

Кристаллография, 2019, T. 64, № 2, стр. 209-219
Новый борофосфат натрия и хрома Na{Cr[BP2O7(OH)3]}: синтез, кристаллическая структура, особенности водородных связей и сравнительная кристаллохимия
Н. А. Ямнова 1, *, С. М. Аксенов 2, **, Е. Ю. Боровикова 1, А. С. Волков 1, О. А. Гурбанова 1, О. В. Димитрова 1, П. К. Бёрнс 2
1 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
2 Университет Нотр-Дам
Саут-Бенд, США
* E-mail: natalia-yamnova@yandex.ru
** E-mail: aks.crys@gmail.com
Поступила в редакцию 21.11.2017
После доработки 21.11.2017
Принята к публикации 02.02.2018
Аннотация
Методами рентгеноструктурного анализа и ИК-спектроскопии изучен новый борофосфат натрия и хрома, полученный методом гидротермального синтеза в системе Na2O–P2O5–B2O3–Cr2O3–Н2О. Параметры моноклинной ячейки: a = 10.4220(3), b = 8.2468(2), c = 9.2053(3) Å, β = 116.568(4)°, V = 707.63(4) Å3, пр. гр. С2/с. Структура расшифрована и уточнена до итогового фактора расходимости R1 = 2.23% в анизотропном приближении смещений атомов с использованием 1311 отражений, I > 2σ(I). Новое соединение родственно семейству щелочных борофосфатов с общей формулой A{M[BP2O7(OH)3]} (A – щелочной или щелочноземельный элемент; M – переходный метал) и характеризуется кристаллохимической формулой (Z = 4) Na{Cr[BP2O7(OH)3]}, где квадратными скобками выделен состав борофосфатного аниона, а фигурными – микропористого каркаса. Установлено присутствие альтернативных систем сильных симметричных водородных связей, а локализация всех позиций атомов водорода позволила детально их проанализировать. Обнаружен дополнительный пик электронной плотности, который можно интерпретировать как статистически распределенный протон H+. ИК-спектр характерен для соединений с сильной водородной связью и подтверждает статистическое распределение протона по подпозициям вокруг центра инверсии. Предполагается, что представители данного семейства могут обладать протонной проводимостью.
ВВЕДЕНИЕ
Борофосфаты представляют обширный класс соединений, который насчитывает более 250 представителей [1–5]. Их главной структурной особенностью является наличие анионных мотивов, образованных тетраэдрически координированными атомами фосфора и атомами бора либо в тетраэдрической, либо, что встречается реже, в треугольной координации. Для борофосфатов, как и боратов, силикатов и боросиликатов, характерно различное формирование (от островных до каркасных) анионных мотивов, а два варианта координационного окружения бора заметно увеличивают возможное количество структурных типов. Активное изучение борофосфатов в последнее десятилетие послужило стимулом к разработке современной систематики данного класса, в основе которой лежит степень полимеризации структурных блоков (олигомеров) [2, 5].
Борофосфаты рассматривают как перспективный класс материалов для применения в современных технологиях [6–8]. Так, соединения переходных элементов привлекают интерес благодаря потенциальным магнитным свойствам. В отличие от боратов и фосфатов борофосфаты хрома изучены плохо, к настоящему времени известны всего четыре фазы (табл. 1), в трех из которых – Cr2(BΔP3O12), Cs2Cr3(BtP4O14)(P4O13) и Na10K5{Na[Cr8O4(BtP2O10)4(P(O,OH)4)4]} · 10H2O (Δ – треугольная координация атома бора; t – тетраэдрическая) – установлены сильные антиферромагнитные взаимодействия между центрами Cr3+–Cr3+ при низких температурах.
Таблица 1.
Сравнительные характеристики борофосфатов хрома
| Соединение | Пр. гр., Z | Параметры ячейки | Литература | ||
|---|---|---|---|---|---|
| a, Å | b, Å, β, град | c, Å, γ, град | |||
| Cr2(B(PO4)3) | P63/m, 2 | 7.944 | 7.944 | 7.344, 120 | [9, 10] |
| Cs2Cr3(BP4O14)(P4O13) | P21/c, 4 | 14.792 | 15.819, 92.45 | 9.704 | [11] |
| Na8(Cr4B12P8O44(OH)4)(P2O7) · 8(H2O) | I23, 6 | 20.024 | 20.024 | 20.024 | [12] |
| Na10K5{Na[Cr8O4(BP2O10)4(P(O,OH)4)4]} · 10H2O | Pmnn, 2 | 10.463 | 15.049 | 18.721 | [13] |
Настоящая работа посвящена гидротермальному синтезу новой фазы Na{Cr[BP2O7(OH)3]} и ее изучению методами рентгеноструктурного анализа.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез и химический состав. Монокристаллы Na{Cr[BP2O7(OH)3]} получены гидротермальным методом в системе Na2O–P2O5–B2O3–Cr2O3–Н2О при равном соотношении компонентов. Синтез проводили при температуре 270–250°C и давлении 80 атм. Нижний предел температуры был ограничен кинетикой гидротермальной реакции, верхний – возможностями аппаратуры. Эксперименты проводили в стандартных автоклавах объемом 16 мл, в качестве защитного покрытия использовали медь. Продолжительность опыта (20 сут) отвечала полному завершению реакции, коэффициент заполнения автоклава выбирали таким образом, чтобы давление оставалось постоянным. Синтезированные кристаллы промывали горячей дистиллированной водой и высушивали при комнатной температуре. Получены кристаллы темно-зеленого цвета размером ~0.1–0.5 мм, имеющие форму утолщенных пластинок.
Присутствие Na, Cr и P в химическом составе синтезированных кристаллов определено с помощью растрового электронного микроскопа Jeol JSM6480LV, оснащенного энергодисперсионным спектрометром INCA Wave 500 (ускоряющее напряжение 20 кВ, сила тока 20 нА, диаметр электронного пучка 3 мкм).
Рентгеноструктурный анализ. Для рентгеноструктурного исследования был отобран обломок кристалла изометричной формы. Экспериментальный набор интенсивностей получен при комнатной температуре в полной сфере обратного пространства с использованием дифрактометра Xcalibur Oxford Diffraction, оснащенного двухкоординатным CCD-детектором. Обработку эксперимента проводили с использованием программы CrysAlis [14]. Характеристики кристалла и условия эксперимента приведены в табл. 2. Структурная модель получена методом charge flipping [15] и уточнена до итогового значения фактора расходимости R1 = 2.23% в анизотропном приближении атомных смещений с использованием 1243 отражений, I > 2σ(I). Все расчеты выполнены по комплексу программ Jana2006 [16]. Итоговые координаты атомов и параметры атомных смещений приведены в табл. 3, характеристики координационных полиэдров – в табл. 4, локальный баланс валентностей [17, 18] – в табл. 5.
Таблица 2.
Кристаллографические характеристики, данные эксперимента и результаты уточнения структуры Na{Cr[BP2O7(OH)3]}
| Т, К | 293(2) |
| Сингония, пр. гр., Z | Моноклинная, С2/с (№ 15), 4 |
| a, b, с, Å | 10.4220(3), 8.2468(2), 9.2053(3) |
| β, град | 116.568(4) |
| V, Å3 | 707.63(4) |
| Размеры кристалла, мм | 0.08 × 0.10 × 0.14 |
| Dx, г/см3 | 2.916 |
| μ, мм–1 | 2.176 |
| Дифрактометр | Xcalibur Oxford Diffraction, ССD-детектор |
| Излучение; λ, Å | MoKα; 0.7107 |
| Тип сканирования | ω |
| F(000) | 612 |
| θmin–θmax, град | 4.37–34.98 |
| Пределы h, k, l | –16 < h < 16, –12 < k < 12, –14 < l < 11 |
| Общее число отражений/число усредненных отражений (N2)/ Rуср/число независимых отражений (N1) (I > 2σ(I)) | 3843/1311/4.90/1243 |
| Метод уточнения | МНК по F 2 |
| Весовая схема | w = 1/(σ2(I) + 0.0004I2) |
| S | 1.07 |
| R1/wR1 | 2.23; 4.69 |
| R2/wR2 | 2.34; 4.75 |
| Δρmin/Δρmax, э/Å3 | –0.20 / 0.27 |
Таблица 3.
Координаты атомов, кратность позиций (Q) и параметры атомных смещений (U) в структуре Na{Cr[BP2O7(OH)3]}
| Пози-ция | x/a | y/b | z/c | Q | Uэкв/изо*, Å2 |
|---|---|---|---|---|---|
| Na | 0 | 0.1338(1) | 0.25 | 4 | 0.0189(3) |
| Cr | 0.25 | 0.25 | 0 | 4 | 0.0066(1) |
| P | 0.2783(1) | 0.4319(1) | 0.3226(1) | 8 | 0.0061(1) |
| B | 0 | 0.5275(2) | 0.25 | 4 | 0.0051(4) |
| O1 | 0.1117(1) | 0.4180(1) | 0.2504(1) | 8 | 0.0096(2) |
| O2 | 0.1715(1) | 0.1018(1) | 0.1111(1) | 8 | 0.0097(2) |
| O3 | 0.3402(1) | 0.3017(1) | 0.4575(1) | 8 | 0.0121(3) |
| O4 | 0.4408(1) | 0.1263(1) | 0.1012(1) | 8 | 0.0091(2) |
| O5 | 0.3249(1) | 0.3983(1) | 0.1909(1) | 8 | 0.0100(2) |
| H1 | 0.270(5) | 0.243(5) | 0.478(7) | 8 | 0.032(12)* |
| H2 | 0.441(4) | 0.072(4) | 0.006(3) | 8 | 0.086(11)* |
Таблица 4.
Межатомные расстояния (Å) в структуре Na{Cr[BP2O7(OH)3]}
| Na | O3 | 2.509(1) × 2 |
| O5 | 2.551(1) × 2 | |
| O1 | 2.616(1) × 2 | |
| O2 | 2.635(1) × 2 | |
| 〈Na–O〉 | 2.578 | |
| Cr | O2 | 1.990(1) × 2 |
| O5 | 1.991(1) × 2 | |
| O4 | 2.050(1) × 2 | |
| 〈Cr–O〉 | 2.010 | |
| P | O5 | 1.520(1) |
| O2 | 1.524(1) | |
| O3 | 1.547(1) | |
| O1 | 1.562(1) | |
| 〈P–O〉 | 1.538 | |
| B | O4 | 1.471(1) × 2 |
| O1 | 1.472(1) × 2 | |
| 〈B–O〉 | 1.472 | |
Таблица 5.
Локальный баланс валентностей в кристаллической структуре Na{Cr[BP2O7(OH)3]}
| Позиция | O1 | O2 | O3 | O4 | O5 | Vкатион |
|---|---|---|---|---|---|---|
| Na | 0.11×2→ | 0.11×2→ | 0.15×2→ | 0.13×2→ | 1.00 | |
| Cr | 0.49×2→ | 0.41×2→ | 0.49×2→ | 2.78 | ||
| P | 1.16 | 1.29 | 1.21 | 1.30 | 4.96 | |
| B | 0.76×2→ | 0.76×2→ | 3.04 | |||
| Н1 | 0.41 + 0.18 | 0.59* | ||||
| Н2 | 0.13 | 0.76 | 0.89 | |||
| Vанион | 2.16 | 1.89 | 1.95 | 1.93 | 1.92 |
ИК-спектроскопия. Порошковый ИК-спектр Na{Cr[BP2O7(OH)3]} получен на ИК-фурье-спектрометре FSM 12011 FTIR (кафедра минералогии, МГУ) при комнатной температуре в диапазоне волновых чисел от 4000 до 400 см–1 c использованием стандартной методики таблетирования с KBr. Разрешение спектра составляло ~ 2 см–1.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Кристаллическая структура. Исследованный в работе борофосфат натрия и хрома изоструктурен соединениям семейства A{M[BP2O7(OH)3]} (A – щелочной или щелочноземельный элемент; M – переходный металл) и характеризуется кристаллохимической формулой (Z = 4) Na{Cr[BP2O7(OH)3]}, где квадратными скобками выделен состав борофосфатного аниона, а фигурными – микропористого каркаса.
В кристаллической структуре Na{Cr[BtP2O7(OH)3]} в формировании открытого (т.е. характеризующегося наличием “висячих” вершин) гетерополиэдрического каркаса участвуют CrO4(OH)2-октаэдры (〈Cr–Ø〉 = 2.010 Å; Ø = O, OH), PO3.5(OH)0.5-тетраэдры (〈P–Ø〉 = 1.538 Å) и BO2(OH)2-тетраэдры (〈B–Ø〉 = 1.472 Å) (рис. 1). Каждый CrØ6-октаэдр связан через экваториальные O2- и O5-вершины с четырьмя PØ4-тетраэдрами, а через апикальные OH(4)-группы – с двумя BØ4-тетраэдрами. Кроме того, один борный и два фосфорных тетраэдра, имеющие общие О1-атомы кислорода, объединяются и формируют борофосфатный тример состава [BP2O7(OH)3]4–.
Рис. 1.
Проекции кристаллической структуры Na{Cr[BP2O7(OH)3]} на плоскости ab (а) и ac (б). Сплошными линиями показаны связи Д–Н, пунктирными – Н···А.
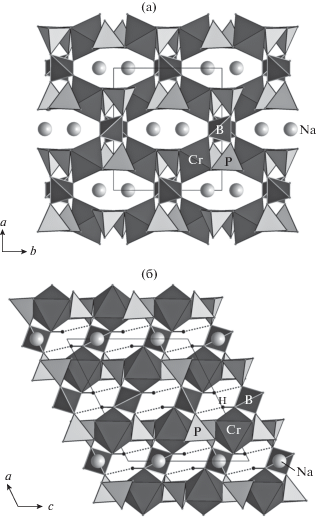
Гетерополиэдрический каркас {Cr[BP2O7(OH)3]}– характеризуется двумя системами пересекающихся каналов, идущих вдоль направлений [001] и [010] соответственно (рис. 1). Канал I характеризуется восьмиугольным сечением и эффективной шириной [19], равной 0.94 × 6.54 Å, а канал II – шестиугольным сечением и эффективной шириной 0.68 × 3.60 Å. Общий отрицательный заряд каркаса компенсируется катионами натрия, которые заполняют каналы I, располагаясь в крупных NaO8-восьмивершинниках (〈Na–O〉 = 2.578 Å).
Водородные связи. В кристаллической структуре Na{Cr[BP2O7(OH)3]} были локализованы все позиции атомов водорода, что позволило детально проанализировать водородные связи (ВС). Геометрические характеристики ВС приведены в табл. 6.
Таблица 6.
Сравнительные геометрические характеристики водородных связей (Д – донор протона, А – акцептор) в структурах борофосфатов c общей формулой A{M[BP2O7(OH)3]}
| Д–Н | А | Д–Н, Å | Н···А, Å | Д···А, Å | Угол ДНА, град | Литература |
|---|---|---|---|---|---|---|
| Na{Cr[BP2O7(OH)3]} | ||||||
| О3–Н1 | О3 | 0.96(6) | 1.555(7) | 2.489(1) | 162(5) | Настоящая работа |
| О4–Н2 | О1 | 0.98(3) | 2.17(3) | 3.053(2) | 148(2) | |
| Na{V[BP2O7(OH)3]} | ||||||
| О5–Н2 | О5 | 1.248(2) | 1.248(2) | 2.495(3) | 180 | [20] |
| О1–Н1 | О5 | 0.70(7) | 2.27(7) | 2.943(3) | 163(7) | |
| Sr{Fe[BP2O8(OH)2]} | ||||||
| О9–Н1 | О3 | 0.89(7) | 2.15(7) | 3.030(6) | 170(6) | Настоящая работа |
| O8 | 2.51(7) | 3.135(1) | 74(4) | |||
| O1 | 2.57(5) | 3.104(1) | 119(5) | |||
| О10–Н2 | О5 | 1.00(6) | 1.78(7) | 2.764(5) | 170(6) | |
| О9–Н1 | О1 | 0.89(6) | 2.57(5) | 3.106(3) | 119(5) | [21] |
| О10–Н2 | О5 | 1.00(5) | 1.78(7) | 2.764(1) | 170(6) | |
Атомы Н2, локализованные в каналах II, образуют ВС, в которых в качестве доноров (Д) выступают О4-вершины ВØ4-тетраэдра, а акцепторов (А) – мостиковые O1-атомы борофосфатного тримера.
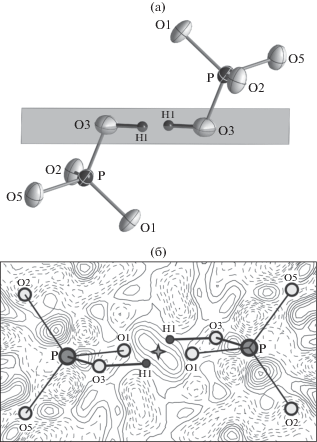
Установлено частичное замещение “висячей” O3-вершины PØ4-тетраэдров ОН-группой, образованной с участием атома водорода Н1, позиция которого статистически заселена на 50% (рис. 2а). Данные атомы водорода занимают расщепленную позицию вокруг центра симметрии с расстоянием между позициями Н1–Н1i = 0.7(1) Å. Таким образом, в структуре присутствуют две альтернативные системы ВС О3–H1···О3i (расстояние O3–O3i = 2.489(3) Å), в которых атом О3 одновременно является и донором, и акцептором. Анализ распределения электронной плотности (рис. 2б) показал, что ВС в структуре частично разупорядочены. На картах разностных синтезов Фурье выявлен “дополнительный“ пик электронной плотности (с координатами (1/4 1/4 1/2)) непосредственно в центре симметрии на расстоянии ~1.24 Å от атома кислорода O3. Наличие дополнительного максимума электронной плотности позволяет предположить, что протон H+ распределен статистически. В [22] показано, что этот пик согласуется с теоретическими и экспериментальными данными [23–25] и может быть объяснен как результат частичного переноса протона H+ от одного атома кислорода O3 к соседнему по схеме: Н1–О3–H···О3i–Н1 ↔ Н1–О3···H+···О3i–Н1 ↔ Н1–О3···H–О3i–Н1 [22].
Рис. 2.
Симметричные водородные связи в структуре Na{Cr[BP2O7(OH)3]} (а) и анализ распределения электронной плотности (б). Шаг изолиний – 0.05 э/Å3.
В изученных ранее изоструктурных соединениях атомы водорода фосфатных групп локализованы непосредственно в центре симметрии при увеличенном расстоянии O···H, равном ~ 1.25 Å (в таком случае вклад протона составляет ~ 0.48 единиц валентности). Анализ суммы валентных усилий на анионах (с учетом расстояний кислород–водород) во всех структурах показал дефицит положительного заряда на “висячей” O3-вершине РØ4-тетраэдра. Все это свидетельствует в пользу статистического разупорядочения атомов водорода вокруг центра симметрии и образования симметричной ВС, как в случае изученного соединения Na{Cr[BP2O7(OH)3]}.
Согласно [21] характер ВС в структуре Sr{Fe3+[BP2O8(OH)2] немного отличается. Исходя из предположений о трехвалентном состоянии атомов железа авторы проанализировали ВС ОН-групп BØ4-тетраэдров. В качестве акцептора H1-атома водорода была выбрана О1-вершина Р1Ø4-тетраэдра (H1···О1 = 2.570 Å). Тем не менее, исходя из детального анализа расположения атомов в кристаллической структуре Sr{Fe3+[BP2O8(OH)2], можно предположить наличие двух акцепторов у H1-атома, характеризующихся более короткими расстояниями H···A (табл. 6): H1···О3 (2.152 Å) и H1···О3 (2.514 Å). Расчет локального баланса валентностей [17, 18] (табл. 7) показал, что два симметрично неэквивалентных Fe1Ø6– и Fe2Ø6-октаэдра заселены преимущественно Fe3+ и Fe2+ соответственно. “Висячая” O3-вершина Р1Ø4-тетраэдра характеризуется дефицитом заряда (1.53 единиц валентности), свидетельствующим о смешанной заселенности кислородом и гидроксильной группой данной позиции, что наблюдается и в других представителях семейства. Таким образом, формулу соединения следует записать в виде (Z = 2): Sr2+{(Fe2+,Fe3+)[BP2O7(OH)2(О,ОН)]}.
Таблица 7.
Локальный баланс валентностей в кристаллической структуре Sr{Fe[BP2O7(OH)3]}
| Позиция | Sr | Fe1 | Fe2 | P1 | P2 | B | H1 | H2 | Vанион |
|---|---|---|---|---|---|---|---|---|---|
| O1 | 0.34 | 0.35×2↓ | 1.35 | 0.07 | 2.11 | ||||
| O2 | 0.17 | 0.54×2↓ | 1.25 | 1.96 | |||||
| O3 | 0.18 | 1.21 | 0.14 | 1.53 | |||||
| O4 | 0.23 | 1.17 | 0.69 | 2.09 | |||||
| O5 | 0.34 | 1.31 | 0.26 | 1.91 | |||||
| O6 | 0.21 | 0.31×2↓ | 1.28 | 1.80 | |||||
| O7 | 0.21 | 0.50×2↓ | 1.23 | 1.94 | |||||
| O8 | 0.27 | 1.12 | 0.74 | 0.08 | 2.21 | ||||
| O9 | 0.47×2↓ | 0.75 | 1.06 | 2.28 | |||||
| O10 | 0.37×2↓ | 0.85 | 0.72 | 1.94 | |||||
| Vкатион | 1.95 | 3.02 | 2.06 | 4.98 | 4.94 | 3.03 | 1.35 | 0.98 |
Симметричные ВС достаточно редки. Среди минералов можно отметить бораты: преображенскит Mg3[B11O15(OH)9] [26], калиборит KMg2H[B6O8(OH)5]2 · 4H2O [27], а также недавно изученный представитель группы миксита–агардит-(Се) CeCu6(AsO4)3(OH)6 · 3H2O [22]. Кроме того, симметричные ВС установлены в синтетических соединениях со структурным типом кренкита: K[Mg(H0.5SO4)2(H2O)2] [28], CsM3+(H1.5AsO4)(H2AsO4) (M = Ga, Cr) [29]. В семействе кристаллов с общей формулой MmHn(XO4)(m + n)/2 · yH2O (M = K, Rb, Cs; X = S, Se, P) наличие протонов H+, связанных с двумя атомами кислорода (акцепторами), в отсутствие атомов кислорода (доноров) создает предпосылки для суперпротонной проводимости [30–32]. Анализ карт распределения электронной плотности во всех случаях показывает расщепление пиков с двумя локальными максимумами [22, 27, 28], что говорит о смещении атомов водорода из центра симметрии. Расчет энергии ВС в кристаллах K3H(SO4) и [K1 – x(NH4)x]3H(SO4) показал два энергетических минимума, разделенных барьером, с энергией 0.29 и 0.37 кДж/моль [33]. Низкая величина энергетического барьера создает предпосылки для переноса протона от одного атома кислорода к другому.
ИК-спектроскопия. ИК-спектр Na{Cr[BP2O7(OH)3]} представлен на рис. 3. Полосы поглощения в области 1150–760 см–1 относятся к валентным колебаниям [PØ4] и [BO2(OH)2]3– тетраэдров [34–37], низкочастотные полосы 860, 766 см–1 могут также соответствовать неплоским деформационным колебаниям P–OH [34, 35]. Острые полосы средней интенсивности 666 и 611 см–1 принадлежат колебаниям мостиковых связей B–O–P [38]. Полосы поглощения в области 550–400 см–1 относятся к деформационным колебаниям тетраэдрических групп [PØ4] и [BO2(OH)2]3– и трансляционным колебаниям Cr3+ в октаэдрах [39].
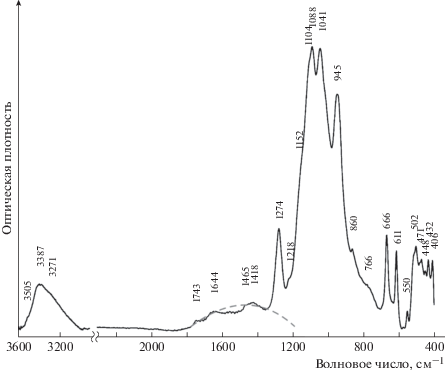
Наиболее интересной частью спектра являются полосы, отвечающие колебаниям гидроксильных групп. Обычно в неорганических соединениях валентные колебания групп OH– находятся в диапазоне 3800–3000 см–1, что соответствует слабым ВС. ИК-спектры соединений с очень сильными ВС (с расстоянием Д···А < 2.5 Å) содержат очень широкие (с шириной на полувысоте более 500 см–1), волнообразные полосы колебаний связи O–H в низкочастотном диапазоне (1600–500 см–1) [40]. Подобные спектры описаны для минералов пектолит-серандитовой группы: Na(Ca,Mn2+)2Si3O8(OH) [41, 42], шейхцерита Na(Mn,Mg,Zn)9[VSi9O28(OH)](OH)3 [43], санероита ${\text{NaMn}}_{5}^{{2 + }}$[Si5O14(OH)](VO3)(OH) [44] и других. Такая специфика спектральной картины объясняется сильным ангармонизмом колебаний и подтверждена теоретическими расчетами [41, 45–47]. Наилучшим способом наблюдения подобных спектров является наблюдение в поляризованном свете ориентированных пластинок [41, 48–50]. В порошковых ИК-спектрах происходит уменьшение интенсивности данных колебаний из-за различной ориентации частиц порошка [51]. Однако благодаря эффекту увеличения интенсивности колебаний OH–-групп с возрастанием силы связи [52] в некоторых случаях удается наблюдать полосы, характерные для сильных ВС, и в порошковых ИК-спектрах [53–56].
Эмпирические зависимости частоты валентного колебания O–H от расстояния Д···А описаны в [53, 57, 58] и представляют собой значительно изогнутые регрессивные кривые. Так, в [57] корреляционная функция представлена в виде
СРАВНИТЕЛЬНАЯ КРИСТАЛЛОХИМИЯ БОРОФОСФАТОВ
Изученное соединение дополняет ряд изоструктурных моноклинных (пр. гр. C2/c) борофосфатов состава A{M[BP2O7(OH)3]} (табл. 8), где A – преимущественно катионы щелочных (Na, K), реже щелочноземельных (Сa, Sr) элементов, а M – трехвалентные (Al, Ga, In) и переходные (Fe, Ni, Cr, V) элементы. Метрические характеристики, а также величины расстояний катион–анион в структуре Na{Cr[BP2O7(OH)3]} сопоставимы с соответствующими значениями для борофосфатов данного ряда. Исследованный Na–Cr-борофосфат близок к Na–V- и Na–Fe-аналогам.
Таблица 8.
Сравнительные характеристики борофосфатов c общей формулой A{M[BP2O7(OH)3]}
| Соединение | Пр. гр., Z | Параметры ячейки | V, Å3 | Литература | ||
|---|---|---|---|---|---|---|
| a, Å α, град |
b, Å β, град |
c, Å γ, град |
||||
| A = Na+ | ||||||
| NaAl[BP2O7(OH)3] | C2/c, | 10.497 | 7.993 | 9.077 | 677.00 | [59] |
| 4 | 90 | 117.26 | 90 | |||
| NaV[BP2O7(OH)3] | C2/c, | 10.415 | 8.236 | 9.196 | 705.63 | [20] |
| 4 | 90 | 116.55 | 90 | |||
| NaCr[BP2O7(OH)3] | C2/c, | 10.422 | 8.247 | 9.205 | 707.63 | Настоящая работа |
| 4 | 90 | 116.55 | 90 | |||
| NaFe[BP2O7(OH)3] | C2/c, | 10.420 | 8.215 | 9.217 | 705.47 | [60] |
| 4 | 90 | 116.60 | 90 | |||
| NaGa[BP2O7(OH)3] | C2/c, | 10.408 | 8.094 | 9.099 | 685.15 | [61] |
| 4 | 90 | 116.64 | 90 | |||
| NaIn[BP2O7(OH)3] | C2/c, | 10.368 | 8.520 | 9.415 | 747.82 | [62] |
| 4 | 90 | 115.95 | 90 | |||
| A = K+ | ||||||
| KGa[BP2O7(OH)3] | C2/c, | 10.863 | 8.162 | 9.305 | 737.80 | [63] |
| 4 | 90 | 116.59 | 90 | |||
| A = Ca2+ | ||||||
| CaFe[BP2O7(OH)3] | C2/c, | 10.233 | 8.239 | 9.159 | 687.61 | [64] |
| 4 | 90 | 117.07 | 90 | |||
| CaNi[BP2O7(OH)3] | C2/c, | 10.252 | 8.336 | 9.175 | 702.71 | [65] |
| 4 | 90 | 116.34 | 90 | |||
| A = Sr2+ | ||||||
| SrFe[BP2O8(OH)2] | C2/c, | 10.320 | 8.309 | 9.506 | 730.15 | [21] |
| 4 | 90 | 116.40 | 90 | |||
| P$\bar {1}$, | 6.670 | 6.693 | 9.389 | 364.74 | ||
| 2 | 109.83 | 102.07 | 103.15 | |||
Основные различия заключаются в особенностях ВС (рис. 4). Симметричные ВС в представителях семейства имеют схожую топологию. Однако связи с участием атомов водорода вершин (OH)4 BØ4-тетраэдров имеют различное направление: в структуре Na{Cr[BP2O7(OH)3]} – вдоль оси с, а в структурах остальных членов семейства – вдоль оси а.
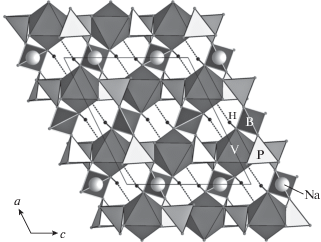
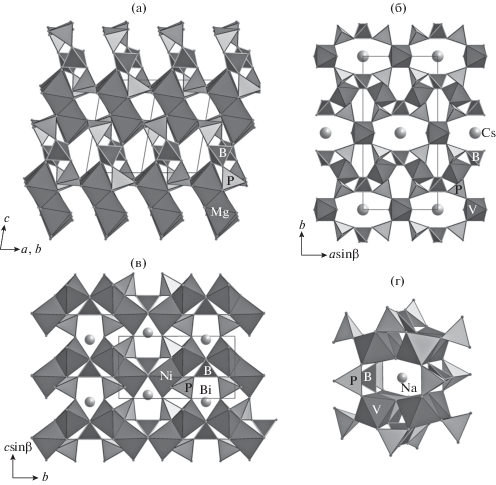
Олигомеры состава [BP2Ø10] являются основной строительной единицей для ряда борофосфатов [2] (табл. 9). Наиболее близка к описанному выше семейству кристаллическая структура Mg2[BP2O7(OH)3], в которой зигзагообразные колонки связанных ребрами MgØ6-октаэдров совместно с изолированными борофосфатными группами образуют гетерополиэдрический каркас со сквозными каналами (рис. 5а). Подобные каркасы характерны также для двух полиморфных модификаций LiCu2[BP2O8(OH)2]. В структуре Cs{(V3(H2O)2)[B2P4O16(OH)4]} в формировании плотного каркаса участвуют изолированные VØ6-октаэдры, связанные общими вершинами с тримерами [BP2O8(OH)2] (рис. 5б). Пустоты каркаса заполнены катионами Cs+. В структуре BiNi2[BP2O10] в гетерополиэдрический каркас объединяются цепочки из [BP2O10]-групп и связанных ребрами NiO6-октаэдров (рис. 5в). Основу структуры Na10K5{Na[Cr8O4(BP2O10)4(P(O,OH)4)4]} · 10H2O составляет изолированный гетерополиэдрический кластер (выделен фигурными скобками), образованный четырьмя тримерами [BP2O10], каждый из которых связан вершинами с двумя четверными кольцами CrO6-октаэдров. “Внешние” вершины октаэдров соединены PØ4-тетраэдрами. В центре кластера расположен атом Na (рис. 5г).
Таблица 9.
Сравнительные характеристики борофосфатов, cодержащих олигомеры [BP2Ø10] (Ø = O,OH)
| Соединение | Пр. гр., Z | Параметры ячейки | Литература | ||
|---|---|---|---|---|---|
| a, Å α, град |
b, Å β, град |
c, Å γ, град |
|||
| Mg2[BP2O7(OH)3] | P1 | 6.452 | 6.455 | 8.360 | [66] |
| 2 | 82.50 | 82.56 | 80.98 | ||
| LiCu2[BP2O8(OH)2] | C2/c | 15.097 | 4.762 | 9.659 | [67] |
| 4 | 90 | 91.02 | 90 | ||
| LiCu2[BP2O8(OH)2] | P212121 | 5.319 | 7.935 | 17.465 | [68] |
| 4 | 90 | 90 | |||
| Cs{(V3(H2O)2)[B2P4O16(OH)4]} | C2/m | 9.588 | 18.408 | 5.035 | [69] |
| 2 | 90 | 110.67 | 90 | ||
| Bi{Ni2BP2O10} | P21/m | 5.068 | 11.281 | 6.322 | [70] |
| 2 | 90 | 106.94 | 90 | ||
| Na10K5{Na[Cr8O4(BP2O10)4(P(O,OH)4)4]} · 10H2O | Pmnn | 10.463 | 15.049 | 18.721 | [13] |
| 2 | |||||
ВЫВОДЫ
Методом рентгеноструктурного анализа изучен борофосфат натрия и хрома Na{Cr[BP2O7(OH)3]} – новый представитель семейства A{M[BP2O7(OH)3]}. Анализ распределения электронной плотности позволил по-новому взглянуть на особенности водородных связей в структурах семейства. В структуре нового борофосфата натрия и хрома Na{Cr[BP2O7(OH)3]} установлены смещение атома водорода из центросимметричной позиции и образование альтернативных систем симметричных водородных связей, что также подтверждается специфическим характером ИК-спектра. Обнаруженный дополнительный пик электронной плотности в центре симметрии указывает на статистическое распределение протона H+, которое может быть результатом его частичного переноса от одного атома кислорода О(3) к соседнему О(3i). Это свидетельствует о том, что исследованный борофосфат натрия и хрома, как и другие представители семейства A{M[BP2O7(OH)3]}, может обладать протонной проводимостью.
Работа выполнена при финансовой поддержке Совета при Президенте РФ по грантам и финансовой поддержке молодых кандидатов наук (гранты № МК-8033.2016.5, МК-7926.2016.5).
Список литературы
Kniep R., Engelhardt H., Hauf C. // Chem. Mater. 1998. V. 10. P. 2930.
Ewald B., Huang Y.-X., Knip. R. // Z. Anorg. Allg. Chem. 2007. V. 663. P. 1517.
Гурбанова О.А., Белоконева Е.Л. // Кристаллография. 2007. Т. 52. № 4. С. 651.
Yakubovich O., Steele I., Massa W., Dimitrova O. // Z. Kristallogr. 2013. B. 228. S. 509.
Li M., Verena-Mudring A. // Cryst. Growth Des. 2016. V. 16. P. 2441.
Maspoch D., Ruiz-Molina D., Veciana J. // Chem. Soc. Rev. 2007. V. 36. P. 770.
Mosner P., Vesely D., Koudelka L. // Pigments Resin Technol. 2008. V. 37. P. 16.
Wang G., Valldor M., Lorbeer Ch., Mudring A.-V. // Eur. J. Inorg. Chem. 2012. V. 18. P. 3032.
Janson O., Chen S., Tsirlin A.A. et al. // Phys. Rev. B. 2013. V. 87. P. 064417.
Mi J.-X., Zhao J.-T., Mao S.-Y. et al. // Z. Kristallogr. New Cryst. Struct. 2000. B. 215. S. 201.
Zhang W., Cheng W., Zhang H. et al. // Inorg. Chem. 2010. V. 49. P. 2550.
Yang T., Sun J., Li G. et al. // Chem. Eur. J. 2008. V. 14. P. 7212.
Liu W., Zhang L., Su G. et al. // Dalton Trans. 2010. V. 39. P. 7262.
Oxford Diffraction. CrysAlisPro. Oxford Diffraction Ltd, Abingdon, Oxfordshire, UK (2009).
Palatinus L., Chapuis G. // J. Appl. Cryst. 2007. V. 40. P. 786.
Petřiček V., Dušek M., Palatinus L. // Z. Kristallogr. B. 2014. B. 229. № 5. S. 345.
Brown I.D., Altermatt D. // Acta Cryst. B. 1985. V. 41. P. 244.
Brown I.D. The Chemical Bond in Inorganic Chemistry. Oxford University Press, 2002. 230 p.
McCusker L.B., Liebau F., Engelhardt G. // Micropor. Mesopor. Mater. 2003. V. 58. P. 3.
Zhang L.-R., Zhang H., Borrmann H., Kniep R. // Z. Kristallogr. New Cryst. Struct. 2002. B. 217. S. 477.
Menezes P.W., Hoffmann S., Prots Y. et al. // Z. Anorg. Allg. Chem. 2009. B. 635. S. 1153.
Aksenov S.M., Chukanov N.V., Göttlicher J. et al. // Phys. Chem. Minerals. 2018. V. 45. P. 39.
Arnold D.W., Cangshan X., Neumark D.M. // J. Phys. Chem. 1995. V. 102. P. 6088.
Huang X., Braams B.J., Carter S., Bowman J.M. // J. Am. Chem. Soc. 2004. V. 126. P. 5042.
Kaledin M., Moffitt J.M., Clark C.R., Rivzi F. // J. Chem. Theory Comput. 2009. V. 5. P. 1328.
Burns P.C., Hawthorne F.C. // Can. Mineral. 1994. V. 32. P. 387.
Burns P.C., Hawthorne F.C. // Can. Mineral. 1994. V. 32. P. 885.
Maciček J. // Acta Cryst. C. 1994. V. 50. P. 1185.
Schwendtner K., Kolitsch U. // Acta Cryst. C. 2005. V. 61. P. i90.
Макарова И.П. // ФТТ. 2015. Т. 57. № 3. С. 432.
Макарова И.П., Гребенев В.В., Васильев И.И. и др. // Кристаллография. 2015. Т. 60. № 4. С. 552.
Mikheykin A.S., Chernyshov D.Yu., Makarova I.P. et al. // Solid State Ionics. 2017. V. 305. P. 30.
Choundhury R.R., Chitra R., Selezneva E.V., Makarova I.P. // Acta Cryst. B. 2017. V. 73. P. 863.
Čejka J., Sejkora J., Plášil J. et al. // J. Raman Spectr. 2011. V. 42. P. 1154.
Farmer V.C. The Infrared Spectra of Minerals, Monograph 4, 1st ed., Mineralogical Society, London, 1974.
Frost R.L., Xi Y., Scholz R. et al. // Vibr. Spectr. 2013. V. 66. P. 69.
Sejkora J., Novotn’y P., Novak M. et al. // Can. Mineral. 2005. V. 43. P. 1393.
Baykal A., Kizilyalli M., Gözel G., Kniep R. // Cryst. Res. Technol. 2000. V. 35. P. 247.
Borovikova E.Yu., Boldyrev K.N., Aksenov S. M. et al. // Opt. Mater. 2015. V. 49. P. 304.
Hadzi D. // Pure Appl. Chem. 1965. V. 11. P. 435.
Hammer V.M.F., Libowitzky E., Rossman G.R. // Am. Mineral. 1998. V. 83. P. 569.
Jacobsen S.D., Smyth J.R., Swope R.J., Sheldon R.I. // Am. Mineral. 2015. V. 85. P. 745.
Brugger J., Krivovichev S., Meisser N. et al. // Am. Mineral. 2006. V. 91. P. 937.
Camara F., Bittarello E., Ciriotti M. et al. // Mineral. Mag. 2015. V. 79. P. 171.
Hadzi D., Bratos S. // The Hydrogen Bond—Recent Developments in Theory and Experiments / Eds. Schuster P. et al. Amsterdam: North-Holland Publishing Company, 1976. P. 565.
Kreevoy M.M., Marimanikkuppam S., Young V.G. et al. // Ber: Bunsenges. Phys. Chem. 1998. V. 102. P. 370.
Nyfeler D., Hoffmann C., Armbruster T. et al. // Am. Mineral. 1997. V. 82. P. 841.
Beran A., Libowitzky E. Giester G. // Mineral. Petrol. 1997. V. 61. P. 223.
Đorđević T., Karanović L. // J. Solid State Chem. 2008. V. 181. P. 2889.
Krickl R., Wildner M. // Eur. J. Mineral. 2009. V. 21. P. 65.
Libowitzky E., Rossman G.R. // Phys. Chem. Minerals. 1996. V. 23. P. 319.
Athokpam B., Ramesh S.G., McKenzie R.H. // Chem. Phys. 2017. V. 488–489. P. 43.
Novak A. // Struct. Bond. 1974. V. 18. P. 177.
Daniels P., Krosse S., Werding G., Schreyer W. // Contrib. Mineral. Petrol. 1997. V. 128. P. 261.
Brugger J., Krivovichev S., Meisser N. et al. // Am. Mineral. 2006. V. 91. P. 937.
Iwamoto K., Mochizuki D., Osada S. et al. // J. Chem. Phys. 2015. V. 143. P. 024503.
Libowitzky E. // Monatshe. Chem. 1999. V. 130. P. 1047.
Mikenda W. // J. Mol. Struct. 1986. V. 147. P. 1.
Koch D., Kniep R. // Z. Kristallogr. New Cryst. Struct. 1999. B. 214. S. 441.
Boy I., Cordier G., Eisenmann B., Kniep R. // Z. Naturforsch. 1998. B. 53b. S. 165.
Huang Y.-X., Mao S.-Y., Mi Jinxiao et al. // Z. Kristallogr. New Cryst. Struct. 2001. B. 216. S. 15.
Huang Y.-X., Mi J., Mao S.-Y. et al. // Z. Kristallogr. New Cryst. Struct. 2002. V. 217. P. 7.
Mi J., Huang Y.-X., Mao S.-Y. et al. // Z. Kristallogr. New Cryst. Struct. 2002. B. 217. S. 167.
Menezes P.W., Hoffmann S., Prots Yu., Kniep R. // Z. Kristallogr. New Cryst. Struct. 2008. B. 223. S. 335.
Menezes P.W., Hoffmann S., Prots Yu., Kniep R. // Z. Kristallogr. New Cryst. Struct. 2006. B. 221. S. 429.
Hauf C., Boy I., Kniep R. // Z. Kristallogr. New Cryst. Struct. 1999. B. 214. S. 3.
Zheng J., Zhang A. // Acta Cryst. E. 2009. V. 65. P. i40.
Yang M., Li X., Yu J. et al. // Dalton Trans. 2013. V. 42. P. 6298.
Engelhardt H., Borrmann H., Schnelle W., Kniep R. // Z. Anorg. Allg. Chem. 2000. B. 626. S. 1647.
Zhang W.-L., He Z.-Z., Xia T.-L. et al. // Inorg. Chem. 2012. V. 51. P. 8842.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография