

Кристаллография, 2019, T. 64, № 3, стр. 494-502
Применение алгоритма DBSCAN для выявления гидрофобных кластеров в структурах белков
А. А. Лашков 1, *, С. В. Рубинский 1, П. А. Эйстрих-Геллер 1
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
* E-mail: alashkov83@gmail.com
Поступила в редакцию 13.08.2018
После доработки 28.09.2018
Принята к публикации 08.10.2018
Аннотация
Гидрофобные ядра и гидрофобные кластеры играют важную роль в фолдинге глобулярных белковых макромолекул, являясь каркасом для функционально важных остатков белков-ферментов. Разработана программа, предназначенная для кластеризации в структурах белков аминокислотных остатков, учитывающая их гидрофобность и использующая алгоритм DBSCAN. Дано описание программы, обозначены ее основные возможности и области применения.
ВВЕДЕНИЕ
Гидрофобные ядра и гидрофобные кластеры играют важную роль в фолдинге белковых глобулярных макромолекул, являясь каркасом для функционально важных аминокислотных остатков (а.о.) белков-ферментов [1, 2]. В случаях лигандов амфифильной природы (например, липидов, полипептидов) сами гидрофобные кластеры включаются в функционально значимые области молекул [3], и взаимодействие с ними необходимо учитывать, например, при поиске и ранжировании решений молекулярного докинга и рациональном дизайне молекул биологически активных соединений [4, 5]. В состав этих кластеров входят главным образом а.о. с неполярными боковыми цепями. В [6, 7] показано, что локализацию и схожесть гидрофобных областей можно использовать для классификации похожих по функции и происхождению белков. В [8] объясняется, как, используя данные о структуре гидрофобных областей, осуществить дизайн фермента с заданными свойствами. Следовательно, расположение и состав гидрофобных кластеров в молекулах белков важны с практической точки зрения.
При поиске гидрофобных ядер главной задачей является кластеризация гидрофобных элементов (отдельных а.о. или групп атомов). Для этого существует специализированное программное обеспечение. Веб-сервис Clud [9] применяет для кластерного анализа алгоритм построения графа взаимодействия гидрофобных групп. Программа использует в качестве объектов кластеризации атомы, которые могут принадлежать в среднем гидрофильным а.о. (Arg, Glu и т.д.). В программе не автоматизирован подбор параметров (порога расстояния между группами и минимального размера кластера) и жестко задано минимальное расстояние между любыми типами гидрофобных атомов (2.7 Å). Кроме того, отсутствует какая-либо оценка качества кластеризации. Веб-сервис Qgrid [10] осуществляет иерархическую кластеризацию заряженных и гидрофобных а.о. Недостатком предложенного подхода является отсутствие в программе весовых коэффициентов, определяющих разницу гидрофобных свойств а.о.
Авторами разработана программа кластеризации положения гидрофобных остатков в структурах белков, использующая алгоритм DBSCAN (Density-Based Spatial Clustering of Applications with Noise – плотностный алгоритм пространственной кластеризации с присутствием шума) [11].
МЕТОДЫ ИССЛЕДОВАНИЯ
DBSCAN относится к детерминированным алгоритмам четкой кластеризации [11, 12]. Его основополагающими идеями являются нахождение плотности распределения точек в пространстве и кластеризация точек на основании схожей плотности точек в кластерах. В отличие от кластеризации k-средних в алгоритме DBSCAN могут быть учтены кластеры произвольной формы, что является значительным преимуществом в рамках решаемой задачи определения гидрофобных кластеров и ядер в белковых молекулах.
В качестве параметров кластеризации (так называемых гиперпараметров) алгоритм принимает радиус ε-окрестности (eps) и минимальное количество соседей (“min_samples”). В описываемой программе eps определяется как максимальное расстояние (в Å) между центрами масс гидрофобных а.о., при котором они являются соседними в одном кластере. Отношение min_samples/eps3 пропорционально пороговой плотности распределения точек – центров масс гидрофобных а.о. Алгоритмическая сложность DBSCAN в О-нотации [13] – O(Nlog(N)).
В рассматриваемой программе на вход алгоритма DBSCAN, реализованного посредством библиотеки scikit-learn [14], подаются не координаты центров масс гидрофобных а.о., а матрица попарных расстояний между ними. Это сделано для увеличения скорости последующего автоподбора гиперпараметров.
В качестве весовых коэффициентов в программе могут быть использованы различные шкалы гидрофобности а.о. [15, 16] (табл. 1). Кроме того, для кластеризации электрически заряженных а.о. реализована функция расчета весовых коэффициентов как модулей частичных зарядов боковых групп, отдельно для положительно и отрицательно заряженных а.о. Модули частичных зарядов рассчитывали по формулам, которые выводятся из уравнения Гендерсона–Гассельбаха [17]:
Таблица 1.
Относительная гидрофобность аминокислотных остатков и данные для расчета
| Тип а.о. | Индекс гидропатии | Весовой коэффициент (шкала гидрофобности “Hydropathy”) | ${\Lambda \mu }_{{{\text{int}}}}^{{{\text{ex}}}}$, кДж | Весовой коэффициент (шкала гидрофобности “Nanodroplet”) |
|---|---|---|---|---|
| Ile | 4.5 | 2.500 | –9.73 | 1.289 |
| Ala | 1.8 | 1.000 | –9.58 | 1.269 |
| Phe | 2.8 | 1.556 | –9.23 | 1.223 |
| Leu | 3.8 | 2.111 | –8.66 | 1.147 |
| Trp | – | – | –8.62 | 1.142 |
| Val | 4.2 | 2.333 | –8.26 | 1.094 |
| Met | 1.9 | 1.056 | –7.65 | 1.013 |
| Pro | – | – | –7.55 | 1.000 |
| Cys | 2.5 | 1.398 | –5.63 | 0.746 |
| Gly | – | – | –4.57 | 0.605 |
| Thr | – | – | –4.06 | 0.538 |
| Ser | – | – | –3.56 | 0.472 |
На вход алгоритма подаются весовые коэффициенты, рассчитываемые для каждого гидрофобного а.о. путем нормировки шкал гидрофобности по минимальному значению.
На выходе имеем метку (номер) кластера, к которому принадлежит этот а.о. Если а.о. не принадлежит ни одному кластеру (“шумовая точка”), ему ставится в соответствие метка “–1”. Кроме того, каждый а.о., находящийся в определенном кластере, классифицируется либо как принадлежащий, либо как не принадлежащий ядру кластера. Критериями качества кластерного анализа в программе используются силуэтный индекс (коэффициент) [18] и индекс Калинского–Харабаза [19], реализованные в пакете scikit-learn.
Индекс Калинского–Харабаза представляет собой отношение матриц внутренней и внешней дисперсии:
где B и W – матрица внутрикластерной и внешней (межкластерной) дисперсии соответственно, k – количество кластеров, n – количество точек. Индекс меняется от нуля в положительную сторону. Чем выше значение индекса, тем оптимальнее структура кластеров. Так как дисперсии определяются через квадраты расстояний от точки данных до точки-центроида (центра кластера), то индекс может работать некорректно с кластерами очень вытянутой и сложной формы [19]. Однако у него простая реализация, высокое быстродействие, вычислительная сложность O(n), и он подходит для большинства глобулярных белков, имеющих не слишком вытянутую форму глобул или структурных доменов.Силуэтный индекс (Si) определяется как среднее арифметическое мер несхожести элементов кластера с элементами из соседних кластеров по формуле
Как показано в [12, 20], для алгоритма кластеризации DBSCAN и кластеров сложной формы лучше использовать оценки компактности и отделимости кластеров на основе средних расстояний между элементами (например, силуэтный индекс), а не расстояний между элементом и центром кластера (индекс Калинского–Харабаза). В то же время индекс Калинского–Харабаза правильно оценивает области кластеров с наибольшей плотностью, а именно они и представляют наибольший интерес с точки зрения структурной организации белков.
Одним из основных недостатков алгоритма DBSCAN является сильная зависимость результатов кластеризации от гиперпараметров – eps и min_samples [11]. В программе Hydrocluster реализован подбор указанных гиперпараметров, основанный на максимизации метрик силуэтного индекса или индекса Калинского–Харабаза. Подбор осуществляется простым перебором их значений в заданных пользователем границах с последующей сортировкой результатов кластеризации по критерию максимизации значения соответствующего индекса. Перебор параметров распараллелен на несколько процессов для ускорения расчетов на многоядерных архитектурах. Для реализации параллелизма используется модуль multiprocessing стандартной библиотеки Python.
Программа Hydrocluster написана на языке программирования Python3 [21]. Работа программы протестирована в операционных системах GNU/Linux (Debian, Ubuntu, Fedora) и Microsoft Windows (XP и более поздние версии). Необходимым условием работы является установленный интерпретатор Python3 (версия не ниже 3.4) и дополнительные пакеты, описанные ниже. Hydrocluster работает только с реализацией CPython [22], сторонние реализации (PyPy, Jython) не поддерживаются.
Координаты атомов, их тип и описание а.о. загружаются из файла форматов PDB, mmCIF или напрямую из Protein Data Bank [23]. Для загрузки пространственных структур и открытия CIF-файлов использован пакет BioPython [24, 25]. С помощью библиотеки NumPy [26, 27] для каждого а.о., тип которого присутствует в таблице относительной гидрофобности, рассчитывается центр масс.
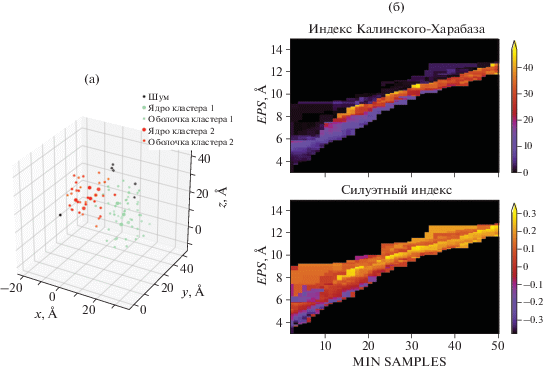
Для отображения графической информации используется модуль matplotlib [28]. Программа рисует трехмерную координатную сетку с точками, определяющими положение центров масс кластеризуемых а.о. (рис. 1a). При использовании функции автоподбора гиперпараметров строятся цветовые карты (рис. 1б).
Рис. 1.
Распределение точек – центров масс а.о. (а) (цветами обозначены кластеры, черным – шумовые точки, eps = = 12.5 Å, min_samples = 48), цветовые карты подбора гиперпараметров (eps и min_samples) (б). Структура ID PDB: 1ATG, таблица гидрофобности на основе индекса гидропатии [19].
На текущий момент в программе реализовано несколько вариантов пользовательского интерфейса: один графический и два интерфейса командной строки. Графический интерфейс пользователя работает на основе модуля tkinter стандартной библиотеки Python. Один из модулей командной строки реализует функции, доступные через графический интерфейс пользователя, второй предназначен для пакетной загрузки и анализа структур из Protein Data Bank по текстовому списку ID PDB. Результаты расчетов в этом случае сохраняются в виде файлов (графиков, автоматически генерированных скриптов для программы молекулярной графики PyMol [29]) и в реляционной базе данных формата SQLite [30] для их последующего анализа.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Тестовая выборка содержит данные для 1181 природного белка и их комплексов, пространственная организация которых определена методом рентгеноструктурного анализа с разрешением лучше, чем 2.6 Å. Используемые шкалы гидрофобности и их нормировка представлены в табл. 1.
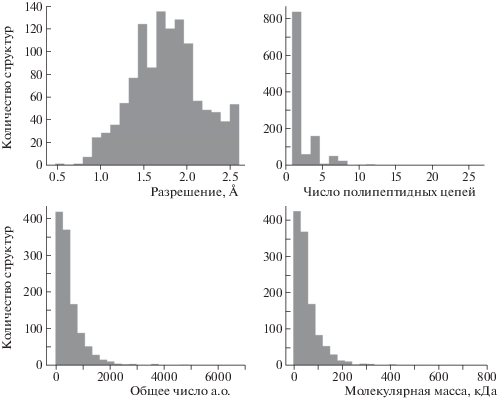
Статистические данные по структурам приведены в табл. 2, а гистограммы распределения основных характеристик – на рис. 2. Эта выборка подверглась обработке программой Hydrocluster в режиме автоподбора гиперпараметров в диапазонах eps 3.0–15.0 Å, min_samples 2–50. Использованы две шкалы гидрофобности и две оценочные функции. Первая основана на индексе гидропатии [15]. Для расчета весового коэффициента здесь брали а.о. только с индексом гидропатии больше нуля, за единицу принимали индекс гидропатии аланина, остальные весовые коэффициенты рассчитывали по отношению к нему (табл. 1). Вторая из рассматриваемых шкал весовых коэффициентов получена [16] в результате измерения контактного угла наноразмерной капли воды и монослоя из гомополипептида соответствующего а.о. Для расчета весового коэффициента в этом случае брали а.о. только с отрицательной разницей химического потенциала аминокислоты в монослое и в наноразмерной капле воды (${\Lambda \mu }_{{{\text{int}}}}^{{{\text{ex}}}} < 0$). Кроме того, из таблицы исключен тирозин, так как для него экспериментально определенный угол контакта близок к 0°. Коэффициенты нормированы на ${\Lambda \mu }_{{{\text{int}}}}^{{{\text{ex}}}}$ пролина (табл. 1).
Таблица 2.
Описательная статистика основных характеристик структур из выборки, используемой для апробации программы Hydrocluster
| Разреше-ние, Å | Число полипеп-тидных цепей | Число а.о. | Молеку-лярная масса, кДа | |
|---|---|---|---|---|
| Среднее | 1.76 | 2.6 | 580.0 | 64.69 |
| σ | 0.39 | 2.2 | 638.1 | 70.94 |
| Минимум | 0.48 | 1 | 16 | 16.27 |
| Первый квартиль | 1.50 | 1 | 227 | 25.62 |
| Медиана | 1.77 | 2 | 392 | 43.71 |
| Третий квартиль | 2.00 | 3 | 686 | 75.65 |
| Максимум | 2.60 | 26 | 6796 | 764.02 |
Рис. 2.
Гистограммы распределения основных характеристик структур из выборки, используемой для апробации программы Hydrocluster. Рисунок выполнен с использованием программы Jupyter Notebook [35 ] и библиотек Matplotlib [27] и Pandas[36 ] .
Гистограммы распределения числа кластеров по выборке приведены на рис. 3, а статистика распределения – в табл. 3. Как видно, при переходе от таблицы гидрофобности “Nanodroplet” к таблице “Hydropathy” распределение размывается и растет доля структур с большим числом кластеров гидрофобных а.о. независимо от выбора оценочной функции. Это объясняется, по-видимому, тем, что в таблице “Nanodroplet” больше а.о., и на границе ядер кластеров образуется широкая зона постепенного понижения плотности распределения а.о. В результате алгоритм объединяет эти кластеры. В то же время переход от силуэтного индекса в качестве оценочной функции к индексу Калинского–Харабаза приводит к увеличению количества структур с большим числом кластеров независимо от используемой таблицы гидрофобности. Вызвано это тем, что в случае более компактных шарообразных кластеров индекс Калинского–Харабаза более высокий, и алгоритм разбивает кластеры неправильной формы на такие кластеры. Отметим, что для глобулярных белков такой автоподбор параметров может считаться оправданным.
Рис. 3.
Гистограммы распределения оптимального числа кластеров в выборке структур, используемой для апробации программы Hydrocluster, оценки: с использованием силуэтного коэффициента для таблиц “nanodroplet” (а) и “hydro-pathy” (б), на основе индекса Калинского–Харабаза для таблиц “nanodroplet” (в) и “hydropathy” (г).
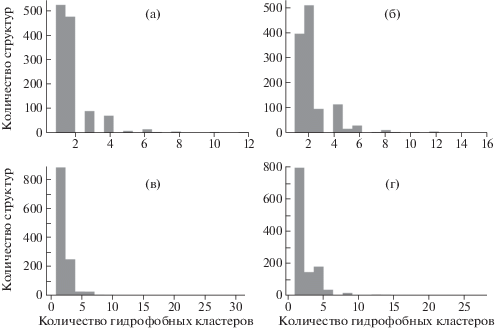
Таблица 3.
Описательная статистика числа гидрофобных кластеров и процентное отношение гидрофобных а.о., не отнесенных ни к одному кластеру (шумовые точки)
| NS | HS | NC | HC | |
|---|---|---|---|---|
| Число гидрофобных кластеров | ||||
| Среднее | 1.9 | 2.2 | 2.3 | 2.7 |
| σ | 1.3 | 1.6 | 1.8 | 2.4 |
| Минимальное | 1 | 1 | 1 | 1 |
| Максимальное | 12 | 16 | 32 | 28 |
| Шумовые точки, % | ||||
| Среднее | 9.89 | 9.26 | 19.23 | 15.16 |
| σ | 14.98 | 14.25 | 19.28 | 17.97 |
| Минимальное | 0 | 0 | 0 | 0 |
| Первый квартиль | 0.66 | 0 | 2.26 | 0 |
| Медиана | 2.93 | 3.16 | 13.24 | 8.04 |
| Третий квартиль | 11.92 | 11.93 | 31.48 | 25.11 |
| Перцентиль 90% | 33.33 | 29.89 | 48.09 | 43.48 |
| Максимальное | 94.73 | 93.24 | 94.26 | 83.08 |
Для каждой из таблиц гидрофобности и каждой из функций оценки рассчитан процент а.о., классифицированных алгоритмом DBSCAN как шум, не входящий ни в один из кластеров. Гистограммы распределения приведены на рис. 4, а его статистические параметры – в табл. 3. Особого внимания заслуживают значения перцентилей и, в частности, перцентиля 90%. При переходе от таблицы гидрофобности “Nanodroplet” к таблице “Hydropathy” растет доля структур с большим процентом шумовых гидрофобных а.о. независимо от выбора оценочной функции. Это объясняется рассмотренными особенностями таблицы “Nanodroplet”. Это же может влиять и на число кластеров.
Рис. 4.
Гистограммы распределения процентного отношения шумовых точек в выборке, используемой для апробации программы Hydrocluster, оценки: с использованием силуэтного коэффициента для таблиц “nanodroplet” (а) и “hydro-pathy” (б), на основе индекса Калинского–Харабаза для таблиц “nanodroplet” (в) и “hydropathy” (г).
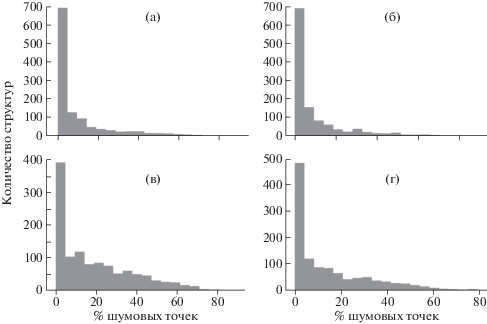
Что касается зависимости числа шумовых точек от оценочной функции, используемой для подбора параметров кластеризации, то при оценке по индексу Калинского–Харабаза доля структур с высоким процентом шумовых точек больше, чем при использовании силуэтного индекса. Связано это с тем, что индекс Калинского для кластеров неправильной формы ниже, чем для кластеров шарообразной формы и окружающего их “облака” шумовых точек, поскольку точки, отстоящие далеко от центра кластера, расцениваются как выбросы.
В табл. 4 показано распределение структур (из набора 1181) по числу полипептидных цепей и кластеров в зависимости от используемой таблицы гидрофобности и оценочной функции подбора параметров кластеризации. Больше половины составляют структуры, в которых число кластеров совпадает с числом цепей. Этот результат объясняется как структурно (одна цепь содержит один домен), так и тем, что гидрофобные а.о. разных цепей в среднем больше удалены друг от друга, чем а.о. разных доменов одной цепи. Однако там, где домены одной цепи четко отделимы друг от друга, результаты кластеризации вполне объективно отражают доменную структуру глобулярных белков.
Таблица 4.
Число структур из выборки, соответствующих определенным условиям
| Условие | NS | HS | NC | HC |
|---|---|---|---|---|
| NCHAIN = N_CLUSTERS | 658 | 632 | 644 | 595 |
| NCHAIN < N_CLUSTERS | 127 | 237 | 253 | 348 |
| NCHAIN > N_CLUSTERS | 396 | 312 | 284 | 238 |
| NCHAIN = 1 | 394 | |||
| NCHAIN ≠ N_CLUSTERS ∧ NCHAIN = 1 | 75 | 114 | 129 | 181 |
| N_CLUSTERS = 1 ∧ NCHAIN ≠ 1 | 199 | 114 | 67 | 37 |
| NCHAIN < N_CLUSTERS ∧ NCHAIN ≠ 1 | 52 | 123 | 124 | 167 |
Анализируя табл. 4, можно повторить выводы, сделанные ранее: по-видимому, при использовании таблицы “Nanodroplet” из-за ee большего размера алгоритм DBSCAN не всегда четко определяет разницу в плотностях, соответствующую одному кластеру. Из-за этого доля структур с числом кластеров большим, чем число цепей, меньше, чем в случае использования индекса гидропатии. В то же время автоподбор параметров на основе индекса Калинского–Харабаза (по сравнению с силуэтным индексом) приводит к решениям с плотными кластерами, число которых почти всегда больше, чем число цепей.
В качестве примера кластеризации гидрофобных а.о. в структуре, содержащей одну полипептидную цепь, возьмем пространственную структуру муреин-лигазы (MurF) из Thermotogamaritima в комплексе с аденозин-5´-дифосфатом (IDPDB: 3ZL8, разрешение 1.65 Å, Rfree = 22%, Rwork = 16.4%, молекулярная масса 48 кДа, число а.о. в структуре 427) [31]. Этот фермент наряду с другими типами муреин-лигаз (MurC, MurD, MurE) участвует в синтезе пептидогликана – важнейшего компонента клеточной стенки как грамположительных, так и грамотрицательных микроорганизмов [32]. Все четыре упомянутые муреин-лигазы топологически схожи друг с другом, несмотря на низкую гомологию аминокислотных последовательностей. Каждый из ферментов состоит из трех доменов: N-концевого домена с укладкой по Россману, ответственного за связывание субстрата; центрального домена, аналогичного АТФ-связывающим доменам АТФаз и ГТФаз; и С-концевого домена, аналогичного по укладке дигидрофолатредуктазе, связывающей приходящую аминокислоту [33] (рис. 5а).
Рис. 5.
Структура муреин-лигазы (MurF) из Thermotogamaritima в комплексе с аденозин-5´-дифосфатом (IDPDB: 3ZL8) в ленточном представлении (домены показаны в соответствии с [31])) (а) и расположение ядер кластеров гидрофобных а.о. (насыщеннее) и “оболочек” кластеров (светлее) (б).
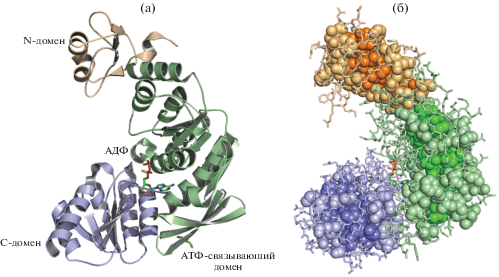
Рассмотрим результат работы программы Hydrocluster c опциями: HTABLE “Hydropathy”, автоподбор параметров кластеризации по силуэтному коэффициенту: оптимальное число кластеров – три, при eps = 12.7 Å и min_samples = 50 силуэтный индекс 0.415, доля шумовых точек 4.76%.
Кластеризация c автоподбором параметров по индексу Калинского–Харабаза тоже выделила три кластера (рис. 5б), однако результаты несколько иные, чем в предыдущем случае: eps = 8.0 Å, min_samples = 16, доля шумовых точек 12.50%.
В обоих случаях кластеры в основном расположены в пределах соответствующих доменов, что согласуется с представлением о том, что в глобулярных белках домены являются независимыми единицами фолдинга. Однако в случае автоподбора гиперпараметров по индексу Калинского–Харабаза в сравнении с силуэтным индексом наблюдается больший процент гидрофобных а.о., не вошедших ни в один из кластеров, но в то же время кластеры более плотные и компактные с большим количеством а.о. в ядре кластера.
В качестве примера кластеризации структуры, содержащей несколько полипептидных цепей, возьмем пространственную структуру гомодимера фактора резистентности к фузидовой кислоте (FusB) золотистого стафилококка (ID PDB: 4ADN, разрешение 1.65 Å, Rfree = 20.2%, Rwork = = 16.3%, молекулярная масса – две цепи по 25 кДа, число а.о. в структуре 420 (две цепи по 210 а.о.)) [34]. Фузидовая кислота – бактериостатический антибиотик, блокирующий фактор элонгации Gз золотистого стафилококка, однако введение плазмиды, кодирующей FusB, вызывает в клинических изолятах золотистого стафилококка резистентность к фузидовой кислоте. В [34] описаны структура и функция белка FusB. Каждая из двух цепей гомодимера FusB состоит из двух доменов: N-концевого, представляющего собой пучок с четырьмя α-спиралями, и C-терминального αβ-домена, содержащего цинковый палец (рис. 6а). Результат работы программы Hydrocluster c пространственной структурой FusB c опциями HTABLE “Hydropathy”, “автоподбор параметров кластеризации по силуэтному коэффициенту” приведен на рис. 6б. Оптимальное число кластеров – четыре, eps = 8.4 Å и min_samples = 12, доля шумовых точек 5.52%. Как видно из рис. 6, кластеры в основном расположены в пределах соответствующих доменов, что опять же согласуется с современными представлениями об устройстве структурно-функциональных доменов глобулярных белков.
Рис. 6.
Структура гомодимера фактора резистентности к фузидовой кислоте (FusB) золотистого стафилококка (ID PDB: 4ADN) в ленточном представлении (домены показаны в соответствии с [34]) (а) и расположение ядер кластеров гидрофобных а.о. (насыщеннее) и “оболочек” кластеров (светлее) (б).
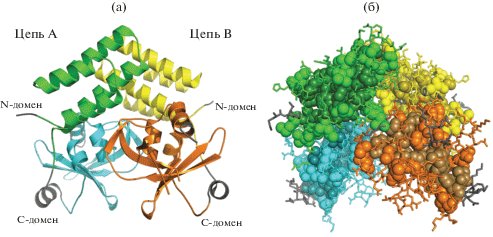
ЗАКЛЮЧЕНИЕ
Разработано программное обеспечение для кластерного анализа положения гидрофобных аминокислотных остатков в структурах белков с использованием алгоритма DBSCAN–Hydrocluster. Описаны программа и используемые ею алгоритмы. Программное обеспечение было протестировано на репрезентативной выборке структур белков, депонированных в Protein Data Bank. Результаты кластеризации оценивали как статистически, так и визуально, используя молекулярную графику. Даны оценки применения таблиц относительной гидрофобности а.о. и критериев автоматического подбора параметров кластеризации, реализованных в программе. Программа опубликована на сервисе GitHub под лицензией GPLv3 (https://github.com/alashkov83/hydrocluster).
Работа выполнена при поддержке Министерства науки и высшего образования РФ в рамках Государственного задания ФНИЦ “Кристаллография и фотоника” РАН.
Список литературы
Kalinowska B., Banach M., Wisniowski Z. et al. // J. Mol. Model. 2017. V. 23. P. 204.
Banach M., Kalinowska B., Konieczny L. et al. // J. Proteomics Bioinformatics. C. 2016. V. 9. P. 264.
Budiman T., Tadokoro C., Angkawidjaja C. et al. // FEBS J. 2012. V. 279. P. 976.
Efremov R.G., Chugunov A.O., Pyrkov T.V. et al. // Curr. Med. Chem. 2007. V. 14. P. 393.
Pyrkov T.V., Chugunov A.O., Krylov N.A. et al. // Bioinformatics. 2009. V. 25. P. 1201.
Gadzala M., Kalinowska B., Banach M. et al. // Heliyon. 2017. V. 3. P. 235.
Karyagina A., Ershova A., Tiptov M. et al. // J. Bioinform. Comput. Biol. 2006. V. 4. P. 357.
Munson M., O’Brien R., Sturtevant J.M. et al. // Protein Sci. 1994. V. 3. P. 2015.
Alexeevski A., Spirin S., Alexeevski D. et al. // Biophisika. 2003. V. 48. P. 14.
Ahmad S., Sarai A. // Nucl. Acids Res. 2004. V. 32. № S2. P. W104.
Ester M., Kriegel H.-P., Sander et al. // Proc. of 2nd Intern. Conf. on Knowledge Discovery and Data Mining. Portland. Oregon: AAAIPress, 1996. P. 226.
Сивоголовка Е.В. // Информационные системы. 2011. Т. 4. С. 14.
Knuth D.E. // SIGACT News. 1976. V. 8. P. 18.
Pedregosa F., Varoquaux G., Gramfort A. et al. // J. Machine Learning Res. 2011. V. 12. P. 2825.
Kyte J., Doolittle R.F. // J. Mol. Biol. 1982. V. 157. P. 105.
Zhu C.Q., Gao Y.R., Li H., et al. // Proc. NAS. 2016. V. 113. P. 12946.
Moore D.S. // Biochem. Education. 1985. V. 13. P. 10.
Rousseeuw P.J. // J. Comput. Appl. Math. 1987. V. 20. P. 53.
CalińskiT., Harabasz J. // Commun. Statistics. 1974. V. 3. P. 1.
Sivogolovka E. // Databases and Information Systems. 2013. V. 249. P. 95.
Oliphant T.E. // Comput. Sci. Eng. 2007. V. 9. P. 10.
Cao H.X., Gu N.J., Ren K.X. et al. // Proc. of the Federated Conf. on Computer Science and Information Systems. 2015. V. 5. P. 435.
Berman H.M., Westbrook J., Feng Z. et al. // Nucl. Acids Res. 2000. V. 28. P. 235.
Cock P.J., Antao J., Chang T. et al. // Bioinformatics. 2009. V. 25. P. 1422.
Hamelryck T., Manderick B. // Bioinformatics. 2003. V. 19. P. 2308.
H.P. Langtangen. Python Scripting for Computational Science. 2008, Springer, P.483.
Van der Walt S., Colbert C., Varoquaux G. // Comp. Sci. Eng. 2011. V. 13. P. 22.
Hunter J.D. // Comp. Sci. Eng. 2007. V. 9. P. 90.
De Lano W.L., Lam J.W. // Abstr. Papers Am. Chem. Soc. 2005. V. 230. P. 1371.
Swaine M. // Dr. Dobbs J. 2007. V. 32. P. 24.
Favini-Stabil S., Contreras-Martel C., Thielens N., Dessen A. // Environ. Microbiol. 2013. V. 15. P. 3218.
Barreteau H., Kovač A., Boniface A. et al. // FEMS Microbiol. Rev. 2008. V. 32. P. 168.
Smith C.A. // J. Mol. Biol. 2006. V. 362. P. 640.
Guo X., Peisker K., Bäckbro K. et al. // Open Biology. 2012. V. 2. P. 1200.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография