

Координационная химия, 2021, T. 47, № 2, стр. 109-120
Кето-β-дикетиминатные комплексы иттрия и лития [{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]2Y(μ2-Cl)2Li(THF)2 и {[{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]Li(THF)}n. Синтез, молекулярная структура и исследование каталитической активности комплексов в полимеризации ε-капролактона
Г. Г. Скворцов 1, А. В. Черкасов 1, Д. Л. Ворожцов 2, Е. С. Щегравина 2, А. А. Трифонов 1, 3, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Нижегородский государственный университет им. Н.И. Лобачевского
Нижний Новгород, Россия
3 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
* E-mail: trif@iomc.ras.ru
Поступила в редакцию 06.05.2020
После доработки 27.05.2020
Принята к публикации 01.06.2020
Аннотация
Реакция β-дикетимината лития [{2,6-Me2C6H3N=CMe}2CH]Li с бензофеноном в толуоле при 25°C протекает с образованием координационного комплекса [{2,6-Me2C6H3N=CMe}2CH]Li(Ph2C=O) (I). По реакции трет-Bu(C=O)Cl с [{2,6-Me2C6H3N=CMe}2CH]Li синтезирован новый кето-β-дикетимин {2,6-Me2C6H3N=C(Me)}2CHC(трет-Bu)=O (II). Металлированием кето-β-дикетимина II н-бутиллитием в THF при 0°C получен кето-β-дикетиминат лития {[{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]Li(THF)}n (III). Обменная реакция YCl3 c III (мольное соотношение 1 : 2, THF) приводит к образованию бис(кето-дикетиминатного) комплекса иттрия [{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]2Y(μ2-Cl)2Li(THF)2 (IV). Молекулярное строение комплексов I, III, IV установлено методом РСА (CIF files CCDC № 2001131 (I), 2001132 (III), 2001133 (IV) соответственно)). Показано, что в кристаллическом состоянии IV существует в виде ate комплекса с одной молекулой LiCl. Комплексы I, III, IV являются катализаторами полимеризации с раскрытием цикла ε-капролактона в толуоле при 25°С.
В настоящее время N,N- и N,O-содержащие лиганды, различающиеся числом донорных группировок, а также длиной и природой мостика между координационными сайтами, являются одним из наиболее широко используемых в химии РЗЭ классов нециклопентадиенильных лигандов. Так, амидные, амидинатные и кетиминатные лиганды с варьирующимися дентатностью и стерическими свойствами были использованы в химии производных РЗЭ в качестве стабилизирующего координационного окружения в [1–8]. Интерес к лигандам нециклопентадиенильного типа вызван в первую очередь тем, что благодаря их применению в последние годы удалось синтезировать ряд реакционноспособных соединений d-переходных и редкоземельных металлов (РЗМ), проявляющих каталитическую активность в полимеризации диенов, метилметакрилата, а также полимеризации с раскрытием цикла рац-лактида и ε-капролактона, в реакциях гидрирования и гидросилилирования олефинов, сополимеризации эпоксидов с CO2 [6–14]. Кроме того, роль лигандного окружения в случае электроположительных РЗМ, имеющих большие ионные радиусы, в значительной мере ионные связи металл–лиганд, а также склонных к реакциям обмена лигандами (равновесие Шленка), исключительно велика. Хелатный лиганд отвечает за подавление реакций перераспределения лигандов, обеспечивает кинетическую устойчивость комплекса. В этой связи встает задача синтеза полидентатных N,N- и N,O-лигандов, способных образовывать с ионом металла наряду с прочной ковалентной связью лабильные координационные связи, за счет которых достигается необходимое насыщение координационной сферы иона металла в комплексе.
В настоящей работе сообщается о синтезе нового тридентатного кето-β-дикетиминатного лиганда {2,6-Me2C6H3N=C(Me)}2CHC(трет-Bu)=O (II) и исследовании возможных типов координации его аниона [{2,6-Me2C6H3N=C(Me)}2CC(трет- Bu)=O]– с катионами лития и иттрия, а также об изучении каталитической активности дикетиминатных и кетодикетиминатных комплексов [{2,6-Me2C6H3N=CMe}2CH]Li(Ph2C=O) (I), {[{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]Li(THF)}n (III), [{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]2 YCl2Li(THF)2 (IV) в полимеризации с раскрытием цикла ε-капролактона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу и выделению продуктов проводили в вакуумной аппаратуре с использованием стандартной техники Шленка. Тетрагидрофуран после осушки гидроксидом калия перегонялся над бензофенонкетилнатрием. Гексан и толуол сушили кипячением и перегонкой над металлическим натрием. Дейтеропиридин (C5D5N) сушили гидридом кальция, дегазировали и конденсировали в вакууме. Дейтеробензол (C6D6) сушили металлическим натрием, дегазировали и конденсировали в вакууме. {2,6-Me2C6H3N=CMe}2CH2 [11], YCl3 [15], были получены в соответствии с опубликованными методиками. Бензофенон, ε-капролактон, C5D5N, C6D6, CDCl3, 2,6-диметиланилин – коммерческие реактивы (Acros). ИК-спектры регистрировали на приборе Bruker-Vertex 70. Образцы соединений готовили в атмосфере сухого аргона в виде суспензий в вазелиновом масле. Спектры ЯМР 1H, 13C, 7Li, HSQC 1H–13C регистрировали на приборах Bruker Avance III и Bruker DRX-200 (25°С, C5D5N, C6D6, CDCl3). Химические сдвиги приведены в миллионных долях по отношению к известным сдвигам остаточных протонов дейтерированных растворителей. Элементный анализ выполняли на приборе Perkin-ElmerSeries II CHNS/O Analyser 2400. Содержание иттрия определяли методом комплексонометрического титрования (Трилон Б) с использованием ксиленолового оранжевого в качестве индикатора [16].
Синтез дифенилкетонат (2,6-диметилфенил)-4-(((2,6-диметилфенил)имино)пент-2-ен-2-ил)анилида лития (I). К раствору {2,6-Me2C6H3N=CMe}2CH2 (0.800 г, 2.61 ммоль) в 20 мл толуола при 0°C добавляли н-бутиллитий (2.40 мл, 2.78 ммоль, 1.16 M раствор в гексане). Реакционную смесь перемешивали 1 ч при 0°C, добавляли бензофенон (0.476 г, 2.61 ммоль) и перемешивали при 25°C в течение 12 ч. Толуол удаляли в вакууме и твердый остаток растворяли в теплом гексане (30 мл). Красные кристаллы I, полученные при медленном концентрировании гексанового раствора, сушили в вакууме 30 мин. Выход 0.970 г (75%).
| Найдено, %: | C 82.23; | H 7.35; | N 5.40. |
| Для C34H35N2OLi (М = 494.58) | |||
| вычислено, %: | C 82.57; | H 7.13; | N 5.66. |
Спектр ЯМР 1H (400 МГц; 25°С; C6D6; δ, м.д., J, Гц): 1.88 (с., 6 H, CH3C=N); 2.25 (с., 12 H, C6H3(CH3)2); 5.05 (с., 1 H, CH); 6.90–7.14 (м., 16 H, C6H3(CH3)2, (C6H5)2C=O). Спектр ЯМР 13C (100 MГц; 25°C; C6D6; δ, м.д.): 19.1 (C6H3(CH3)2); 22.9 (CH3C=N); 93.2 (CH); 121.9, 128.2, 128.5, 130.9, 131.1, 133.3, 136.7, 153.2, 163.3 (C6H3(CH3)2, CH3C=N, (C6H5)2C=O); 200.7 (C6H5)2C=O). Спектр ЯМР 7Li (155.5 MГц; 25°C; C6D6; δ, м.д.): 3.0. ИК-спектр (ν, см–1): 1667 с, 1648 c, 1626 с, 1597 с, 1557 с, 1325 c, 1277 c, 1176 c, 1088 c, 1076 с, 1025 c, 976 c, 941 с, 924 c, 823 c, 812 c, 761 с, 701 c, 639 с, 628 с, 611 с, 540 с, 487 с.
Синтез 5-((2,6-диметилфенил)имино)-4-(1-((2,6-диметилфенил)имино)этил)-2,2-диметилгексан-3-она (II). К раствору {2,6-Me2C6H3N=CMe}2CH2 (2.000 г, 6.50 ммоль) в 35 мл толуола при 0°C добавляли н-бутиллитий (6.20 мл, 7.20 ммоль, 1.16 M раствор в гексане). Реакционную смесь перемешивали 1 ч при 0°C и добавляли раствор трет-Bu(C=O)Cl (0.868 г, 7.20 ммоль) в 10 мл толуола. Реакционную смесь перемешивали 48 ч, раствор декантировали от осадка LiCl и удаляли растворители в вакууме. Твердый остаток промывали гексаном (10 мл) и сушили в вакууме 1 ч. Кетодикетимин II выделяли в виде белого порошка с выходом 2.030 г (80%).
| Найдено, %: | C 79.67; | H 8.95; | N 6.87. |
| Для C26H34N2O (М = 390.57) | |||
| вычислено, %: | C 79.96; | H 8.77; | N 7.17. |
Тпл = 103°С. Масс-спектр (ЭУ, 70 эВ), m/z (Iотн (%)): 390.57 [M]+ (20). Спектр ЯМР 1H (мажорный изомер (69%); 400 МГц; 25°С; CDCl3; δ, м.д.; J, Гц): 1.30 (с., 9 H, C(CH3)3); 1.65 (с., 6 H, CH3C=N); 2.17 (с., 12 H, C6H3(CH3)2); 6.87–7.08 (м., 6 H, C6H3(CH3)2); 13.12 (с., 1 H, NH). Спектр ЯМР 1H (минорный изомер (31%), 400 МГц, 25°С, CDCl3, δ, м.д., J/Гц): 1.36 (с., 9 H, C(CH3)3); 1.78, 2.02, 2.11 (с., вместе 18 H, CH3C=N, C6H3(CH3)2); 5.42 (с., 1 H, CH); 6.87–7.08 (м., 6 H, C6H3(CH3)2). Спектр ЯМР 13C (оба изомера; 100 MГц; 25°C; CDCl3; δ, м.д.): 18.5, 18.7, 19.5, 19.7 (CH3C=N, C6H3(CH3)2); 26.6, 28.7 (C(CH3)3); 46.3, 47.1 (C(CH3)3); 68.7 (CH, кетодиимин); 108.1 (CH3C=С); 123.2, 124.8, 128.1, 128.2, 131.9, 142.8, 158.5, 211.1, 217.4 (C6H3(CH3)2, CH3C=N, трет-BuC=O). ИК-спектр (ν, см–1): 3304 ср, 1697 c, 1656 c, 1593 с, 1302 c, 1254 c, 1233 c, 1196 c, 1181 с, 1092 c, 1061 c, 1033 с, 986 c, 918 c, 854 cр, 829 с, 805 c, 793 с, 762 c, 683 с, 649 ср, 598 ср, 579 ср, 526 ср, 511 ср, 461 ср.
Синтез тетрагидрофуранат (2,6-диметилфенил)(3-(1-((2,6-диметилфенил)имино)этил)-5,5-диметил-4-оксогекс-2-ен-2-ил)анилида лития (III). К раствору II (0.810 г, 2.07 ммоль) в гексане (40 мл) при 0°C добавляли н-бутиллитий (2.00 мл, 2.32 ммоль, 1.16 M раствор в гексане) и реакционную смесь перемешивали 12 ч при 25°C. Гексан удаляли в вакууме, и твердый остаток сушили в течение 20 мин, затем растворяли в THF (5 мл). Светло-желтые кристаллы III получали при медленной конденсации гексана в концентрированный раствор комплекса в THF при 25°С. Кристаллы промывали холодным гексаном и сушили в вакууме при 25°С в течение 30 мин. Выход светло-желтых кристаллов III 0.720 г (74%).
| Найдено, %: | С 76.55; | H 8.89; | N 5.70. |
| Для C60H82N4O4Li2 (М = 937.17) | |||
| вычислено, %: | C 76.89; | H 8.82; | N 5.98. |
Спектр ЯМР 1H (400 МГц; 25°С; C5D5N; δ, м.д.; J, Гц): 1.57 (уш. С., 9 H, C(CH3)3); 1.64 (м., 4 H, β-CH2, THF); 1.98 (уш. С., 6 H, CH3C=N); 2.06 (уш. c., 12 H, C6H3(CH3)2); 3.67 (м., 4 H, α-CH2, THF); 7.00 (т., 2 H, C6H3(CH3)2, 3JH,H = 7.3 Гц); 7.12 (д., 4 H, C6H3(CH3)2, 3JH,H = 7.3 Гц). Спектр ЯМР 13C (100 MГц; 25°C, C5D5N; δ, м.д.): 19.3 (C6H3(CH3)2); 23.0 (CH3C=N); 26.3 (β-CH2, THF); 29.3 (C(CH3)3); 47.4 (C(CH3)3); 68.3 (α-CH2, THF); 109.0 (CH3C=С), 122.3, 125.8, 128.8, 131.0, 153.0, 160.5, 211.2, 216.6 (C6H3(CH3)2, CH3C=N, трет-BuC=O). Спектр ЯМР 7Li (155.5 MГц; 25°C; C5D5N; δ, м.д.): 2.7. ИК-спектр (ν, см–1): 1648 c, 1541 c, 1292 c, 1251 c, 1197 c, 1154 с, 1094 c, 1053 c, 1008 с, 993 с, 921 c, 896 с, 829 c, 809 c, 790 c, 760 c, 676 с, 646 с, 600 ср, 568 с, 529 с, 503 с.
Синтез бис[(2,6-диметилфенил)(3-(1-((2,6-диметилфенил)имино)этил)-5,5-диметил-4-оксогекс-2-ен-2-ил)анилид]дихлороиттрат(III) дитетрагидрофураната лития (IV). К суспензии YCl3 (0.084 г, 0.43 ммоль) в THF (10 мл) приливали раствор комплекса III (0.402 г, 0.43 ммоль) в THF (15 мл) при 25°C. Реакционную смесь перемешивали в течение 12 ч и тетрагидрофуран удаляли в вакууме. Продукт реакции экстрагировали толуолом (25 мл) и декантировали от нерастворимого осадка. Растворитель удаляли, вещество сушили в вакууме 20 мин и растворяли в тетрагидрофуране (2 мл). Белые кристаллы IV получали при медленной конденсации гексана в концентрированный раствор комплекса в ТHF при 25°С. Кристаллы промывали холодным гексаном и сушили 20 мин в вакууме при 25°С. Выход белых кристаллов IV 0.323 г (67%).
| Найдено, %: | С 66.37; | H 7.95; | N 4.70; | Y 8.03. |
| Для C63.5H89.5N4O4.5Cl2LiY (М = 1147.64) | ||||
| вычислено, %: | C 66.46; | H 7.86; | N 4.88; | Y 7.75. |
Спектр ЯМР 1H (400 МГц; 25°С; C6D6; δ, м.д.; J, Гц): 1.38 (м., 10 H, β-CH2, THF); 1.43 (уш. с, 18 H, C(CH3)3); 2.20, 2.28 (уш. С., 36 H, CH3C=N, C6H3(CH3)2); 3.58 (м., 10 H, α-CH2, THF); 6.92–7.05 (м., 12 H, C6H3(CH3)2). Спектр ЯМР 13C: (100 MГц; 25°C; C6D6; δ, м.д.): 19.6, 20.3 (CH3C=N, C6H3(CH3)2); 25.7 (β-CH2, THF); 30.3 (C(CH3)3); 42.1 (C(CH3)3); 68.6 (α-CH2, THF); 116.7 (CH3C=С), 123.5, 125.1, 129.3, 131.8, 147.9, 158.7, 170.1, 185.2 (C6H3(CH3)2, CH3C=N, трет-BuC=O). Спектр ЯМР 7Li (155.5 MГц; 25°C; C5D5N; δ, м.д.): 5.1. ИК-спектр (ν, см–1): 1681 c, 1627 c, 1608 с, 1530 с, 1334 c, 1271 с, 1251 c, 1218 c, 1188 с, 1096 c, 1073 c, 1047 с, 985 с, 959 с, 918 c, 896 с, 839 c, 817 c, 798 c, 764 c, 748 с, 690 с, 675 с, 646 с, 600 с, 575 ср, 543 с, 516 с, 489 с).
Полимеризация ε-капролактона (общая методика). В главбоксе в инертной атмосфере комплекс I (5.0 мг, 0.01 ммоль) растворяли в 2.5 мл толуола и добавляли ε-капролактон (0.285 г, 2.50 ммоль). Реакционную смесь перемешивали в течение 5 мин при 25°C и отбирали аликвоту для определения степени конверсии мономера методом ЯМР. Затем в реакционную смесь добавляли 1 мл раствора 1.2 М HCl в этаноле и осаждали полимер избытком этанола (20 мл). Твердый остаток отделяли и сушили в вакууме до постоянной массы. Выход определяли весовым методом.
РСА соединений I, III, IV проведен на дифрактометрах Bruker D8 Quest (I) и Rigaku OD Xcalibur (III, IV) (МоKα-излучение, ω-сканирование, λ = = 0.71073 Å, T = 100(2) K). Измерение и интегрирование экспериментальных наборов интенсивностей выполнены с помощью программных пакетов APEX2 [17] и CrysAlisPro [18]. Учет поглощения, решение и уточнение структур проведены с помощью программных пакетов ABSPack (CrysAlisPro), SADABS [19] и SHELX [20]. Структуры решены прямым методом и уточнены полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для неводородных атомов. Все водородные атомы помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв (U(H)изо = 1.5U(C)экв для метильных групп). Кристаллографические данные, а также параметры рентгеноструктурных экспериментов и уточнения структур I, III и IV приведены в табл. 1, избранные длины связей и валентные углы – в табл. 2.
Таблица 1.
Кристаллографические данные, параметры рентгеноструктурных экспериментов и уточнения структур I, III, IV
| Параметр | I | III | IV |
|---|---|---|---|
| М | 494.58 | 937.17 | 1147.64 |
| Кристаллическая система | Триклинная | Моноклинная | Моноклинная |
| Пр. гр. | P1 | P21/n | P21/c |
| a, Å | 9.6948(10) | 13.9888(4) | 16.7492(7) |
| b, Å | 9.7829(10) | 19.2892(5) | 17.6465(8) |
| c, Å | 15.2592(15) | 21.0326(6) | 22.5936(10) |
| α, град | 93.050(2) | 90 | 90 |
| β, град | 91.478(2) | 107.696(3) | 107.192(5) |
| γ, град | 100.707(2) | 90 | 90 |
| V, Å3 | 1419.1(2) | 5406.7(3) | 6379.5(5) |
| Z | 2 | 4 | 4 |
| Размеры кристалла, мм | 0.29 × 0.20 × 0.12 | 0.50 × 0.30 × 0.20 | 0.25 × 0.15 × 0.05 |
| ρ(выч.), г/см3 | 1.157 | 1.151 | 1.195 |
| μ, мм–1 | 0.069 | 0.071 | 1.045 |
| Область сканирования θ, град | 2.44–29.98 | 2.96–30.03 | 2.92–30.03 |
| Количество измеренных рефлексов | 17 394 | 10 8752 | 38 094 |
| Количество независимых рефлексов c I > 2σ(I) | 5919 | 12093 | 9384 |
| Rint | 0.0497 | 0.0667 | 0.0923 |
| Число уточняемых параметров | 349 | 649 | 740 |
| S(F 2) | 1.016 | 1.041 | 1.004 |
| R1 (I > 2σ(I)) | 0.0644 | 0.0581 | 0.0717 |
| wR2 (по всем данным) | 0.1856 | 0.1342 | 0.1818 |
| Остаточной электронная плотность (min/max), е/Å3 | 0.54/–0.30 | 0.71/–0.30 | 0.93/–1.03 |
Таблица 2.
Избранные длины связей (d) и валентные углы (ω) в I, III, IV
| Связь | d, Å | Угол | ω, град |
|---|---|---|---|
| I | |||
| Li(1)–O(1) Li(1)–N(1) Li(1)–N(2) O(1)–C(22) N(1)–C(1) N(2)–(C3) C(1)–C(2) C(2)–C(3) |
1.825(3) 1.897(3) 1.898(3) 1.228(2) 1.314(2) 1.318(2) 1.412(2) 1.407(2) |
O(1)Li(1)N(1) O(1)Li(1)N(2) N(1)Li(1)N(2) C(22)O(1)Li(1) C(1)N(1)C(6) |
129.3(2) 129.8(2) 100.2(2) 169.0(2) 122.0(2) |
| III | |||
| Li(1)–O(2) Li(1)–N(2) Li(1)–N(1) Li(1)–O(3) Li(2)–O(1) Li(2)–N(3) Li(2)–N(4) Li(2)–O(4) O(1)–C(6) O(2)–C(32) N(1)–C(1) N(2)–C(3) C(1)–C(2) C(2)–C(3) |
1.946(3) 1.988(3) 1.994(3) 2.024(2) 1.970(3) 1.986(3) 1.988(3) 2.050(3) 1.228(2) 1.228(2) 1.322(2) 1.314(2) 1.429(2) 1.439(2) |
O(2)Li(1)N(2) O(2)Li(1)N(1) N(2)Li(1)N(1) O(2)Li(1)O(3) N(2)Li(1)O(3) N(1)Li(1)O(3) O(1)Li(2)N(3) O(1)Li(2)N(4) N(3)Li(2)N(4) O(1)Li(2)O(4) |
115.8(2) 119.3(2) 92.3(2) 104.6(2) 111.23(2) 113.6(2) 115.1(2) 113.9(2) 92.2(2) 109.6(2) |
| IV | |||
| Y(1)–O(2) Y(1)–O(1) Y(1)–N(3) Y(1)–N(1) Y(1)–Cl(1) Y(1)–Cl(2) Cl(1)–Li(1) Cl(2)–Li(1) O(1)–C(1) O(2)–C(27) N(1)–C(3) N(1)–C(19) N(2)−C(8) N(2)−C(10) N(3)–C(29) N(3)–C(45) N(4)–C(34) N(4)–C(36) C(1)–C(2) C(2)–C(3) C(27)–C(28) C(28)–C(29) |
2.157(2) 2.171(2) 2.443(3) 2.436(3) 2.659(2) 2.676(2) 2.318(7) 2.344(7) 1.309(4) 1.307(4) 1.306(4) 1.449(4) 1.283(4) 1.430(4) 1.312(4) 1.445(5) 1.285(5) 1.437(5) 1.395(5) 1.473(5) 1.399(5) 1.467(5) |
O(2)Y(1)O(1) O(2)Y(1)N(3) O(1)Y(1)N(3) O(2)Y(1)N(1) O(1)Y(1)N(1) N(3)Y(1)N(1) O(2)Y(1)Cl(1) O(1)Y(1)Cl(1) N(3)Y(1)Cl(1) N(1)Y(1)Cl(1) O(2)Y(1)Cl(2) O(1)Y(1)Cl(2) N(3)Y(1)Cl(2) N(1)Y(1)Cl(2) Cl(1)Y(1)Cl(2) |
106.20(9)
71.1(2) 95.1(2) 92.2(2) 72.6(2) 156.0(2) 161.00(7) 87.95(7) 95.48(7) 104.36(7) 87.89(7) 161.04(7) 101.57(7) 94.72(7) 81.56(3) |
Структуры зарегистрированы в Кембриджском банке структурных данных (CCDC № 2001131 (I), 2001132 (III), 2001133 (IV), ccdc.cam.ac.uk/structures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
С целью получения нового дикетиминаталкоксидного κ3-N,N,O-лиганда скорпионатного типа [{2,6-Me2C6H3N=C(Me)}2CHCPh2O]ˉ мы провели реакцию бензофенона с [{2,6-Me2C6H3N=CMe}2CH]Li, полученным in situ металлированием {2,6-Me2C6H3N=CMe}2CH2 [11] н-бутиллитием в толуоле при 0°C (схема 1 ). Обнаружено, что присоединения дикетимината лития по связи С=О бензофенона не происходит, а реакция приводит к образованию аддукта с бензофеноном [(2,6-Me2C6H3N=CMe)2CH]Li(Ph2C=O) (I), в котором последний выступает в качестве нейтрального лиганда, координированного на ион лития. При удалении растворителя в вакууме и последующей перекристаллизации продукта реакции из гексана аддукт с бензофеноном I был выделен в виде красных кристаллов с выходом 75%. Комплекс I чувствителен к кислороду и влаге воздуха, хорошо растворим в эфирных и ароматических растворителях и умеренно растворим в алифатических углеводородах. Состав и строение комплекса установлены методом элементного анализа, ИК-, ЯМР-спектроскопии и РСА.
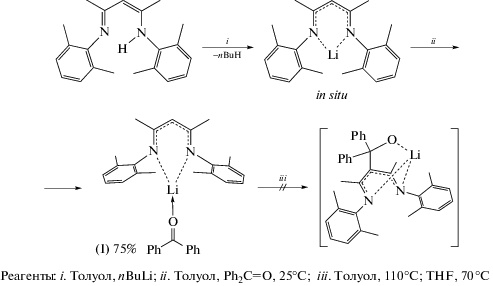
Схема 1 .
Проведение реакции бензофенона c дикетиминатом лития [{2,6-Me2C6H3N=CMe}2CH]Li в более жeстких условиях в растворе толуола (10 ч, 110°C) или THF (7 ч, 70°C) также не привело к желаемому результату, а из реакции после перекристаллизации продукта из гексана был выделен тот же аддукт I (схема 1 ).
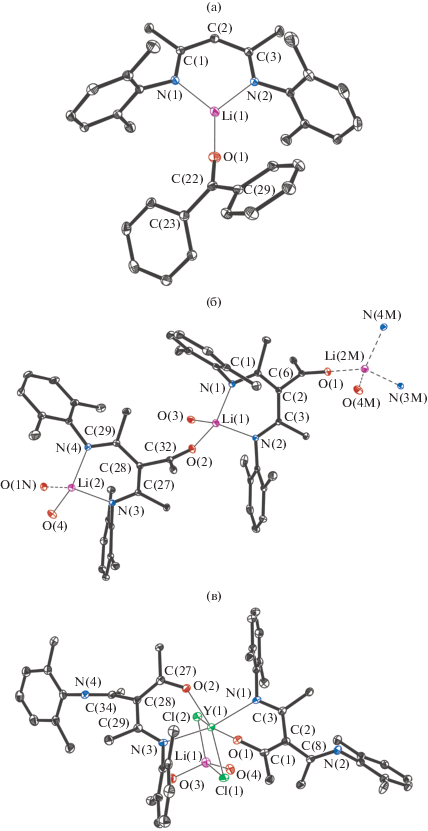
Прозрачные красные кристаллы комплекса I получены медленным концентрированием из раствора гексана при комнатной температуре. Согласно данным РСА, I представляет собой комплекс Li(I), в котором катион металла связан с двумя атомами азота дикетиминатного лиганда и одним атомом кислорода молекулы бензофенона. Таким образом, в I реализуется обычная для лигандов такого типа κ2-N,N-координация. Молекулярная структура комплекса I приведена на рис. 1а. Длины связей Li–N в I (1.897(3), 1.898(3) Å) близки между собой и заметно короче аналогичных величин в кетоиминатных (1.983(8)–2.022(2) Å) [21–23], дикетиминатных (1.955(2)–2.009(4) Å) [24, 25] и трикетиминатных (1.958(2), 1.973(2) Å) [8, 26] комплексах лития. Расстояния N–C (1.314(2)–1.318(2) Å) и C–C (1.407(2)–1.412(2) Å) в металлоцикле LiNCCCN лежат в узких интервалах значений и свидетельствуют о делокализации электронной плотности внутри дикетиминатного фрагмента. Металлоцикл практически плоский: угол между плоскостями NLiN и NCCCN равен 170.4(2)°. Расстояние Li–O в I (1.824(3) Å) значительно короче длины координационной связи Li–O (1.972(6) Å) в дикетиминатном комплексе лития [{Me3SiNCPh}2CH]Li(Ph2CO)2 [27] и сопоставимо с расстоянием Li–O (1.860(3) Å) в трикетиминатном комплексе лития [(2,6-Me2C6H3N=CMe)3C]Li(THF) [26]. Длина связи O(1)–C(22) составляет 1.228(2) Å.
Рис. 1.
Молекулярная структура I (a), III (фрагмент, (б)) и IV (в). Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода, а также метильные заместители трет-бутильных групп и CH2-группы молекул ТHF (б, в) не изображены для ясности.
Кетодикетиминатный κ3-N,N,O-лиганд скорпионатного типа (2,6-Me2C6H3N=CMe)2CH(ButC=O) (II) был синтезирован по реакции трет-Bu(C=O)Cl с дикетиминатом лития [{2,6-Me2C6H3N=CMe}2CH]Li в толуоле и выделен в виде белого порошка с выходом 80% (схема 2 ). Кето-β-дикетимин II охарактеризован методами элементного анализа, ЯМР-, ИК-спектроскопии и масс-спектрометрии.
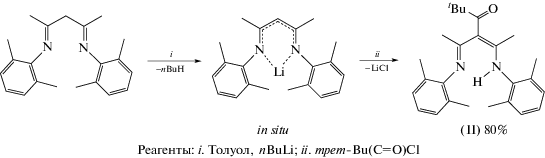
Схема 2 .
Исследование спектров ЯМР 1H и 13С соединения II показало, что в растворе комплекс существует в виде двух прототропных таутомеров: кетоенаминимина (69%) и кетодиимина (31%). Методом ЯМР (двумерный спектр HSQC 1H–13C ЯМР, CDCl3) установлено, что синглет при 5.42 м.д. соответствует метиновому протону α-CH центрального фрагмента СНССС кетодиимина (минорный изомер), а протоны групп NH кетоенаминимина проявляются в виде синглета при 13.12 м.д. Следует отметить, что в спектре ЯМР 13С также наблюдаются два набора сигналов, соответствующих кетоенаминиминной и кетодииминной таутомерным формам соединения II. В ИК-спектре II, присутствуют интенсивная полоса поглощения при 1656 см–1, соответствующая асимметричным колебаниям кратных связей C=N кето-β-дикетиминатного лиганда, а также интенсивная полоса поглощения при 1697 см–1, относящаяся к валентным колебаниям связи С=O кетогруппы. Следует отметить, что ИК-спектр соединения II в СН2Cl2 содержит полосу поглощения в области, характерной для колебаний связи N–H (3304 см–1). Таким образом, исследование строения соединения II методами ЯМР (1Н, 13С) и ИК-спектроскопии позволяет сделать вывод o том, что в растворе оно существует в кетоенаминиминной и кетодииминной формах.
Металлированием кето-β-дикетимина II н-бутиллитием в THF при 0°C был получен кетодикетиминат лития {[{2,6-Me2C6H3N=C(Me)}2CС(трет- Bu)=O]Li(THF)}n (III) (схема 3 ).
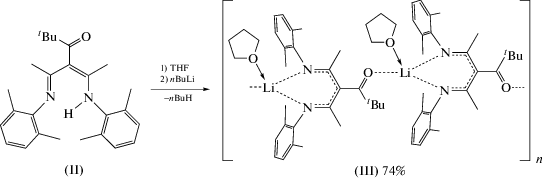
Схема 3 .
Комплекс III был выделен в виде светло-желтых кристаллов с выходом 74%. Кето-β-дикетиминат III чувствителен к влаге и кислороду воздуха, хорошо растворим в ароматических углеводородах, а также в эфирных растворителях.
В спектре ЯМР 1Н диамагнитного комплекса III протоны трет-Bu-заместителей проявляются в виде синглета при 1.57 м.д. Два уширенных синглета при 1.98, 2.06 м.д. соответствуют протонам метильных групп фрагментов (2,6-Me2C6H3N=CMe). Ароматические протоны проявляются в слабом поле в виде триплета (7.00 м.д., 3JH,H = 7.3 Гц) и дублета (7.12 м.д., 3JH,H = 7.3 Гц). Координированные молекулы THF в спектре ЯМР 1H комплекса III дают два мультиплета при 1.64 и 3.67 м.д., относящиеся к β- и α-метиленовым протонам. В спектре ЯМР 7Li комплекса III наблюдается единственный сигнал при 2.7 м.д. (155.5 МГц, 25°С, C5D5N).
Прозрачные светло-желтые кристаллы комплекса III получены медленным конденсированием гексана в концентрированный раствор соединения в THF. Так же как в I, в III реализуется κ2-N,N-координация кетодикетиминатного лиганда катионом лития. Однако РСА показал, что каждый Li+ в III дополнительно связан с атомом кислорода группы C=O кето-β-дикетиминатного лиганда соседней молекулы. Таким образом, комплекс III представляет собой одномерный координационный полимер {[{2,6-Me2C6H3N=C(Me)}2CС(трет- Bu)=O]Li(THF)}n, в котором каждый катион металла связан с двумя атомами азота одного кетодикетиминатного лиганда, атомом кислорода кетогруппы второго и атомом кислорода молекулы THF. Фрагмент кристаллической структуры III приведен на рис. 1б.
Длины связей Li–N в III лежат в узком интервале значений 1.986(3)–1.994(3) Å. Они заметно длиннее аналогичных величин в I (1.897(3), 1.898(3) Å) и сопоставимы с расстояниями Li–N в родственных кетоиминатных [21–23], ди- и трикетиминатных комплексах лития (1.958(2)–2.022(2) Å) [8, 24–26]. Длины связей N–C (1.313(2)–1.322(2) Å) и C–C (1.427(2)–1.439(2) Å) свидетельствуют о делокализации электронной плотности по фрагменту NCCCN. Интересно отметить, что металлоциклы LiNCCCN в III искажены значительно сильнее по сравнению с I. Так, углы между плоскостями NLiN и NCCCN в III равны 154.88(9)° и 158.5(2)°. Длины связей Li(1)–O(2) (1.946(3) Å) и Li(2)–O(1) (1.970(3) Å) в III несколько короче расстояний Li(1)–O(3), Li(2)–O(4) (2.024(2), 2.050(3) Å) и значительно длиннее, чем Li(1)–O(1) в I (1.825(3) Å). Длина связи C=O в кето-дикетиминатном лиганде составляет 1.228(2) Å.
Комплексы РЗМ в N,N-дикетиминатном и N,N,N-трикетиминатном лигандном окружении продемонстрировали достаточно высокую каталитическую активность в полимеризации с раскрытием цикла рац-лактида и ε-капролактона [8, 28]. Для того чтобы изучить влияние координационного окружения на каталитическую активность металлокомплексов, а также возможные типы координации нового хелатного N,N,O-лиганда ионами РЗМ, была проведена реакция III с безводным YCl3 в среде тетрагидрофурана при соотношении реагентов 2 : 1 в течение 12 ч (схема 4 ). После экстракции продукта реакции толуолом и перекристаллизации из смеси THF–гексан комплекс [{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]2-Y(μ2-Cl)2Li(THF)2 (IV) был выделен в виде бесцветных кристаллов с выходом 67%. Комплекс IV охарактеризован методами элементного анализа, ЯМР-, ИК-спектроскопии и представляет собой чувствительное к влаге и кислороду воздуха соединение, хорошо растворимое в ароматических углеводородах и эфирных растворителях.
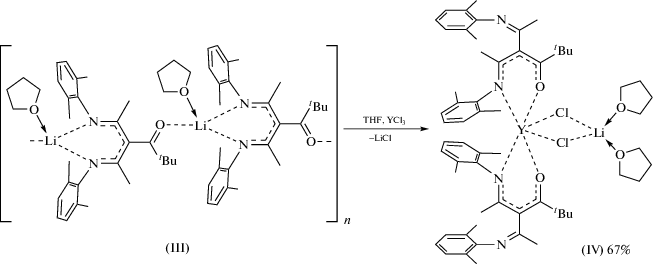
Схема 4 .
Кристаллы комплекса IV были получены медленным охлаждением концентрированного раствора соединения в смеси THF–гексан (1 : 4) до –20°C. РCA показал, что IV представляет собой мономерный ate комплекс, кристаллизующийся в виде сольвата [{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]2YCl2Li(THF)2 ⋅ 1/2THF ⋅ 1/4Hex (молекулярная структура комплекса IV изображена на рис. 1в).
В IV реализуется принципиально отличная от III κ2-N,O-координация кето-дикетиминатного лиганда атомом металла. Так, катион Y3+ в IV связан с двумя атомами кислорода и двумя атомами азота двух кетодикетиминатных лигандов и двумя µ2-мостиковыми хлорными лигандами. Таким образом, КЧ атома иттрия в IV формально равно шести. Оба потенциально тридентатных кетодикетиминатных лиганда в IV координированы c металлоцентром бидентатным образом, тогда как третий координационный сайт каждого из лигандов во взаимодействие с металлом не вовлечен. Катион Li+, в свою очередь, связан с двумя атомами хлора и с двумя атомами кислорода двух молекул ТHF.
Кето-β-дикетиминатный лиганд в IV координирован катионом иттрия несимметрично. Длины связей Y–O равны 2.171(2) и 2.157(2) Å, в то время как расстояния Y–N в IV значительно длиннее (2.436(3), 2.443(3) Å) и несколько превышают соответствующие величины в β-дикетиминатных комплексах иттрия [{HC(2-RC6H4N=CMe)2}- YCl3Li(Et2O)2]2 (2.315(6), 2.317(6), 2.3427(19), 2.377(2) Å; R = изо-Pr, трет-Bu) [29], [HC(PhN=CMe)2]3Y (2.406(4), 2.404(3) Å) [9]. Расстояния Y–Cl составляют 2.659(2) и 2.676(2) Å и сопоставимы с аналогичными расстояниями в β-дикетиминатных производных [{HC(2-RC6H4N=CMe)2}YCl3Li(Et2O)2]2 (2.611(2), 2.660(2), 2.6294(6), 2.6169(6) Å; R = изо-Pr, трет-Bu) [29]. Длины связей Li–Cl равны 2.218(7) и 2.344(7) Å. Делокализация электронной плотности в металлоциклах выражена для IV в меньшей степени, чем для комплексов I и III. Так, расстояния C(1)–O(1) и C(27)–O(2) равны 1.309(4) и 1.307(4) Å соответственно, расстояния N(1)–C(3) и N(3)–C(29) (1.306(4), 1.312(4) Å) незначительно длиннее, чем длины двойных связей N(2)–C(8) и N(4)–C(34) (1.283(5), 1.285(5) Å) [30]. Несмотря на то что длины связей C–C лежат в широком интервале значений 1.395(5)–1.507(5) Å, они скорее характеризуют делокализацию отрицательного заряда в кетоиминатном фрагменте, чем альтернирование расстояний C–С. Металлоциклы YNCCСO сильно искажены: углы между плоскостями NYO и NCCCO составляют 135.3(2)°, 140.60(9)°.
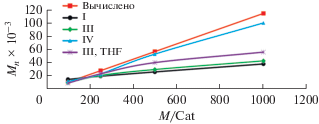
Комплексы I, III, и IV катализируют полимеризацию с раскрытием цикла ε-капролактона в мягких условиях (25°С, толуол). При катализе комплексами лития I, III полная конверсия 1000 экв. мономера достигается за 30 мин. Индексы полидисперсности образцов полученных полимеров имеют средние значения (Mw/Mn = 1.4–2.3), молекулярные массы полимеров лежат в интервале 10 500–43 200 (табл. 3, опыты 1–8). В результате проведенной серии опытов с участием катализаторов I, III было обнаружено, что при высокой степени загрузки мономера (500/1, 1000/1) значения $M_{n}^{{\exp }}$ сильно занижены (рис. 2), что обусловлено, по-видимому, протеканием конкурирующей реакции переэтерификации (табл. 3, опыты 3, 4, 7, 8). При проведении полимеризации ε-капролактона в присутствии комплекса III как в толуоле, так и в полярном тетрагидрофуране, количественная конверсия мономера в обоих случаях достигается за 2–30 мин, однако в среде полярного растворителя при высокой степени загрузки мономера (500/1, 1000/1) были получены образцы полилактонов, характеризующиеся более высокими значениями $M_{n}^{{\exp }}$ (табл. 3, опыты 7, 8, 11, 12). Следует отметить, что бис(кето-дикетиминат)хлоридный комплекс иттрия IV продемонстрировал существенно меньшую каталитическую активность в полимеризации с раскрытием цикла ε-капролактона в сравнении с комплексами лития I и III. Так, при катализе комплексом иттрия IV полная конверсия 1000 экв. мономера достигается за 24 ч. Наряду с хорошим соответствием значений экспериментальной и рассчитанной Mn (рис. 2) при использовании IV были получены образцы полилактонов, характеризующиеся достаточно узким молекулярно-массовым распределением Mw/Mn = 1.6–1.8 и высокой молекулярной массой Mn = 53 300–99 900 (табл. 3, опыты 15, 16).
Таблица 3.
Полимеризация ε-капролактона, инициируемая комплексами I, III, IV*
| Опыт | Комплекс | Растворитель | [М]0/[Cat]0 | t, мин | $M_{n}^{{{\text{calc}}}}$× 10–3 | $M_{n}^{{{\text{exp}}}}$× 10–3 | Mw/Mn |
|---|---|---|---|---|---|---|---|
| 1 | I | Tолуол | 100 | 2 | 11.4 | 15.5 | 2.2 |
| 2 | I | Tолуол | 250 | 5 | 28.5 | 20.3 | 1.8 |
| 3 | I | Tолуол | 500 | 15 | 57.1 | 26.7 | 1.4 |
| 4 | I | Tолуол | 1000 | 30 | 114.1 | 38.6 | 1.7 |
| 5 | III | Tолуол | 100 | 2 | 11.4 | 10.5 | 2.3 |
| 6 | III | Tолуол | 250 | 5 | 28.5 | 21.4 | 2.2 |
| 7 | III | Tолуол | 500 | 15 | 57.1 | 30.3 | 2.1 |
| 8 | III | Tолуол | 1000 | 30 | 114.1 | 43.2 | 2.0 |
| 9 | III | THF | 100 | 2 | 11.4 | 9.9 | 2.0 |
| 10 | III | THF | 250 | 5 | 28.5 | 23.8 | 2.5 |
| 11 | III | THF | 500 | 15 | 57.1 | 40.6 | 1.9 |
| 12 | III | THF | 1000 | 30 | 114.1 | 56.3 | 2.4 |
| 13 | IV | Tолуол | 100 | 120 | 11.4 | 12.2 | 2.1 |
| 14 | IV | Tолуол | 250 | 320 | 28.5 | 24.0 | 1.5 |
| 15 | IV | Tолуол | 500 | 690 | 57.1 | 53.3 | 1.6 |
| 16 | IV | Tолуол | 1000 | 1440 | 114.1 | 99.9 | 1.8 |
* Условия: толуол, [М] = 1.0 моль л–1; ТHF, [М] = 1.0 моль л–1; T = 25°C. Конверсия мономера М определена методом ЯМР 1Н и во всех опытах составляет 100%. Молекулярные массы полимеров $M_{n}^{{{\text{exp}}}}$ определены методом гель-проникающей хромaтографии с использованием полистирольных стандартов. $M_{n}^{{{\text{calc}}}}$ рассчитаны по уравнению: 114.14Y × [M]/[Ln] (Y = выход).
Рис. 2.
Mn vs [M]0/[Cat]0. Полимеризация ε-капролактона. Условия: катализатор I, III, IV; толуол (III, ТHF), 25°C, [M]0 = 1.0 моль л–1.
Таким образом, взаимодействие [{2,6-Me2C6H3N=CMe}2CH]Li с бензофеноном проходит с образованием координационного комплекса [{2,6-Me2C6H3N=CMe}2CH]Li(Ph2C=O) (I), а присоединения дикетимината лития по С=О связи бензофенона и образования дикетиминаталкоксида лития не наблюдается. По реакции пивалоилхлорида трет-Bu(C=O)Cl с дикетиминатом лития [{2,6-Me2C6H3N=CMe}2CH]Li синтезирован кето-β-дикетимин {2,6-Me2C6H3N=C(Me)}2 CHC(трет-Bu)=O (II). По реакции кетодикетимина II с н-бутиллитием был синтезирован комплекс лития {[{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]Li(THF)}n (III). В кристаллическом состоянии комплекс III является координационным полимером. Получен и структурно охарактеризован первый пример комплекса РЗМ с анионным кето-β-дикетиминатным лигандом [{2,6-Me2C6H3N=C(Me)}2CС(трет-Bu)=O]–. По обменной реакции YCl3 c производным лития III синтезирован бис(кетодикетиминат) иттрия [{2,6- Me2C6H3N=C(Me)}2CС(трет-Bu)=O]2YCl2Li(THF)2 (IV), представляющий собой ate комплекс с одной молекулой LiCl. Методом РСА установлено, что в комплексе иттрия IV моноанионный кетодикетиминатный лиганд выступает в роли бидентатного и координируется на ион металла только через атом азота и кислорода, тогда как третий координационный сайт во взаимодействие с металлом не вовлечен, что обусловлено, по-видимому, не ионным радиусом металла, а электронным состоянием лиганда. Комплексы I, III, IV инициируют полимеризацию с раскрытием цикла ε-каполактона в толуоле при 25°С.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Ward B.D., Dubberley S.R., Maisse-Francois A. et al. // Dalton Trans. 2002. P. 4649.
Skinner M.E.G., Mountford P. // Dalton Trans. 2002. P. 1694.
Hultzsch K.C., Hampel F., Wagher T. // Organometallics. 2004. V. 23. P. 2601.
Zeimentz P.M., Arndt S., Elvidge B.R., Okuda J. // Chem. Rev. 2006. V. 106. P. 2404.
Skvortsov G.G., Shavyrin A.S., Kovylina T.A. et al. // Eur. J. Inorg. Chem. 2019. P. 5008.
Trifonov A.A. // Russ. Chem. Rev. 2007. V. 76. P. 1122.
Konkol M., Okuda J. // Coord. Chem. Rev. 2008. V. 252. P. 1577.
Скворцов Г.Г., Черкасов А.В., Трифонов А.А. // Изв. АН. Сер. хим. 2017. V. 9. P. 1665 (Skvortsov G.G., Cherkasov A.V., Trifonov A.A. // Russ. Chem. Bull. (Int. Ed.). 2017. V. 66. P. 1665).
Lazarov B.B., Hampel F., Hultzsch K.C. // Z. Anorg. Allg. Chem. 2007. P. 2367.
Bourget-Merle L., Lappert M.F., Severn J.R. // Chem. Rev. 2002. V. 102. P. 3031.
Budzelaar P.H.M., Gelder R., Gal A.W. // Organometallics. 1998. V. 17. P. 4121.
Barnes D., Brown G.L., Brownhill M. et al. // Eur. J. Inorg. Chem. 2009. P. 1219.
Hamedani N.G., Arabi H., Zohuri G.H. et al. // J. Polym. Sci. A. 2013. V. 51. P. 1520.
Alnajrani M.N., Mair F.S. // Dalton Trans. 2016. V. 45. P. 10435.
Taylor M.D., Carter C.P. // J. Inorg. Nucl. Chem. 1962. V. 24. P. 387.
Lyle S.J., Rahman M.M. // Talanta. 1963. V. 10. P. 1177.
APEX3. Madison (WI, USA): Bruker AXS Inc., 2015.
CrysAlis Pro. Version 1.171.38.46. Rigaku Oxford Diffraction. Wroclaw (Poland): Rigaku Corporation, 2015.
Krause L., Herbst-Irmer R., Sheldrick G.M., Stalke D. // J. Appl. Cryst. 2015. V. 48. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Brehon M., Cope E.K., Mair F.S. et al. // Dalton Trans. 1997. P. 3421.
Gietz T., Boere R.T. // Inorganics. 2017. V. 5. P. 30.
Liu Z., Chen H.-X., Huang D. et al. // J. Organomet. Chem. 2014. V. 749. P. 7.
Carey D.T., Cope-Eatough E.K., Vilaplana-Mafe E. et al. // Dalton Trans. 2003. P. 1083.
Jutzi P., Leszczynska K., Mix A. et al. // Organometallics. 2009. V. 28. P. 1985.
Skvortsov G.G., Fukin G.K., Cherkasov A.V. et al. // Inorg. Chim. Acta. 2020. V. 508. 119623. https://doi.org/10.1016/j.ica.2020.119623
Tong H.-B., Liu D.-S. // Z. Kristallogr. NCS. 2008. V. 223. P. 419.
Shen X., Xue M., Jiao R et al. // Organometallics. 2012. V. 31. P. 6222.
Allen F.A., Connard O., Watson D.G. et al. // Perkin Trans. 1987. V. 2. S1.
Wei X., Cheng Y., Hitchcock P.B., Lappert M.F. // Dalton Trans. 2008. P. 5235.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия