

Кристаллография, 2019, T. 64, № 3, стр. 477-483
Кристаллизации фосфатов кальция из прототипа плазмы крови в присутствии неорганических и органических примесей
1 Омский государственный университет
Омск, Россия
* E-mail: golovanoa2000@mail.ru
Поступила в редакцию 24.08.2017
После доработки 17.11.2017
Принята к публикации 07.12.2017
Аннотация
Представлены результаты исследований процессов кристаллизации в растворах, моделирующих состав плазмы крови человека. Установлено, что полученные твердые фазы состоят из OH-дефицитного водосодержащего карбонатапатита. Исследовано влияние примесей (ионов магния и глутаминовой кислоты) на процесс кристаллизации фосфатов кальция. Выявлено, что присутствие добавок в модельном растворе влияет на фазовый состав образцов. Изучена растворимость синтетических образцов в растворах разной природы и в препарате Верапамил. Установлены кинетические характеристики данного процесса, показана зависимость скорости растворения от условий лабораторных экспериментов.
ВВЕДЕНИЕ
За последние годы увеличился процент патогенного минералообразования в кровеносных сосудах, сердечных клапанах, стентах и грудных имплантатах [1–9]. Это связано с рядом факторов экзогенного и эндогенного характера. Плазма крови предcтавляет собой воду, содержащую главным образом растворенные соли и белки. Химический состав растворимых в плазме крови веществ относительно постоянен, растворимые вещества плазмы cоставляют около 10% массы крови, из них на долю белков приходится около 7%, на долю неорганических солей – 0.9%, остальную часть образуют небелковые органические соединения [10, 11]. В плазме имеются белки, обеспечивающие иммунитет (иммуноглобулины), и белки, участвующие в свертывании крови. Неорганические компоненты плазмы представлены макро- и микроэлементами, неорганический состав плазмы крови достаточно хорошо изучен (табл. 1). В крови постоянно содержится некоторое количество свободных аминокислот (АК). Почти пятую часть содержащихся в плазме АК составляют аланин и глутаминовая кислота (ГК). На сегодняшний день имеются доcтаточно обширные данные о составе плазмы крови, что позволяет проводить моделирование процессов, протекающих в исследуемой среде [12, 13].
Таблица 1.
Минеральный состав плазмы крови человека [10]
| Ион | Среднее значение концентрации (ммоль/л) |
|---|---|
| Ca2+ | 2.35 |
| Na+ | 143 |
| K+ | 4.35 |
| Mg2+ | 0.95 |
| ${\text{NH}}_{4}^{ + }$ | 0.04 |
| Cl– | 103 |
| ${\text{CO}}_{3}^{{2 - }}$ | 0.45 |
| ${\text{CO}}_{3}^{{2 - }}$ | 26 |
| ${\text{Р О }}_{4}^{{3 - }}$ | 1.3 |
По результатам исследования поражения коронарных артерий авторами [14] выделены три независимые фазы атеросклеротического поражения коронарных артерий, сопряженные с кальцификацией атеросклеротической бляшки: фиброзные участки бляшки с незначительным содержанием липидов; заключительная стадия формирования некротических очагов; кровоизлияние и тромбообразование. Кальцификаты указанных зон атеросклеротической бляшки имеют собственные характерные морфологические и композиционные особенности. Единственной кристаллизованной фазой кальцификатов является гидроксиапатит, а начальной стадией петрификации являются скопления микро- и наночастиц гидроксиапатита, при этом механизмы их формирования в толще атеросклеротической бляшки и на поверхности при тромбогенезе различны. Не исключено, что минерализация участков, контактирующих с кровью, является пассивным физико-химическим процессом. Соотношение Са/Р в кальцификатах по данным [14–19] варьирует в значительных пределах. В матричных везикулах это соотношение может быть около 0.66, а в зрелом апатите доходит до 2. Размер кристаллов в процессе созревания биоапатита увеличивается от наноразмерных до 100 и более микрон.
Патогенный апатит не имеет такой тесной связи с обменными процессами в организме, как физиогенный. Таким образом, кристаллохимия природного неорганического апатита невероятно сложна, а структура биоапатита имеет ряд дополнительных особенностей, при этом минерализация тканей изучена недостаточно.
Именно из-за кальцификации сроки функционирования трансплантатов сердечных клапанов ограничены. В связи с этим возникла необходимость в исследовании механизма инициации кальциноза как коллагеновых и мышечных тканей, так и трансплантатов клапанов сердца с целью разработки способов его предотвращения.
Цель работы – изучение кристаллизации фосфатов кальция из прототипов плазмы крови человека в присутствии неорганических и органических добавок, исследование их свойств.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Выбор исходных реагентов и их соотношение в растворе определялись таким образом, чтобы концентрации ионов и ионная сила раствора были максимально приближены к параметрам моделируемой системы плазмы крови человека (табл. 1).
В качестве исходных реагентов использовали соли марки ч.д.а. и х.ч. и дистиллированную воду. Для каждой серии экспериментов были приготовлены растворы, содержащие катионы и анионы, при совместном присутствии которых в данных условиях не образуются малорастворимые соединения. В каждом растворе проводили корректировку рН до физиологического значения (7.4 ± 0.01) путем добавления 30%-ного раствора NaOH или HCl (конц.). После смешения эквивалентных объемов получали раствор с заданным пересыщением и рассчитанной концентрацией компонентов. Кристаллизация твердой фазы осуществлялась в течение разных промежутков времени (от 2 до 12 нед) при варьировании исходного пересыщения (S = 5, 10, 25, 50, 100). Все образцы во время синтеза находились в шкафу БИАТРОН при температуре 36.6°С, соответствующей организму человека.
После вызревания осадка под маточным раствором через определенные временные промежутки твердую фазу отделяли от раствора фильтрованием, высушивали при температуре ~80°С до постоянной массы и полного удаления химически несвязанной воды, взвешивали и исследовали с применением группы физико-химических методов.
Для исследования влияния органических и неорганических добавок в модельную систему вводили выбранные с учетом биохимии плазмы крови человека и физиологической нормы (Сфиз) следующие добавки: ГК и ионы магния, а также с концентрацией выше физиологической нормы: ионы магния (2 и 4 раза), ГК (3 и 5 раз). Выбранные концентрации добавок представлены в табл. 2.
Синтетические твердые фазы исследовали с применением комплекса физико-химических методов. Рентгенофазовый анализ (РФА) осадков проводили методом порошковой дифрактометрии (D8 Advance, Bruker; программа TOPAS 3.0 (Bruker)). Количественный фазовый анализ поликомпонентных образцов, расчет размеров кристаллитов (областей когерентного рассеяния (ОКР)) и идентификацию фаз выполняли с применением базы данных ICDDPDF для порошковой дифракции. Съемку ИК-спектров осуществляли с помощью предварительного таблетирования исходных твердых фаз с KBr (ИК-фурье-спектрометр IRPrestige21, “Shimadzu”). Газообменный потенциометрический метод использовали для определения общего содержания углерода в синтезированных биомиметических аналогах апатитов кардиолитов. Метод основан на прокаливании навески в токе кислорода при температуре 1250–1300°С и последующем поглощении образующегося углекислого газа раствором гидрата окиси калия на газоанализаторе.
В жидкости, отделенной от осадка, определяли остаточное содержание кальция [РД 52.24.40394] и фосфатов [ГОСТ 1830972].
Кинетику растворения полученных твердых фаз изучали в 0.9%-ном растворе NaCl, трис-буфере и препарате Верапамил (представитель группы антагонистов кальция, Ирбитский ХФЗ, Россия). Измерения проводили в термостатируемой ячейке при 37°С в течение 2 ч при постоянном объеме жидкой фазы и перемешивании. В ходе эксперимента контролировали рН и рСа. На основе экспериментальных данных получены кинетические кривые и проведена их математическая обработка по алгоритму, описанному в [20].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Результаты исследования твердых фаз без добавок. Результаты РФА показали, что все образцы, полученные в среде модельного раствора плазмы крови человека при варьировании пересыщений и времени синтеза, представляют собой плохо окристаллизованный апатит (рис. 1). Установлены значения параметра a полученного апатита (9.41–9.48 Å; табл. 3), они близки к значению схожего по параметрам стехиометрического апатита (а = 9.418, с = 8.884 Å, JCPDS N 9-432) или больше него. Из таблицы видно, что полученные значения параметра с (6.874–6.894 Å, табл. 3) могут быть как ниже, так и выше значения, характерного для стехиометрического апатита. Размер ОКР вдоль [001] составляет 18–28 нм. Установленные размеры кристаллитов и значения параметров элементарной ячейки характерны для нестехиометрических водосодержащих биологических карбонатапатитов [21].
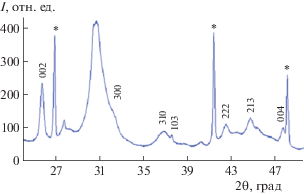
Таблица 3.
Концентрация карбонат-ионов, параметры элементарной ячейки и размеры ОКР синтезированных апатитов
| Образец | CO2/${\text{CO}}_{3}^{{2 - }}$, мас. % | а, Å | с, Å | Размеры ОКР вдоль [001], нм | |
|---|---|---|---|---|---|
| Пересыщение | Время, нед. | ||||
| 25 | 8 | 3.3/4.50 | 9.45 | 6.890 | 18 |
| 10 | 3.19/4.35 | 9.41 | 6.889 | 21 | |
| 12 | 3.18/4.34 | 9.44 | 6.894 | 21 | |
| 50 | 2 | 1.49/2.03 | 9.42 | 6.878 | 22 |
| 4 | 1.78/2.43 | 9.44 | 6.884 | 24 | |
| 8 | 3.55/4.84 | 9.46 | 6.886 | 20 | |
| 10 | 4.06/5.54 | 9.43 | 6.889 | 20 | |
| 12 | 2.98/4.06 | 9.46 | 6.888 | 23 | |
| 100 | 2 | 1.26/1.72 | 9.47 | 6.868 | 27 |
| 4 | 1.46/1.99 | 9.48 | 6.874 | 28 | |
| 8 | 1.95/2.66 | 9.46 | 6.881 | 24 | |
| 10 | 2.61/3.56 | 9.45 | 6.886 | 24 | |
| 12 | 2.02/2.75 | 9.48 | 6.882 | 27 | |
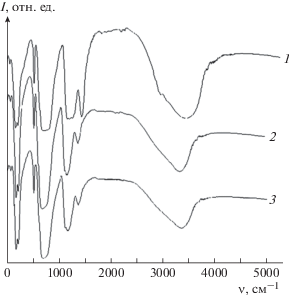
ИК-спектры полученных образцов (рис. 2) содержат полосы поглощения, относящиеся к фосфатным группам (1105, 1070, 1040, 975, 610, 570 и 480 см–1).
Рис. 2.
ИК-спектры синтезированных образцов (пересыщение – 50, время эксперимента: 1 – 2, 2 – 4, 3 – 10, 4 – 12 нед.).
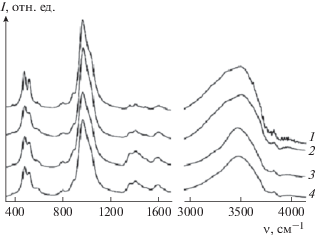
Все полученные спектры содержат интенсивные и одновременно широкие полосы поглощения, относящиеся к воде (~1600 см–1), деформационные колебания (3300–3500 см–1), валентные колебания и слабые полосы гидрофосфат-иона (875 cм–1). Все спектры также содержат полосы, относящиеся к карбонат-иону В-типа (1420 и 1460 см–1). Видно, что полосы гидроксил-иона (640 и 3570 см–1) на всех спектрах отсутствуют. Следовательно, результаты ИК-спектроскопии подтверждают близость синтезированных апатитов к биологическим OH-дефицитным водосодержащим карбонатапатитам [21].
Результаты экспериментов позволяют сделать вывод и о влиянии экспериментальных условий (пересыщение, время эксперимента) на содержание карбонат-иона в апатите (рис. 1, табл. 3). Из анализа следует, что при пересыщении 50 и 100 увеличение времени эксперимента с 2 нед. до 10 ведет к увеличению содержания карбонат-ионов с 2.03–1.72 до 5.54–3.56 мас. % соответственно. А увеличение времени эксперимента до 12 нед. ведет к уменьшению содержания карбонат-ионов от 4.35, 5.54 и 3.56 до 4.34, 4.06 и 2.75 мас. % соответственно. При времени эксперимента 10 нед. видим, что максимальную концентрацию карбонат-ионов содержат апатиты, синтезированные при пересыщении, равном 50. Следовательно, максимальное количество карбонат-ионов (5.54 мас. %) соответствует апатиту, синтезированному при пересыщении 50 в течение 10 нед.
Изучение растворения проведено для моделирования пассивной резорбции твердых фаз в организме человека. Для этого были выбраны пробы с пересыщением 5, 10, 50 и временем синтеза от 4 до 8 нед.
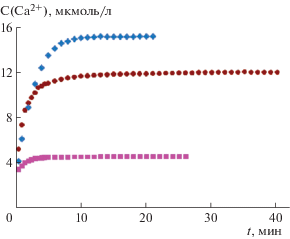
В результате получены кинетические кривые С(Ca2+) = f(τ) (рис. 3, в качестве примера для 0.9%-ного раствора NaCl), из которых видно, что основное насыщение раствора ионами кальция наблюдается на десятой минуте и зависит от начального пересыщения модельного раствора плазмы крови.
Рис. 3.
Кинетическая кривая растворения С(Ca2+) = = f(τ) в 0.9%-ном растворе NaCl образца со временем синтеза 8 нед. и пересыщением:  – 5,
– 5,  – 10, $\blacksquare $ – 50.
– 10, $\blacksquare $ – 50.
Математическая обработка полученных кинетических зависимостей по алгоритму, описанному в [20], позволила определить скорость растворения твердых фаз (табл. 4). Сравнение действия трех растворителей на стадию резорбции твердых фаз выявило, что наибольшая скорость растворения характерна для препарата Верапамил.
Таблица 4.
Скорость растворения образцов в разных растворителях
| Образец | 0.9%-ный раствор NaCl | Трис-буфер | Вера-памил | |
|---|---|---|---|---|
| Время синтеза, недели | Пересы-щение | $\text{v}$, ммоль/(л мин) | ||
| 4 | 5 | 0.264 | 0.184 | 0.161 |
| 10 | 0.345 | 0.152 | 0.454 | |
| 50 | 0.227 | 0.144 | 0.477 | |
| 8 | 5 | 0.144 | 0.178 | 0.376 |
| 10 | 0.285 | 0.142 | 0.265 | |
| 50 | 0.151 | 0.114 | 0.432 | |
Результаты исследования твердых фаз, полученных с участием ГК. По результатам РФА твердых фаз, полученных в присутствии ГК, выявлено, что фазовый состав по сравнению с беспримесными фазами изменяется (рис. 4), кроме карбонатгидроксилапатита (КГА) на дифрактограммах присутствуют пики, характерные для фазы витлокита (пики по шкале 2θ: 17.4; 30.2; 33.7). Но для образцов, полученных с максимальной концентрацией ГК, фаза витлокита отсутствует и идентифицируется только фаза КГА. Для уточнения состава и структуры образцов, полученных в присутствии ГК, использован метод ИК-спектроскопии (рис. 5). В ИК-спектре всех исследованных образцов в области 450–700 см–1 проявляется деформационное колебание О–Р–О в ${\text{Р О }}_{4}^{{3 - }}$, в области 900–1200 см–1 – валентное колебание Р–О в ${\text{Р О }}_{4}^{{3 - }}$. Области 1500–1650 и 2700–2250 см–1 определяются деформационными колебаниями аминогруппы, что подтверждает адсорбцию АК на поверхности твердой фазы.
Рис. 4.
Дифрактограмма образца с S = 5, временем синтеза 8 нед. и добавкой ГК (Сфиз): * – пик гидроксилапатита, ** – пик витлокита.
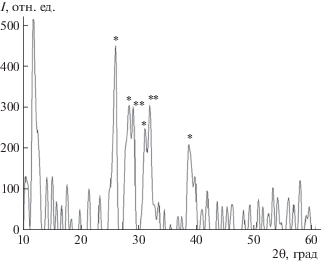
Рис. 5.
ИК-спектр образцов синтезированных в присутствии ГК при варьировании концентрации: 1 – Cфиз, 2 – Cфиз3, 3 – Cфиз5.
В ИК-спектрах образцов зарегистрированы полосы поглощения в спектральных областях 1416–1547 и 850–900 см–1, относящиеся соответственно к валентным и деформационным колебаниям связей С–О карбонат-ионов. Видно, что характерная для гидроксилапатита интенсивность полос колебания Н2О в области 3400–3550 см–1 очень хорошо выражена, но с увеличением концентрации ГК происходит изменение интенсивности полосы, это связано с уменьшением количества ионов OH– в структуре за счет их замещения на ${\text{CO}}_{3}^{{2 - }}$-группу. Так, образец, полученный при физиологической концентрации ГК, является карбонатгидроксилапатитом В-типа, а образцы, синтезированные при большей концентрации добавки ((ГК) Cфиз3 и Cфиз5), относятся к карбонатгидроксилапатиту А-типа.
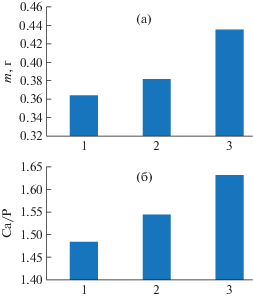
Для всех образцов определены масса осадка и соотношение Са/Р (рис. 6). С увеличением концентрации добавки масса осадка и Са/Р-коэффициент возрастают, что хорошо согласуется с результатами РФА и ИК-спектроскопии.
Рис. 6.
Зависимость массы образцов (а) и соотношение Са/Р (б) от концентрации ГК в исходном растворе при S = 5: 1 – Cфиз, 2 – Cфиз3, 3 – Cфиз5.
Для твердых фаз получены кинетические кривые растворения и проведена их обработка (табл. 5).
Таблица 5.
Скорость растворения образцов, полученных в присутствии глутаминовой кислоты (S = 5, время синтеза 8 нед.)
| Образец с добавкой ГК | 0.9%-ный раствор NaCl | Трис-буфер |
|---|---|---|
| $\text{v}$, ммоль/(л мин) | ||
| Сфиз | 0.603 | 0.749 |
| Сфиз3 | 0.306 | 0.477 |
| Сфиз5 | 0.390 | 0.479 |
Анализ кинетических параметров показал, что увеличение концентрации добавки ГК ведет к снижению скорости растворения твердой фазы. Это связано с изменением фазового состава образцов, так как рост концентрация добавки ГК в исходном модельном растворе плазмы крови приводит к снижению содержания фазы витлокита и увеличению фазы КГА в составе твердой фазы, известно, что витлокит является более растворимым соединением, чем КГА.
Результаты исследования твердых фаз, полученных с участием ионов магния. Для изучения влияния неорганических компонентов на процессы кристаллизации выбран магний-ион. Он представляет особый интерес, так как является ингибитором кристаллизации в плазме крови человека [12, 13]. Чтобы установить кристаллизацию в модельном растворе плазмы крови человека, варьировали концентрацию магний-ионов и время синтеза.
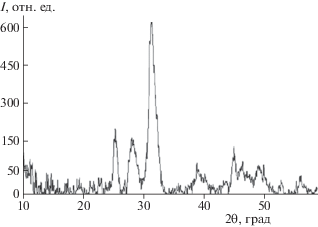
В результате анализа рентгенограмм установили, что фазовый состав образцов включает в себя КГА и витлокит. Полуколичественный анализ показал преобладание витлокита (рис. 7).
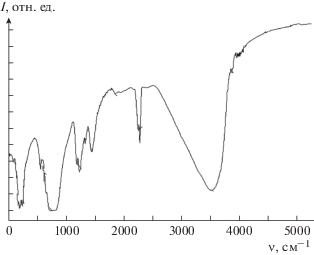
Интерпретация полученных ИК-спектров показала, что во всех исследованных образцах (рис. 8) в области 564–566 см–1 проявляются валентные ассиметричные колебания связей Р–О–Р иона ${\text{Р О }}_{4}^{{3 - }}$; в области 500–650 см–1 – деформационные асимметричные колебания связей Р–О в ионе ${\text{Р О }}_{4}^{{3 - }}$; в области 1000–1100 см–1 – валентные симметричные колебания связей Р–О иона ${\text{Р О }}_{4}^{{3 - }}$. В спектрах образцов регистрируются полосы поглощения в спектральных областях 1400–1500 и 850–900 см–1, относящихся соответственно к валентным и деформационным колебаниям связей С–О карбонат-ионов. Отметим, что характерная для гидроксилапатита интенсивность полос колебания Н2О в области 3400–3500 см–1 очень хорошо выражена. Следовательно, осадок содержит карбонатгидроксилапатит В-типа.
Таким образом, в присутствии ионов магния из модельного раствора плазмы крови синтезированы образцы, состоящие из смеси фаз витлокита и карбонатгидроксилапатита В-типа.
Было исследовано растворение в препарате Верапамил образцов с пересыщением 50 и временем синтеза 8, 10 нед. и с концентрацией ионов магния, увеличенной в 2 и 4 раза от физиологической нормы. При обработке кинетических кривых установлено, что скорость растворения зависит как от времени синтеза, так и от концентрации добавки в исходном растворе плазмы крови (табл. 6).
Таблица 6.
Кинетические параметры растворения образцов с добавкой ионов магния (пересыщение 50) в препарате Верапамил
| Образец | Кинетическое уравнение | $\text{v}$, ммоль/(л мин) | |
|---|---|---|---|
| Время синтеза, нед. | Концен-трация добавки | ||
| 8 | Сфиз2 | С = 81.28 – 0.282х | 0.282 |
| 10 | Сфиз2 | С = 80.07 – 0.326х | 0.326 |
| 8 | Сфиз4 | С = 93.42 – 0.339х | 0.339 |
Видно, что растворение в препарате Верапамил повышается с увеличением добавки ионов магния и времени синтеза. Это связано с изменением фазового состава, а именно с увеличением в составе образца более растворимой и метастабильной фазы витлокита, что находится в согласии с результатами РФА и ИК-спектроскопии.
Сравнение резорбции беспримесных образцов с твердыми фазами, полученными в присутствии ионов магния, показало, что скорость их растворения в препарате Верапамил выше. Поскольку данный препарат относится к группе лекарств антагонистов кальция, его действие направленно на резорбцию фосфатов кальция, а не магния, а витлокит, обнаруженный в составе твердых фаз, содержит порядка 1.94% магния.
ВЫВОДЫ
В ходе синтеза из модельного раствора плазмы крови человека получен нестехиометрический водосодержащий карбонатгидроксилапатит, совпадающий по составу с кальцификатом человека.
Выявлено, что пересыщение 50 и 100 и увеличение времени эксперимента с 2 нед. до 10 ведут к увеличению содержания карбонат-ионов, их максимальное количество (5.54 мас. %) соответствует апатиту, синтезированному при пересыщении 50 в течение 10 нед.
Установлено, что влияние препарата Верапамил на резорбцию беспримесных образцов в 2 раза выше, чем в 0.9%-ном растворе NaCl и трис-буфере.
Изучено влияние добавок на процессы образования фосфатов кальция в модельном растворе плазмы крови и выявлено, что присутствие глутаминовой кислоты способствует образованию карбонатгидроксилапатитов В и А типа в зависимости от ее концентрации, ее увеличение способствует снижению резорбции образцов. Образцы с добавкой ионов магния состоят из КГА и витлокита, увеличение концентрации добавки способствует увеличению скорости растворения в препарате Верапамил, но при этом для образцов с добавкой магния отмечено снижение скорости растворения по сравнению с беспримесными образцами.
Работа выполнена при частичной финансовой поддержке Российского фонда фундаментальных исследований (грант № 15-29-04839 офи_м).
Список литературы
Golovanova O.A., Frank-Kamenetskaya O.V., Punin Y.O. // Russian J. General Chem. 2011. V. 81. P. 1392.
Tetsuo Kodaka, Ryoichi Mori, Akihiko Hirayama et al. // Clin. Electron Microscopy Society. 2003. V. 36. P. 272.
Becker A., Epple M., Mueller K.M. // Inorg. Biochem. 2004. V. 98. P. 2032.
Kazuyuki Yahagi, Frank D. Kolodgie, Fumiyuki Otsuka // Nat. Rev. Cardiol. 2015. V. 10. P. 1038.
Титов А.Т., Ларионов П.М., Щукин В.С. и др. // Поверхность. рентген., синхротр. и нейтр. исслед. 2001. № 3. С. 74.
Власов Д.Ю., Зеленская М.С., Баринова К.В. и др. Биоминералогия. Луцьк. Украина. 2008. 26 с.
Indulekha Pillai C.L., Shen Li, Milagros R. et al. // Cell. Stem. Cell. 2017. V. 20(2). P. 218.
Титов А.Т., Ларионов П.М., Зайковский В.И. // Поверхность. рентген., синхротр. и нейтр. исслед. 2000. № 7. С. 66.
Ламанова Л.М. // Вестн. Томск. гос. ун-та. 2010. № 337. С. 194.
Марри Р. Биохимия человека. М.: Миp: БИНОМ, 2009. Т. 2. 414 с.
Березов Т.Т., Коровин М.А. Биологическая химия. М.: Медицина, 2002. 704 с.
Голованова О.А., Солодянкина А.А. // Кристаллография. 2017. Т. 62. № 2. С. 338.
Голованова О.А., Солодянкина А.А. // Кристаллография. 2017. Т. 62. № 3. С. 505.
Gibson I.R., Bonfield W., Sayer M. // J. Biomed. 2002. V. 13. P. 697.
Яхно Т.А., Яхно В.Г., Соколов А.В. // Биофизика. 2005. Т. 50. № 4. С. 726.
Lars-Fride Olsson, Karin Sandin, Rolf Odselius et al. // Inorg. Chem. 2007. V. 26. P. 4123.
Solodyankina A., Nikolaev A., Frank-Kamenetskaya O., Golovanova O. // J. Mol. Struct. 2016. V. 1119. № 5. P. 484. org/. doi 10.1016/j.molstruc.2016.04.080
Grazielle V. Nogueira, Landulfo Silveira, Jr. Airton A. et al. // Biomed. Opt. 2005. V. 3. P. 10.
Pigozzi F. // Sports Med. Phys. Fitness. 2011. V. 51. P. 260.
Голованова О.А., Цыганова А.А., Чиканова Е.С. // Физика и химия стекла. 2016. Т. 42. № 6. С. 798.
Frank-Kamenetskaya O., Kol’tsov A. // Mol. Struct. 2011. V. 9. P. 9.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография