔE | ΔEZPE | ||
| IIа (S) IIb (S) IIс (S) |
5.6 3.4 0.0 |
5.8 3.6 0.0 |
5.4 3.0 0.0 |
5.4 2.9 0.0 |
IIIа (S) IIIb (S) IIIс (S) |
3.1 0.0 2.9 |
3.4 0.0 2.4 |
1.4 0.0 6.1 |
1.7 0.0 5.8 |
| IIа (Se) IIb (Se) IIс (Se) |
4.5 2.6 0.0 |
4.7 2.6 0.0 |
2.3 0.5 0.0 |
2.4 0.5 0.0 |
IIIа (Se) IIIb (Se) IIIс (Se) |
3.0 0.0 2.4 |
3.1 0.0 1.9 |
2.0 0.0 6.6 |
2.4 0.0 6.2 |
Таблица 4.
Рассчитанные (DFT-метод) геометрические параметры координационных узлов CdN2O2, CdN2O2Y, CdN2O2Y2 стереоизомеров IIа, IIb, IIс и IIIа, IIIb, IIIс комплексов CdL2 на основе азометинов II и III (Y = S, Se)
| Стереоизомеры IIа, IIb, IIс комплексов CdL2 (Y = S, Se) | ||||||
|---|---|---|---|---|---|---|
| CdL2 (Y, КЧ) Y = S, Se |
Cd–N, Å | Cd–O, Å | ∠NCdO, град |
∠NCdN, град |
∠OCdO, град |
Cd–Y, Å |
| IIа (S, КЧ 4) | 2.226 | 2.154 | 88.2 | 136.6 | 129.597.2 | |
| IIb (S, КЧ 5) | 2.294 (2.232) |
2.201 (2.176) |
83.5 (88.1) |
136.2 | 109.0 | 2.913 |
| IIс (S, КЧ 6) | 2.319 | 2.219 | 82.5 | 159.5 | 97.2 | 2.911 |
| IIа (Se, КЧ 4) | 2.220 | 2.156 | 87.9 | 137.1 | 128.2 | |
| IIb (Se, КЧ 5) | 2.296 (2.237) |
2.197 (1.183) |
83.9 (87.4) |
134.3 | 107.2 | 2.977 |
| IIс (Se, КЧ 6) | 2.318 | 2.227 | 82.4 | 171.1 | 94.2 | 2.968 |
| Стереоизомеры IIIа, IIIb, IIIс комплексов CdL2 (Y = S, Se) | ||||||
| IIIа (S, КЧ 4) | 2.248 | 2.134 | 85.9 | 114.9 | 115.4 | |
| IIIb (S, КЧ 5) | 2.342 (2.298) |
2.187 (2.155) |
79.1 (83.9) |
102.0 | 100.0 | 2.811 |
| IIIс (S, КЧ 6) | 2.368 | 2.226 | 77.3 | 164.6 | 90.7 | 2.835 |
| IIIа (Se, КЧ 4) | 2.242 | 2.134 | 86.1 | 116.0 | 119.4 | |
| IIIb (Se, КЧ 5) | 2.357 (2.301) |
1.191 (2.165) |
78.8 (83.4) |
101.1 | 106.1 | 2.854 |
| IIIс (Se, КЧ 6) | 2.387 | 2.233 | 76.6 | 166.7 | 89.5 | 2.903 |
В табл. 4 собраны рассчитанные (DFT-метод) геометрические параметры координационных узлов тетра-, пента- и гексакоординированных стереоизомеров комплексов CdL2 на основе азометинов II и III (Y = S, Se).
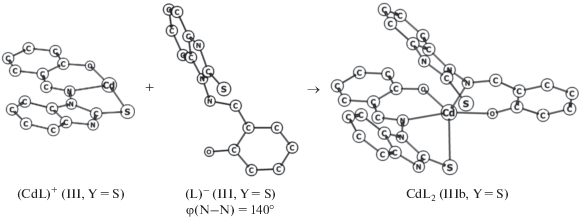
Для оценки возможности формирования наиболее выгодных по энергии гексакоординированного изомера IIс для комплексов CdL2 на основе азометинов II и пентакоординированного изомера IIIb для комплексов CdL2 на основе азометинов III в рамках постадийной модели механизма реакции образования бис-лигандных комплексов ML2 [24, 25] проведены квантово-химические расчеты молекулярной структуры катионов (CdL)+, как результата связывания ионом металла аниона первого лиганда. Согласно расчетам, Y-донорный центр первого лиганда участвует в дополнительной координации Y→Cd (Y = S, Se) при формировании катионов (CdL)+ на основе азометинов II и III. Это учтено в модели второй стадии формирования комплексов ML2 (рис. 4, 5), в которой при стартовом расстоянии (5 Å) между атомом кадмия катиона (CdL)+ и атомом азота аниона второго лиганда (L)– локализованными оказались, с одной стороны, гексакоординированный изомер IIс, с другой – пентакоординированный изомер IIIb.
Рис. 4.
Циклические фрагменты рассчитанных (DFT-метод) структур катиона (CdL)+, аниона (L)– и гексакоординированного изомера IIc комплекса CdL2 (Y = S).
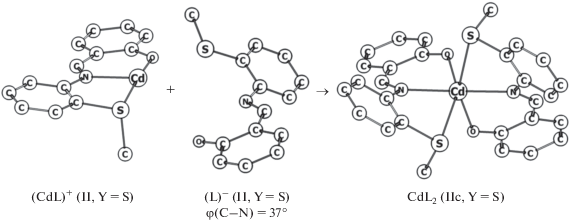
Рис. 5.
Циклические фрагменты рассчитанных (DFT-метод) структур катиона (CdL)+, аниона (L)– и пентакоординированного изомера IIIb комплекса CdL2 (Y = S).
Таким образом, гексакоординированный изомер IIc комплексов Cd(II) на основе азометинов II и пентакоординированный изомер IIIb комплексов Cd(II) на основе азометинов III являются, с одной стороны, исходными изомерами для последующих межконфигурационных переходов (как продукты реакции (CdL)+ + (L)– → CdL2 (рис. 4, 5)). С другой стороны, эти же изомеры наиболее выгодны по полной энергии (табл. 3), что позволяет заключить о предпочтительности гексакоординации в комплексах CdL2 на основе азометинов II и пентакоординации в комплексах CdL2 на основе азометинов III.
Следует отметить, что выявленная (в случае комплексов CdL2 на основе азометинов II и III) зависимость структуры предпочтительного стереоизомера (с гекса- и пентакоординацией соответственно) от особенностей строения лигандов аналогична проанализированнай ранее при исследовании бис-лигандных комплексов Ni(II) на основе азометинов II и III [6]. С учетом этого формирование пентакоординации кадмия в исходном изомере комплексов CdL2 на основе азометинов III (рис. 5) можно отнести к высокой степени акопланарности аниона (L)– (диэдральный угол поворота φ бензимидазольного фрагмента вокруг связи N–N равен 140°). В результате донорный атом Y выведен из реакционного пространства аниона (L)–, что придает ему четко выраженный (N,O)-хелатный тип. Такое строение аниона (L)– азометинов III способствует проявлению бидентатности вторым лигандом при формировании исходного (пентакоординированного) изомера комплекса CdL2 (рис. 5). В отличие от азометинов III для азометинов II строение аниона (L)– более уплощенное (диэдральный угол поворота φ фенильного фрагмента вокруг связи C–N равен 37°). В этом случае атом Y находится вместе с другими донорными атомами N и O в реакционном пространстве аниона (L)–, что способствует проявлению им тридентатности при формировании исходного (гексакоординированного) изомера комплекса CdL2 (рис. 4).
Таким образом, квантово-химическое исследование механизма формирования с учетом межконфигурационных переходов для тетра-, пента- и гексакоординированных стереоизомеров бис-лигандных комплексов Zn(II) и Cd(II) на основе (N,O,S(Se))-тридентатных азометинов позволило установить, что для комплексов цинка предпочтительна тетракоординация (в виде псевдотетраэдра), а для комплексов кадмия пента- или гексакоординация в зависимости от особенностей строения лигандов. Для азометиновых лигандов с N-фенилтио(селено)эфирным заместителем в комплексах CdL2 предпочтительна гексакоординация, в отличие от пентакоординации, предпочтительной для комплексов CdL2 на основе азометинов с тио(селено)бензимидазольными фрагментами.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Garnovskii A.D., Nivorozhkin A.L., Minkin V.I. // Coord. Chem. Rev. 1993. V. 126. № 1. P. 1.
Bourget-Merle. L., Lappert M.F., Severn J.R. // Chem. Rev. 2002. V. 102. № 6. P. 3031.
Garnovskii A.D., Vasilchenko I.S., GarnovskiiD.A., Kharisov B.I. // J. Coord. Chem. 2009. V. 62. № 2. P. 151.
Харабаев Н.Н., Стариков А.Г., Минкин В.И. // Докл. РАН. 2014. Т. 458. № 5. С. 555.
Харабаев Н.Н., Стариков А.Г., Минкин В.И. // Коорд. химия. 2015. Т. 41. № 7. С. 387 (Kharabayev N.N., Starikov A.G., Minkin V.I. // Russ. J. Coord. Chem. 2015. V. 41. № 7. P. 421). https://doi.org/10.1134/ S1070328415070039
Харабаев Н.Н. // Коорд. химия. 2019. Т. 45. № 8. С. 485 (Kharabayev N.N. // Russ. J. Coord. Chem. 2019. V. 45. № 8. P. 673). https://doi.org/10.1134/S1070328419080050
Ali M.A., Mirza A.H., Fong G.A. // Transition Met. Chem. 2004. V. 29. P. 613.
Ali M.A., Bakar H.J.H.A., Mirza A.H. et al. // Polyhedron. 2008. V. 27. P. 71.
Patra A., Sarkar S., Chakraborty R. et al. // J. Coord. Chem. 2010. V. 63. P. 1913.
Hashimoto Y., Yashinari N., Naruse D. et al. // Inorg. Chem. 2013. V. 52. P. 14368.
Mirza A.H., Hamid M.H.S.A., Aripin S. et al. // Polyhedron. 2014. V. 74. P. 16.
Pastor-Medrano J., Jancik V., Bernabe-Pabio E. et al. // Inorg. Chim. Acta. 2014. V. 412. P. 52.
Patra C., Bhanja A.K., Sen C. et al. // RSC Advances. 2016. V. 6. P. 53378.
Lee S.-G., Park K.-M., Habata Y., Lee S.-S. // Inorg. Chem. 2013. V. 52. P. 8416.
Li L., Li W., Yang S. et al. // J. Coord. Chem. 2013. V. 66. P. 2948.
Nogueira V.S., Bresolin L., Nather C. et al. // Acta Crystallogr. E. 2015. V. 71. P. m234.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09. Revision D.01. Wallingford (CT, USA): Gaussian, Inc., 2013.
Parr R., Yang W. Density-Functional Theory of Atoms and Molecules. N.Y.: Oxford Univ. Press, 1989. 333 p.
Becke A.D. // Phys. Rev. A. 1988. V. 38. P. 3098.
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. P. 785.
Burke K., Wagner L.O. // Int. J. Quantum Chem. 2013. V. 113. № 2. P. 96.
Tsipis A.C. // Coord. Chem. Rev. 2014. V. 272. P. 1.
Zhurko G.A., Zhurko D.A. Chemcraft. Version 1.6. URL: http://www.chemcraftprog.com.
Харабаев Н.Н. // Журн. общ. химии. 2017. Т. 87. № 4. С. 756.
Харабаев Н.Н. // Коорд. химия. 2017. Т. 43. № 12. С. 709 (Kharabayev N.N. // Russ. J. Coord. Chem. 2017. V. 43. № 12. P. 807). https://doi.org/10.1134/S107032841712003X
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия