

Кристаллография, 2019, T. 64, № 2, стр. 242-247
НОВЫЙ ПЕНТАБОРАТ ЦЕЗИЯ С АНИОННЫМ РАДИКАЛОМ ЛЕНТОЧНОГО ТИПА
Л. В. Шванская 1, *, А. В. Сапегина 1
1 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: lshvanskaya@mail.ru
Поступила в редакцию 22.03.2018
После доработки 09.04.2018
Принята к публикации 10.05.2018
Аннотация
Методом дифракции рентгеновских лучей исследована кристаллическая структура нового гидратированного гидроксил-пентабората цезия, полученного из раствора-расплава борной кислоты при температуре 180°С. Соединение кристаллизуется в триклинной сингонии, пр. гр. P$\bar {1}$, параметры: a = 7.8107(5), b = 9.1929(8), c = 12.3553(11) Å, α = 98.98(1)°, β = 106.32(1)°, γ = 91.10(1)°, V = 839.1(1) Å3, Z = 4. Структура уточнена до фактора расходимости R1 = 5.8% в анизотропном приближении тепловых смещений всех атомов, кроме водорода, с учетом немероэдрического двойникования осью второго порядка, совпадающей с осью b. Компоненты двойника составили 0.546(1) и 0.454(1). Проанализированы особенности водородных связей Cs[B5O7(OH)2] · 0.5H2O по сравнению с изоструктурным рубидиевым аналогом. Установлено закономерное увеличение параметров a и c элементарных ячеек пентаборатов семейства A[B5O7(OH)2] · 0.5H2O при увеличении радиуса катиона A в ряду NH4 → Rb → Cs; незначительное изменение параметра b обусловлено жесткостью пентаборатных цепочек, вытянутых вдоль данного направления.
ВВЕДЕНИЕ
Анионные радикалы кристаллических структур шести изученных на сегодняшний день пентаборатов цезия построены из блоков, состоящих из одного центрального тетраэдра BO4 и четырех связанных с ним вершинами треугольников. В водосодержащих полиморфных α- [1] и β-модификациях [2] Cs[B5O6(OH)4] · 2H2O пентаборатные анионные радикалы изолированы. Основное различие кристаллических структур полиморфов заключается в том, что в β-фазе атом бора, центрирующий кислородный тетраэдр, расположен в частном положении – на поворотной оси второго порядка, поэтому внутри блока [B5O6(OH)4] только три атома бора являются независимыми, в то время как в α-модификации пять независимых полиэдров бора. Схема для борных треугольников и тетраэдров в таком случае будет выглядеть как 5 : 2(2Δ + 0.5Т) и 5 : (4Δ + 1Т) для β- и α-модификаций соответственно. Четыре полиморфные модификации (α, β, γ и δ) безводного пентабората CsB5O8 обладают каркасными структурами, различающимися конфигурацией каналов, вмещающих катионы цезия. Среди известных пентаборатов щелочных металлов выделим изоструктурные соединения рубидия и аммония А[B5O7(OH)2] · 0.5H2O [3, 4], полученные в гидротермальных условиях и методом раствор-расплавного синтеза. Их кристаллические структуры содержат ленточные мотивы из пентаборатных блоков, топологически идентичные блокам в структуре минерала лардереллита NH4[B5O7(OH)2] · H2O [5]. Отметим, что в природных условиях кристаллизуются и другие щелочные водосодержащие пентабораты, все островные: сборгит Na[B5O6(OH)4] · 3H2O [6], сантит K[B5O6(OH)4] · 2H2O [7], аммониоборит (NH4)2[B5O6(OH)4]2 · H2O [8], а также Rb- и Cs-раманиты A[B5O6(OH)4] · 2H2O [9], найденные в расплавных и флюидных включениях кварца пегматитов. По схожести их дифрактограмм с дифрактограммами синтетических аналогов установлена изоструктурность Rb-раманита и сантита (пр. гр. Aba2); Cs-раманит изоструктурен β-модификации синтетического Cs[B5O6(OH)4] · 2H2O (пр. гр. C2/c).
Интерес к гидратированным пентаборатам вызван возможностью их технологического применения в качестве преобразователей лазерных частот. Так, нелинейно-оптические свойства выявлены у полярных представителей семейства Me[B5O6(OH)4] · 4H2O, Me = K [10], Cs, NH4 [11].
В настоящей работе изложены результаты синтеза и рентгеноструктурного исследования нового водосодержащего пентабората цезия с анионным радикалом ленточного типа.
ЭКСПЕРИМЕНТ И УТОЧНЕНИЕ СТРУКТУРЫ
Бесцветные прозрачные кристаллы удлиненно-пластинчатого габитуса нового соединения были получены из раствора-расплава борной кислоты H3BO3 при исследовании фазообразования в многокомпонентной системе Cs–Cu–P–B–O. Исходные реактивы H3BO3 : CsH2PO4 : CuO, взятые в мольном соотношении 10 : 1 : 2, тщательно перетирали и помещали в автоклав, футерованный фторопластом. Нагрев осуществляли до 180°С, расплав выдерживали в течение 7 сут с последующим охлаждением до комнатной температуры. Кристаллы извлекали из общей массы серо-голубого цвета механическим способом. По результатам рентгенофазового анализа округлые выделения темно-синего и темно-серого цвета с металлическим блеском были диагностированы как аморфные фазы. По предварительным рентгеноструктурным данным параметры элементарной ячейки выделенной кристаллической фазы были равны: a = 7.7999(5), b = 9.1767(9), c = 12.3342(11) Å, α = 98.985(8)°, β = 106.344(7)°, γ = 91.099(7)°, V = = 837.1 Å3. Качественный рентгеноспектральный анализ монокристаллов (Jeol JSM-6480LV, энергодисперсионная приставка INCA 350) показал наличие атомов Cs, B, O в их составе. На основе данных предварительного фазового исследования и химического анализа было высказано предположение, что полученное соединение является цезиевым аналогом щелочных пентаборатов Rb[B5O7(OH)2] · 0.5H2O и NH4[B5O7(OH)2] · 0.5H2O, синтезированных ранее в мягких гидротермальных условиях и раствор-расплавным методом [3, 4] соответственно. Дальнейшая расшифровка структуры и ее уточнение подтвердили это предположение.
Экспериментальный набор интенсивностей дифракционных отражений получен на рентгеновском дифрактометре Xcalibur S фирмы Oxford Diffraction c двумерным ССD-детектором при комнатной температуре. Зарегистрированные интенсивности отражений скорректированы с использованием программного обеспечения дифрактометра с учетом фактора Лоренца и поляризационного фактора. Анализ экспериментального набора рентгеновских отражений с помощью программного комплекса CrysAlisPro [12] показал, что изучаемый кристалл является немероэдрическим двойником, компоненты которого связаны псевдоосью второго порядка, проходящей вдоль направления [010]. Отражения от разных компонентов двойника были разделены в новом наборе. Введена эмпирическая поправка на поглощение. Все вычисления проводились в рамках программного пакета WinGX 32 [13]. Использованы кривые атомного рассеяния и поправки на аномальную дисперсию из [14]. Кристаллическая структура решена прямыми методами в пр. гр. P$\bar {1}$ и уточнена с помощью программы SHELX [15] на основе интенсивностей, разделенных для каждого двойникового компонента в файле отражений HKLF5, до R-фактора 0.0586 (для 7529 независимых отражений с I > 2σ(I)) в анизотропном приближении тепловых смещений всех атомов, кроме атомов водорода. Положения четырех независимых атомов Н, образующих гидроксильные группы, и две позиции, относящиеся к молекуле воды, были выявлены на картах разностного синтеза Фурье и уточнены в изотропном приближении. Длину связи O–H фиксировали при эмпирическом значении 0.90 Å. Основные кристаллографические характеристики, данные эксперимента и результаты уточнения структуры нового пентабората цезия приведены в табл. 1. Уточненные координаты базисных атомов и межатомные расстояния, рассчитанные на их основе, приведены в табл. 2–4 соответственно. Расчет локального баланса валентностей, выполненный с использованием алгоритма и параметров, предложенных в [16, 17], подтвердил наличие групп OH и H2O. Установленной кристаллической структуре нового цезиевого пентабората соответствует кристаллохимическая формула Cs[B5O7(OH)2] · 0.5H2O.
Таблица 1.
Кристаллографические характеристики, данные эксперимента и результаты уточнения структуры Cs[B5O7(OH)2] · 0.5H2O
| М | 341.996 |
| Симметрия, пр. гр., Z | Триклинная, P$\bar {1}$, 2 |
| a, b, c, Å | 7.8107(5), 9.929(8), 12.553(11) |
| α, β, γ, град | 98.984(7), 106.324(7), 91.096(6) |
| V, Å3 | 839.1(1) Å3 |
| Dx, г/см3; μ, мм–1 | 2.707; 4.44 |
| Излучение, λ, Å | MoKα, 0.71069 (графитовый монохроматор) |
| Т, K | 298(2) |
| Размер образца, мм | 0.0796 × 0.0516 × 0.0366 |
| Дифрактометр | Xcalibur SCCD |
| Тип сканирования | Ω |
| Tmin, Tmax | 0.97, 1.00 |
| θmax, град | 30.586 |
| Пределы h, k, l | –10 ≤ h ≤ 10, –12 ≤ k ≤ 12, –17 ≤ l ≤ 17 |
| Число отражений: измеренных/наблюдаемых с I > 2σ(I) | 14 576/7529 |
| Метод уточнения | МНК по F2 |
| Число уточняемых параметров | 305 |
| Весовая схема | 1/[σ2($F_{o}^{2}$) + (0.1P)2 + + 0.00P] |
| R1/wR2 | 0.0586/0.1153 |
| Коэффициент двойникования (BASF) | 0.454(1) |
| S | 0.608 |
| Δρmin/Δρmax, э/Å3 | –2.155/1.627 |
Таблица 2.
Координаты базисных атомов и эквивалентные тепловые параметры в структуре Cs[B5O7(OH)2] · 0.5H2O
| Атом | x/a | y/b | z/c | Uэкв, Å2* |
|---|---|---|---|---|
| Cs1 | 0.9925(1) | 0.0574(1) | 0.2542(1) | 0.018(0) |
| Cs2 | 0.4011(1) | 0.7063(1) | 0.2092(1) | 0.021(0) |
| B1 | 0.4956(15) | 0.2422(13) | 0.4650(9) | 0.011(2) |
| B2 | 0.8638(14) | 0.8243(13) | 0.4378(10) | 0.011(2) |
| B3 | 0.9135(15) | 0.6602(13) | 0.0727(10) | 0.013(3) |
| B4 | 0.9039(16) | 0.6578(14) | 0.2705(10) | 0.016(3) |
| B5 | 0.5452(15) | 0.2470(13) | 0.1040(10) | 0.013(2) |
| B6 | 0.6777(13) | 0.0411(14) | 0.4396(9) | 0.009(2) |
| B7 | 0.7282(16) | 0.4757(14) | 0.1060(10) | 0.015(3) |
| B8 | 0.3230(14) | 0.0503(14) | 0.0669(10) | 0.013(2) |
| B9 | 0.0813(15) | 0.6437(14) | 0.4699(10) | 0.014(3) |
| B10 | 0.5011(15) | 0.1382(13) | 0.2678(10) | 0.009(2) |
| O1 | 0.6185(9) | 1.1448(8) | 0.5124(5) | 0.013(2) |
| O2 | 1.0508(9) | 0.6039(8) | 0.3562(6) | 0.015(2) |
| O3 | 0.7696(9) | 0.5338(9) | 0.2167(6) | 0.016(2) |
| O4 | 0.9904(9) | 0.7557(8) | 0.5137(6) | 0.015(2) |
| O5 | 0.5960(9) | 0.2367(8) | 0.2168(6) | 0.016(2) |
| O6 | 0.6159(9) | 0.3522(8) | 0.0580(5) | 0.014(2) |
| O7 | 0.4110(9) | 0.1528(8) | 0.0275(6) | 0.015(2) |
| O8 | 0.7917(9) | 0.5388(8) | 0.0296(6) | 0.015(2) |
| O9 | 1.2050(10) | 0.5731(8) | 0.5407(6) | 0.017(2) |
| O10 | 0.9763(9) | 0.7113(8) | 0.1854(6) | 0.015(2) |
| O11 | 0.8230(9) | 0.7804(8) | 0.3247(6) | 0.012(2) |
| O12 | 0.1708(12) | 0.3856(11) | 0.2242(7) | 0.037(2) |
| O13 | 0.4276(9) | 1.2319(8) | 0.3503(6) | 0.014(2) |
| O14 | 0.4471(10) | 1.3414(9) | 0.5426(6) | 0.020(2) |
| O15 | 0.9758(9) | 0.7275(8) | –0.0014(6) | 0.016(2) |
| O16 | 0.2046(9) | –0.0486(9) | –0.0134(6) | 0.019(2) |
| O17 | 0.6271(9) | 1.0389(8) | 0.3251(5) | 0.013(2) |
| O18 | 0.7917(9) | 0.9431(8) | 0.4882(6) | 0.013(2) |
| O19 | 0.3545(8) | 0.0494(8) | 0.1799(5) | 0.013(2) |
| H1 | 0.135(9) | –0.112(8) | 0.008(7) | 0.08(6) |
| H2 | 0.375(9) | 1.414(6) | 0.522(10) | 0.08(6) |
| H3 | 0.916(13) | 0.671(9) | –0.068(4) | 0.04(4) |
| H4 | 1.225(19) | 0.617(12) | 0.614(3) | 0.08(6) |
| H5 | 0.252(8) | 0.319(7) | 0.243(12) | 0.08(6) |
| H6 | 0.116(9) | 0.446(9) | 0.267(6) | 0.06(5) |
Таблица 3.
Межатомные расстояния (Å) в структуре Cs[B5O7(OH)2] · 0.5H2O
| Cs1-девятивершинник | Cs2-девятивершинник | ||
| O16 | 2.928(7) | O14 | 3.067(8) |
| O4 | 3.071(7) | O11 | 3.214(7) |
| O10 | 3.153(7) | O6 | 3.225(6) |
| O18 | 3.156(6) | O12 | 3.247(7) |
| O11 | 3.196(7) | O10 | 3.245(7) |
| O12 | 3.207(7) | O3 | 3.298(7) |
| O17 | 3.222(7) | O8 | 3.368(7) |
| Ow | 3.414(11) | O17 | 3.395(7) |
| O5 | 3.486(7) | Ow | 3.481(11) |
| 〈Cs1–O〉 | 3.204 | 〈Cs2–O〉 | 3.282 |
| B1-треугольник | B4-тетраэдр | ||
| O13 | 1.354(12) | O11 | 1.465(14) |
| O14 | 1.356(13) | O10 | 1.468(14) |
| O1 | 1.396(13) | O3 | 1.472(14) |
| 〈B1–O〉 | 1.369 | O2 | 1.478(13) |
| B2-треугольник | 〈B4–O〉 | 1.471 | |
| O11 | 1.336(13) | B10-тетраэдр | |
| O18 | 1.381(13) | O12 | 1.464(13) |
| O4 | 1.393(13) | O17 | 1.465(13) |
| 〈B2–O〉 | 1.370 | O5 | 1.476(13) |
| B3-треугольник | O13 | 1.478(13) | |
| O10 | 1.343(13) | 〈B10–O〉 | 1.471 |
| O15 | 1.367(14) | B7-треугольник | |
| O8 | 1.387(13) | O3 | 1.333(14) |
| 〈B3–O〉 | 1.366 | O6 | 1.367(14) |
| B5-треугольник | O8 | 1.378(13) | |
| O5 | 1.357(13) | 〈B7–O〉 | 1.359 |
| O7 | 1.376(13) | B8-треугольник | |
| O6 | 1.376(13) | O12 | 1.349(13) |
| 〈B5–O〉 | 1.370 | O16 | 1.355(13) |
| B6-треугольник | O7 | 1.380(13) | |
| O17 | 1.355(12) | 〈B8–O〉 | 1.361 |
| O18 | 1.367(12) | B9-треугольник | |
| O1 | 1.384(13) | O2 | 1.349(14) |
| 〈B6–O〉 | 1.369 | O9 | 1.359(13) |
| O4 | 1.389(14) | ||
| 〈B9–O〉 | 1.366 | ||
Таблица 4.
Геометрическая характеристика водородных связей в структуре соединения Cs[B5O7(OH)2] · · 0.5H2O
| D–H···A | Расстояние, Å | Угол D–H···A, град | ||
|---|---|---|---|---|
| D–H | H···A | D···A | ||
| O16–H1···O15 | 0.900(2) | 1.88(3) | 2.749(10) | 160(8) |
| O14–H2···O9 | 0.900(2) | 2.03(5) | 2.873(10) | 156(12) |
| O15–H3···Ow2 | 0.900(2) | 1.84(4) | 2.695(11) | 158(9) |
| O9–H4···O5 | 0.900(2) | 2.36(7) | 3.178(10) | 152(13) |
| Ow–Hw1···O13 | 0.900(2) | 1.90(6) | 2.736(10) | 153(13) |
| Ow–Hw2···O2 | 0.900(2) | 1.85(3) | 2.734(11) | 165(9) |
АНАЛИЗ МЕЖАТОМНЫХ РАССТОЯНИЙ И ОПИСАНИЕ КРИСТАЛЛИЧЕСКОЙ СТРУКТУРЫ
Исследование кристаллической структуры нового пентабората цезия Cs[B5O7(OH)2] · 0.5H2O подтвердило его изоструктурность с изученными ранее рубидиевым [3] и аммонийсодержащим аналогами [4], поэтому анализ межатомных расстояний проведен при сопоставлении данных о трех соединениях.
Независимый фрагмент кристаллической структуры цезиевого пентабората показан на рис. 1. Атомы бора занимают 10 независимых позиций; две из них имеют тетраэдрическую координацию по кислороду, а восемь характеризуются треугольной координацией. В треугольниках расстояния B–O варьируются в пределах 1.333(14)–1.396(13) Å с несколько меньшим разбросом значений по сравнению с рубидиевым аналогом (1.332–1.409 Å) и чуть большим по сравнению с аммонийсодержащим пентаборатом (1.337–1.391 Å). Тетраэдр бора практически не искажен. Расстояния B–O типичны для бора в тетраэдрической координации и лежат в интервалах 1.465(14)–1.478(13) и 1.464(13)–1.478(13) Å для B1 и B2 соответственно, со средним значением 1.471 Å для обоих тетраэдров. Среднее расстояние B–O несколько больше, чем соответствующее значение 1.469(4) Å для NH4-аналога. Заметим, что в структуре рубидиевого аналога боратные тетраэдры сильнее искажены и характеризуются большим разбросом расстояний B–O 1.465–1.503 Å.
Рис. 1.
Координация катионов в кристаллической структуре Cs[B5O7(OH)2] · 0.5H2O: независимый фрагмент, эллипсоиды тепловых колебаний атомов представлены с вероятностью 80%.
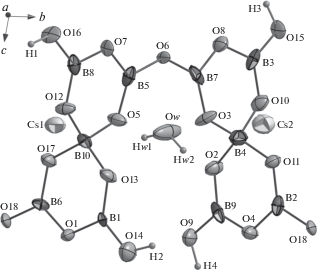
Двойные пентаборатные фрагменты, обобществляя по два атома кислорода, образуют зигзагообразные ленты, вытянутые вдоль направления [010] (рис. 2а). Висячие вершины треугольников BO3 этих лент протонированы и образуют гидроксильные группы O16–H1, O14–H2, O15–H3 и O9–H4. Гидроксильные группы O16–H1 и O14–H2 играют роль доноров, участвуя в водородной связи с атомами кислорода O15 и O9 соседних пентаборатных лент, в результате чего образуются полярные слои (рис. 2б), чередующиеся вдоль оси с. Молекула воды располагается в пустотах слоя пентаборатных лент и является донором двух водородных связей с атомами кислорода O2 и O13, общими для борных тетраэдров и треугольников соседних лент (рис. 3). Акцепторная функция этой молекулы воды Ow выполняется за счет ее участия в водородной связи с гидроксогруппой H3–O15, координированной с атомом бора B3 ленты соседнего слоя (рис. 3). Таким образом, молекула воды укрепляет слой из пентаборатных лент и связывает его с одним из двух соседних слоев. Вследствие полярности рассматриваемого слоя его связь с другим, противоположным, слоем осуществляется за счет образования водородной связи гидроксильной группы H4–O9 с атомами кислорода O5. Согласно табл. 4 водородная связь, образованная H4, самая слабая, все остальные можно отнести к средним. По сравнению с рубидиевым аналогом [3] в кристаллической структуре исследуемого соединения установлено иное положение атома водорода Hw2 молекулы воды (рис. 3, вставка). В исследованной структуре молекула воды закреплена в пустоте тремя водородными связями: Ow–Hw1···O13, Ow–Hw2···O2 и O15–H3···O2. В Rb[B5O7(OH)2] · · 0.5H2O наблюдаются две водородные связи: Hw2–Ow···O11 и O5–H2···Ow. В структуре аммониевого аналога атомы молекулы воды распределены по двум позициям; топология водородных связей аналогична наблюдаемой в исследуемом соединении.
Рис. 2.
Фрагмент кристаллической структуры Cs[B5O7(OH)2] · 0.5H2O: a – пентаборатная цепочка, вытянутая вдоль направления [010]; б – слой соединенных водородными связями пентаборатных цепочек, параллельный плоскости ab и вмещающий катионы цезия и молекулы воды.
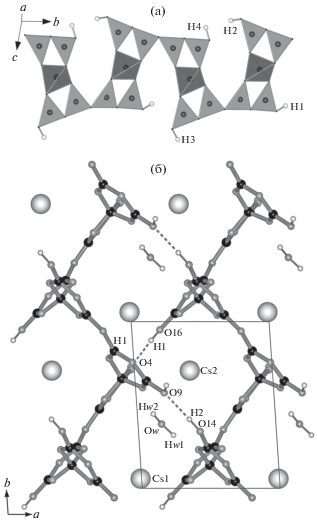
Рис. 3.
Проекция кристаллической структуры Cs[B5O7(OH)2] · 0.5H2O на плоскость xz. На вставке – конфигурация молекулы воды в кристаллической структуре Rb[B5O7(OH)2] · 0.5H2O.
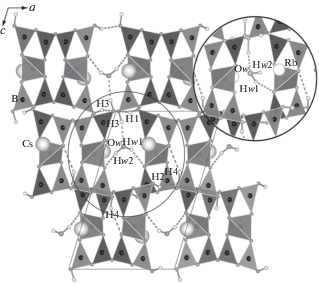
Катионы цезия занимают две независимые позиции, располагаясь в пространстве между пентаборатными цепочками (рис. 2 и 3), и окружены девятью ближайшими атомами кислорода (рис. 1). Расстояния Сs1–O и Сs2–O варьируются в пределах 2.927(7)–3.486(7) Å (среднее 3.204 Å) и 3.066(8)–3.481(10) Å (среднее 3.282 Å) соответственно. Эти значения закономерно увеличены по сравнению с расстояниями Rb1–O и Rb2–O (средние значения 3.085 и 3.188 Å) в кристаллической структуре рубидиевого пентабората.
Закономерно, что в ряду соединений А[B5O7(OH)2] · 0.5H2O при увеличении ионного радиуса щелочного катиона NH4 → Rb → Cs наблюдается увеличение объемов их элементарных ячеек (табл. 5). Увеличиваются также параметры a и c, в то время как параметр b для всех членов ряда меняется незначительно. Это связано с жесткостью основного структурного фрагмента обсуждаемых соединений – пентаборатных цепочек, вытянутых вдоль направлений b их кристаллических структур.
Таблица 5.
Кристаллографические характеристики изоструктурных соединений с общей формулой А[B5O7(OH)2] · 0.5H2O (A = NH4, Rb, Cs)
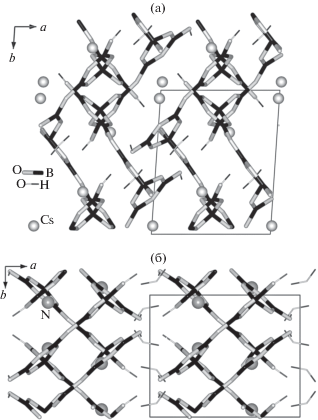
В [3, 4] было показано, что изоструктурные соединения А[B5O7(OH)2] · 0.5H2O (А = NH4, Rb) родственны минералу лардереллиту NH4[B5O7(OH)2] · H2O, так как основой кристаллических структур обоих типов являются зигзагообразные пентаборатные цепочки, вытянутые вдоль осей b. Основные различия обсуждаемых структурных типов связаны с конфигурацией и топологией взаимного расположения указанных цепочечных фрагментов и характером заполнения пустот между ними. В кристаллической структуре лардереллита соседние пентаборатные блоки [B5O10] в цепочке симметрийно связаны винтовой осью второго порядка. В структурах синтетических пентаборатов симметрийно независимым является двойной пентаборатный фрагмент [B10O20]. В элементарной ячейке лардереллита цепочки, связанные осью второго порядка, вдоль оси b перекрещиваются (рис. 4б). В структурах А[B5O7(OH)2] · 0.5H2O (А = NH4, Rb, Сs) аналогичные цепочки, размноженные центром инверсии, параллельны друг другу (рис. 4а). Это обусловливает наличие в структуре лардереллита чередующихся вдоль направления [001] слоев, перпендикулярных оси a, пространство между которыми занято молекулами воды, что определяет пластинчатый габитус кристаллов вдоль направления [100]. Подобное обширное пространство отсутствует в кристаллической структуре синтетических пентаборатов (рис. 3, 4). Уменьшение количества молекул воды вдвое по сравнению с лардереллитом, взаимное смещение пентаборатных цепочек в плоскости ab и дополнительное их укрепление водородными связями приводят к тому, что перпендикулярно направлению [010] располагаются наиболее плотноупакованные слои (рис. 3). С этим согласуется таблитчатая форма кристаллов исследуемого соединения, для которых было установлено, что уплощенность связана с развитием граней [010]. Таблитчатая форма описана и для кристаллов аммонийсодержащего пентабората [4]. Наблюдаемый изометричный облик кристаллов Rb[B5O7(OH)2] · 0.5H2O, аналога [3], может быть связан с условиями их роста, например, в условиях пересыщения.
Рис. 4.
Проекция пентаборатных цепочек на плоскость xy в кристаллических структурах Cs[B5O7(OH)2] · 0.5H2O (а) и NH4[B5O7(OH)2] · · H2O (б).
Авторы выражают благодарность В.О. Япаскурту за рентгеноспектральный анализ образца и Н.В. Зубковой за проведение рентгеновского эксперимента.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (грант № 18-03-00908).
Список литературы
Behm H. // Acta Cryst. C. 1984. V. 40. P. 1114.
Penin N., Seguin L., Gerand B., et al. // J. Alloys Compd. 2002. V. 334. P. 97.
Belokoneva E.L., Borisova T.A., Dimitrova O.V. // Crystallogr. Rep. 2003. V. 48. № 4. P. 583.
Li L.Y., Li G.B., Xiong M., et al. // Acta Cryst. C. 2003. V. 59. № 11. P. 1115.
Merlino S., Sartori F. // Acta Cryst. B. 1969. V. 25. № 11. P. 2264.
Merlino S. // Acta Cryst. B. 1972. V. 28. № 12. P. 3559.
Merlino S., Sartori F. // Contrib. Mineral. Petrol. 1970. V. 27. № 2. P. 159.
Merlino S., Sartori F. // Science. 1971. V. 171. № 3969. P. 377.
Thomas R., Davidson P., Hahn A. // Am. Mineral. 2008. V. 93. № 7. P. 1034.
Dmitriev V.G., Gurzadyan G.G., Nikogosyan D.N. // Handbook of Nonlinear Optical Crystals. Berlin–Heidelberg: Springer, 1997. P. 67.
Becker P., Held P., Bohaty L. // Cryst. Res. Technol. 2000. V. 35. P. 1251.
CrysAlisPro. Crysalis Software System: Oxford Diffraction Ltd. Abingdon, UK. 2007.
Farrugia L.J. // J. Appl. Cryst. 1999. V. 32. P. 837.
Prince E. // International Tables for Crystallography. IUCr, 2004. Tables 4.2.6.8; 6.1.14.
Sheldrick G.M. // Acta Cryst. C. 2015. V. 71. P. 3.
Brown I.D., Altermatt D. // Acta Cryst. B. 1986. V. 41. P. 244.
Brown I.D. // Chem. Rev. 2009. V. 109. P. 6858.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография