

Кристаллография, 2019, T. 64, № 3, стр. 465-476
Рост кристалла посредством неавтономной фазы: следствия для распределения элементов в рудных системах
В. Л. Таусон 1, 2, *, С. В. Липко 1, К. Ю. Арсентьев 1, 3, Н. В. Смагунов 1
1 Институт геохимии СО РАН
Иркутск, Россия
2 Иркутский научный центр СО РАН
Иркутск, Россия
3 Лимнологический институт СО РАН
Иркутск, Россия
* E-mail: vltauson@igc.irk.ru
Поступила в редакцию 23.05.2017
После доработки 10.01.2018
Принята к публикации 24.01.2018
Аннотация
Рассмотрены явления, связанные с ростом кристалла в многокомпонентных системах, близких к природным. Показано, что распределение редкоземельных элементов в магнетите и гематите, благородных металлов в пирите и магнетите контролируется поверхностными неавтономными фазами (ПНФ). С ними связано возрастание коэффициентов разделения и сокристаллизации элементов. Зависимость ПНФ от физико-химических условий роста позволяет говорить о типоморфизме минеральных поверхностей. Эволюция ПНФ в процессе роста кристалла объясняет ряд особенностей минеральных ростовых систем, в частности существование высоко детерминированных зависимостей равномерно распределенной примеси несовместимого элемента от удельной поверхности кристалла, а также образование нано- и микровключений и микрозональности в кристалле. Результаты важны для теории рудообразования и практики оценки экономического потенциала рудных месторождений в плане определения “скрытой” металлоносности и выработки рациональной технологии переработки рудного сырья.
ВВЕДЕНИЕ
Сообщества минералов обладают рядом трудно объяснимых особенностей, без понимания природы которых адекватность методов моделирования их образования (как экспериментальных, так и компьютерных) представляется сомнительной. До сих пор непонятно, по какой причине в природных условиях на протяжении геологических отрезков времени (миллионы, десятки и сотни миллионов лет) проявляют устойчивость многофазные минеральные ассоциации, не подчиняющиеся Правилу фаз даже в его расширенных вариантах [1, 2]. Особенно это касается так называемых микропарагенезисов, состоящих из кристаллов минералов микронных размеров. Само появление таких ассоциаций в результате единого ростового процесса объяснить трудно, поскольку неясно, как происходит отбор компонентов при росте конкретного индивида в такой многофазной системе. К сожалению, ростовая практика мало помогает в решении этого вопроса. В отличие от геохимиков, которых интересуют процессы в сложных системах (мультисистемах), “…ростовики рассматривают модифицирующие вещества как нежелательные примеси и затрачивают большие усилия, для того чтобы удалить их из исходных материалов” [3]. Как следствие, многофазный рост изучен в настоящее время крайне слабо.
Рассмотрим минеральный парагенезис, содержащий минералы с общим компонентом (скажем, парагенезис сульфидов). При наличии градиента температуры кристаллизация минеральной ассоциации происходит в режиме постоянного, но разного для каждой из совместно растущих фаз пересыщения, задаваемого произведением растворимости наиболее растворимой в этих условиях фазы. В микрогетерогенной системе, где фазы имеют еще и избыток поверхностной энергии, более растворимые минералы с более мелкими кристаллами будут расти медленно, и в итоге они должны уступить место фазам с меньшей растворимостью. Однако эксперименты показывают, что этого не происходит, и все кристаллы ассоциации имеют близкие размеры. Обычно близость размеров кристаллов связывают с приостановкой нуклеации на поздних стадиях кристаллизации в связи с расходом растворенного вещества [4]. Однако это не имеет места при стационарном термоградиентном росте, когда все компоненты находятся в избытке и сохраняются в шихте. Точнее говоря, в первом случае [4] расход материала приводит к снижению разности химических потенциалов до величины, не обеспечивающей преодоления энергетического барьера зародышеобразования, а во втором случае постоянство разности химических потенциалов сохраняется на протяжении всего ростового процесса. Значит, существует особый механизм, с помощью которого система сосуществующих фаз регулирует распределение компонентов, осаждающихся на поверхностях растущих кристаллов. В [5] применительно к механизму регулирования системой соотношения компонентов при образовании смешанных кристаллов обоснованы следующие положения:
– на поверхность осаждаются разнообразные ансамбли частиц, характеризующиеся некоторым статистическим распределением по составу;
– выживают только “свои” ансамбли, состав которых удовлетворяет условиям;
– остальные ансамбли, будучи “чужеродными”, растворяются, инициируя высаливание “своих” ансамблей”.
Но возникает вопрос: каким образом система различает “свои” и “чужие” ансамбли? Если это чисто статистический отбор, то с одинаковой вероятностью должен происходить и рост грани путем присоединения “своих” ансамблей, и ее отравление “чужеродными” ансамблями.
Следующий вопрос касается примесных элементов, коэффициентов их распределения между фазами, часто используемых для оценки уровня содержания элемента в среде роста, а также сегрегации примесей с образованием включений собственных фаз. До сих пор неясно, как ведут себя совместимые и несовместимые элементы в процессе роста кристалла в многокомпонентной системе. Каковы причины существования так называемых скрытых форм элементов, которые принято называть “невидимыми” и трудно идентифицировать [6, 7], и, как следствие, – наличия “скрытой” металлоносности, представляющей очевидный практический интерес для оценки и разработки рудных месторождений [8]. Традиционно считается, что чем ниже скорость роста (V), тем однороднее кристалл. Это следует из известного выражения для эффективного коэффициента распределения примеси [9], отличие которого от равновесного определяется так называемым ростовым числом Пекле Vδ/Di, где δ – толщина поверхностного слоя кристалла, Di – коэффициент поверхностной диффузии i-й примеси [10]. С другой стороны, предлагается модель, в которой, напротив, быстрый рост способствует равномерному распределению примеси, тогда как медленный приводит к ее агрегации в наночастицы за счет поверхностной диффузии атомов [11]. Следовательно, вопрос изучен слабо и, вероятно, правы те авторы, которые считают, что “…фундаментальная мульти-масштабная природа явления роста кристалла исключает какой-либо общий подход к его моделированию…” [12].
В настоящей работе рассмотрим в едином ключе природу названных выше особенностей и покажем, что они могут быть связаны с участием поверхностных неавтономных фаз (ПНФ) в ростовых процессах.
МЕТОДЫ ИССЛЕДОВАНИЯ
Особенность предлагаемой методологии в том, что эксперимент в ней дополняется изучением природных объектов. Это придает результатам необходимую общность, позволяющую наметить пути их практического использования. Ниже будут представлены результаты синтеза кристаллов магнетита (Fe3O4, Mt) и гематита (α-Fe2O3, Hm) в присутствии редкоземельных элементов (РЗЭ), пирита (FeS2) с примесями Au, Pt и Ag в присутствии As и без него, магнетита с Au, Pt и Pd. Данные по распределению благородных металлов (БМ) сопоставляются с природными объектами – пиритом и арсенопиритом месторождения Дегдекан (Северо-Восток России).
Методика эксперимента. Кристаллы получали по стандартной методике гидротермального термоградиентного синтеза [13] в автоклавах из нержавеющей стали объемом около 200 см3, оснащенных титановыми вкладышами (ВТ-6, ВТ-8) объемом около 50 см3 с пассивированной поверхностью. Использовали хорошо зарекомендовавшие себя при синтезе различных минеральных кристаллов растворы на основе хлорида аммония. Применяли внутренний пробоотбор для получения данных о составе высокотемпературного флюида [14]. Температура в зоне роста кристаллов составляла 450 либо 500°С, давление – 100 МПа (1 кбар). Опыты проводили в две стадии. На первой стадии длительностью 4 сут при 450°C и 3 сут при 500°С поддерживался изотермический режим для гомогенизации шихты и обеспечения близких к равновесным условий при последующей термоградиентной перекристаллизации, проводившейся с 15°-ным перепадом температуры (по внешней стенке автоклава) в течение 20 и 15 сут при 450 и 500°С соответственно. Эксперименты завершали закалкой автоклавов в холодной проточной воде со скоростью ∼5°C/c. В качестве шихты при синтезе Hm и Mt использовали FeO, Fe2O3 марки “ч. д. а.”, карбонильное железо. В опытах с РЗЭ Ce, Eu, Er и Yb добавляли в форме оксидов в количестве 1% каждого от массы шихты, составлявшей 5 г. В опытах с БМ добавляли по 1 мас. % Au, Pt, Pd. Концентрация минерализатора (NH4Cl марки “ос. ч.”) составляла 5 либо 10 мас. %. При синтезе пирита шихта массой 6 г состояла из элементных веществ – Fe, S, Ag, Pt ($ \pm $As). Растворы готовили на основе NH4Cl, добавляя в ряде случаев 0.5–1% HCl либо NaOH. Размеры синтезированных кристаллов Mt и Hm достигают 2–2.5 мм в диаметре, пирита – 3–5 мм. Mt представлен в основном уплощенными октаэдрами, реже двойниками и кубооктаэдрами. Пирит имеет кубические формы, но нередко усложнение морфологии происходит за счет развития граней {111}, {110}, {hk0}. Для анализа по возможности отбирали кубические формы, минимально усложненные гранями октаэдра. Это важно при вычислении точного размера кристалла и удельной поверхности. Однако в опытах с добавлением As кристаллы были в основном представлены более сложными многогранниками. Их форма отличалась обилием вициналей с малой площадью и точнее аппроксимировалась полусферой. Отбор кристаллов определенной формы, в принципе, может повлиять на усредненные данные по их химическому составу. Специальный анализ вопроса показал, что тенденция изменения зависимости среднего содержания равномерно распределенной примеси от удельной поверхности сохраняется для синтезированных в одинаковых условиях кристаллов разной формы [15]. Более того, точки, отвечающие разной морфологии, образуют единую кривую и для природных кристаллов при условии использования соответствующих коэффициентов формы (например, пентагондодекаэдры и октаэдры пирита на месторождении Дукат [16]). Тем не менее следует учитывать, что различие поверхностных наноструктур на гранях разных и даже одинаковых простых форм может приводить к двум разным тенденциям поведения примеси при изменении удельной поверхности среднего кристалла, что также наблюдалось для некоторых образцов [16].
Природный материал. Месторождение Дегдекан расположено в углеродизированных породах и принадлежит к золото-кварцевой формации руд. Продуктивная Au-сульфидная минерализация сформировалась на начальной стадии гидротермального процесса. Главный рудный минерал – пирит, менее распространен арсенопирит. Эти два сосуществующих в одной пробе минерала в виде идиоморфных кристаллов с чистыми, ровными гранями отобраны для изучения распределения в них БМ. Проба отобрана на участке Верный из кварцевого прожилка в углеродизированных и слабо сульфидизированных алевритистых аргиллитах. Кристаллы пирита в подавляющем большинстве кубической формы, псевдоромбические кристаллы арсенопирита близки к немного искаженным параллелепипедам, что позволяет при расчетах их удельной поверхности использовать тот же коэффициент формы, что и для куба.
Методы анализа. Анализ полученных кристаллов проводили методами атомно-абсорбционной спектрометрии (ААС), в том числе в версии статистических выборок аналитических данных для монокристаллов (СВАДМ), масс-спектрометрии с индуктивно связанной плазмой и лазерной абляцией (ЛА-ИСП-МС), сканирующей электронной микроскопии с энергодисперсионной спектрометрией (СЭМ-ЭДС), атомно-силовой микроскопии (АСМ), рентгеновской фотоэлектронной спектроскопии (РФЭС).
Концентрации БМ определяли методом ААС с электротермической атомизацией на приборе М503 фирмы Perkin-Elmer (США) с графитовым атомизатором HGA-74 в ЦКП “Изотопно-геохимических исследований” ИГХ СО РАН. Au и Ag определяли напрямую из аликвотной части растворов, полученных после кислотного разложения кристаллов. Определение Pt, Pd и Ru проводили после предварительного экстракционного концентрирования металла и отделения от элементов матрицы [17]. Пределы обнаружения составляли 0.3 и 0.5 мкг/л для Au и Ag, 5 для Pd и 50 мкг/л для Pt и Ru. Точность определения ±10–12 отн. %. Полученные данные обрабатывали по процедуре СВАДМ [17, 18], суть которой состоит в том, чтобы путем последовательного “просеивания” представительных выборок данных анализа единичных кристаллов (монокристаллов) разного размера выделить составляющие полной концентрации элемента, отвечающие определенным формам его нахождения.
Анализы методом ИСП МС растворов и кристаллов выполнены в ЦКП “Ультрамикроанализ” ЛИН СО РАН на приборе Agilent 7500ce (США). При определении РЗЭ в кристаллах Mt и Hm использовалась платформа лазерной абляции New Wave Research UP-213. Параметры эксперимента ЛА-ИСП-МС: мощность плазмы 1400 Вт, скорость потока несущего газа 1.18 л/мин, плазмообразующего 15 л/мин, охлаждающего 1 л/мин, энергия лазера 0.16–0.19 мДж, частота 10 Гц, диаметр лазерного пятна 55 мкм. Время накопления на один канал 0.15 с, время съемки 27 с. При расчете концентраций использовали стандартный образец NIST 612 (National Institute of Standards and Technology). Пределы обнаружения РЗЭ точно не определяли, согласно литературным данным они находятся на уровне десятых ppm [18]. Погрешность анализа оценивается как 15 отн. %.
Исследование методом СЭМ выполнено на растровом электронном микроскопе FEI Company Quanta 200 с энергодисперсионной (ЭДС) приставкой EDAX (США) в ЛИН СО РАН. Микрофотосъемка проводилась в режиме высокого вакуума при ускоряющем напряжении 30 кВ. Максимальное разрешение прибора с вольфрамовым катодом и стандартным детектором вторичных и обратнорассеянных электронов составляет 3.5 нм. ЭДС-приставка позволяет проводить микроанализ элементов в широком диапазоне (от Be доU) с разрешением 127 эВ и пределом обнаружения 0.5 мас. %. Приставка EDAX с программным обеспечением EdaxGenesis позволяет проводить не только качественный, но и количественный анализ по методу трех поправок.
Исследования методом АСМ проведены в ИГХ СО РАН и ЛИН СО РАН. Изображения получены в контактной моде на сканирующем зондовом мультимикроскопе СММ-2000 отечественного производства. Применялись кантилеверы из нитрида кремния фирмы Veeco (США) с радиусом закругления зонда 10 нм. Максимальная контролируемая разрешающая способность в плоскости XY около 2.5 нм, по направлению Z – 1 нм.
Рентгеновские фотоэлектронные спектры образцов получали на фотоэлектронном спектрометре SPECS (Германия) с энергоанализатором PHOIBOS 150 MCD 9 (Красноярский региональный центр коллективного пользования СО РАН). Спектры записывали при возбуждении излучением магниевого анода рентгеновской трубки (MgKα, 1253.6 эВ), мощность составляла 180 Вт, напряжение на трубке 12.5 кВ. Обзорный спектр записывали с шагом 0.5 эВ при энергии пропускания энергоанализатора 20 эВ. Спектры высокого разрешения отдельных элементов (узкие сканы) снимали, как правило, с шагом 0.05 эВ при энергии пропускания 8 эВ. В качестве внутреннего стандарта для учета электростатической подзарядки использовалась линия С 1s (285.0 эВ) углеводородных контаминантов. Ионное травление проводили с использованием растровой ионной пушки PU-IQE112/38, работавшей с ускоряющим напряжением 2.5 кВ и ионным током 30 мкА. Разложение спектров выполняли с помощью программы CasaXPS после вычитания нелинейного фона по Ширли, форму пиков описывали симметричной гаусс-лоренцевской функцией. При интерпретации спектров использовали базу данных РФЭС NIST, справочник [19] и работы [20, 21].
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТОВ
Mt и Hm с РЗЭ. В геохимии на РЗЭ возлагаются большие надежды как на универсальные индикаторы генезиса пород и минералов. Ce-аномалия в ряду РЗЭ рассматривается как редокс-сенсор окислительного состояния магматических систем [22], отношение Ce4+/Ce3+ считается чувствительным индикатором летучести кислорода [23]. Вместе с тем коэффициенты распределения РЗЭ минерал–флюид в гидротермальных системах неизвестны или ненадежны; неясно, от чего и в какой мере они зависят и каковы пределы их вариаций. Ниже рассмотрим первые результаты по определению коэффициентов сокристаллизации РЗЭ с Fe и их форм нахождения в Mt и Hm. Сe и Eu представляли в этих опытах легкие, Er и Yb – тяжелые РЗЭ. Информация о содержании элементов в минералах и сосуществующих с ними флюидах представлена в табл. 1. Применяли две концентрации раствора-минерализатора NH4Cl – 5 и 10 мас. %. В табл. 2 рассчитаны коэффициенты сокристаллизации (D) РЗЭ и Fe, которые обнаруживают превосходное согласие в обоих использованных растворах для Mt и несколько худшее для Hm. При оценке этих параметров использовали два подхода. В первом ЛА-ИСП-МС анализ проводили на срезах кристаллов, запрессованных в эпоксидную шашку и отполированных, т.е. полученные данные относились к объемным содержаниям РЗЭ (табл. 2). Эти данные показывают, что значения D низкие, на уровне (1–2) × 10–5–(1–2) × 10–4 для Mt и (1–4) × 10–5–(2–8) × 10–3 для Hm, причем имеется явная тенденция их повышения для тяжелых РЗЭ – примерно на порядок величины для Mt и на 2 порядка для Hm.
Таблица 1.
Содержания элементов в кристаллах Mt и Hm и сосуществующих с ними флюидах при 450°С и 1 кбар по данным ИСП МС (растворы) и ЛА-ИСП-МС (кристаллы)
| Опыт | Минерал | Раствор | Содержание элементов, ppm | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| в кристаллах | в растворе из ловушки | ||||||||||
| Ce | Eu | Er | Yb | Ce | Eu | Er | Yb | Fe | |||
| 1 | Mt | 5%NH4Cl | 0.45 | 0.36 | 0.23 | 0.22 | 600 | 234 | 30.3 | 19.3 | 9880 |
| 2 | Mt | 10%NH4Cl | 0.26 | 0.30 | 0.83 | 1.23 | 407 | 210 | 136 | 77.7 | 11 290 |
| 3 | Hm | 5%NH4Cl | 3.79 | 2.67 | 1.51 | 2.59 | 296 | 54.8 | 6.7 | 3.7 | 2340 |
| 4 | Hm | 10%NH4Cl | 0.99 | 2.20 | 2.25 | 4.20 | 1190 | 135 | 11.5 | 7.3 | 9380 |
Таблица 2.
Коэффициенты сокристаллизации РЗЭ и Fe в Mt и Hm при 450°С и давлении 1 кбар в растворах хлорида аммония
| Опыт | Минерал | D = (РЗЭ/Fe)крист/(РЗЭ/Fe)р-р | |||
|---|---|---|---|---|---|
| Ce | Eu | Er | Yb | ||
| 1 | Mt | 10–5 | 2.1 × 10–5 | 10–4 | 1.6 × 10–4 |
| 2 | Mt | 10–5 | 2.2 × 10–5 | 9.5 × 10–5 | 2.5 × 10–4 |
| 3 | Hm | 4.3 × 10–5 | 1.6 × 10–4 | 7.5 × 10–4 | 2.3 × 10–3 |
| 4 | Hm | 1.1 × 10–5 | 2.2 × 10–4 | 2.6 × 10–3 | 7.7 × 10–3 |
Во втором случае проводили анализ естественных граней, что оказалось возможным благодаря тому, что размеры полученных кристаллов были достаточно велики. Анализ показал сильное обогащение поверхности РЗЭ относительно объема (табл. 3). Измерения с помощью различных методов микроскопии глубины ямок после испарения материала лазером показало в среднем ∼20 мкм, так что можно полагать, что удаляемый лазером материал с поверхности значительно разбавлен материалом объема. Тем не менее поверхностное обогащение даже в этом варианте составляет ∼2 порядка величины (табл. 3). К сожалению, из всех РЗЭ удалось зафиксировать РФЭС-пики, надежно превосходящие фон, только для Ce. Анализ обзорных спектров показал, что его содержания в поверхностном слое могут достигать нескольких атомных процентов. Узкие спектры указывают на две формы элемента – III и IV, с заметным преобладанием первой. В Hm содержания Ce в поверхности настолько велики, что хорошо прописываются оба пика спин-орбитального дублета 3d5/2‒3/2 (рис. 1). Важно отметить, что травление Ar+ до ~60 нм мало влияет на соотношение валентных форм в Mt и совсем не влияет на него в Hm (табл. 4). Следовательно, присутствие двух форм Ce не является артефактом, вызванным травлением. Кроме того, эффект обогащения поверхности РЗЭ не вызван формированием диффузионной зоны или обычной адсорбцией (тогда бы наблюдалось резкое снижение содержания Ce после нескольких удаленных атомных слоев), а валентное состояние Ce не так жестко связано с ${{f}_{{{{{\text{O}}}_{2}}}}}$, как принято считать, поскольку отношение Ce3+/Ce4+ примерно одинаково для Mt и Hm. Последнее обстоятельство существенно для генетических интерпретаций и нуждается в объяснении. Состав поверхности Mt согласуется с данными РФЭС работы [20]. Отношение Fe3+/Fe2+ составляет ∼1.1 (табл. 5), в спектре O 1s зафиксированы O2– и OH– в соотношении 60:40 в Mt и 80:20 в Hm. Полученные данные показывают, что составы ПНФ на Mt и Hm подобны друг другу и отвечают оксигидроксидной композиции [17]: $\operatorname{Fe} _{{1 + x}}^{{3 + }}{\kern 1pt} \operatorname{Fe} _{{1--x}}^{{2 + }}$[${\text{O}}_{{1 + y}}^{{2--}}({\text{OH}})_{{1--y}}^{--}$]$V_{{2--y + x}}^{--}$, где V– – катионные вакансии.
Таблица 3.
Содержания РЗЭ на естественных поверхностях кристаллов Mt и Hm по данным ЛА-ИСП-МС (в слое ∼20 мкм) и их отношения к содержаниям в объеме кристаллов
| Опыт | Минерал | Спов, ppm | Спов/Соб | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Ce | Eu | Er | Yb | Ce | Eu | Er | Yb | ||
| 1 | Mt | 24 | 26 | 55 | 44 | 53 | 72 | 239 | 199 |
| 2 | Mt | 33 | 64 | 93 | 74 | 127 | 214 | 112 | 60 |
| 3 | Hm | 433 | 60 | 21 | 24 | 114 | 22 | 14 | 9 |
| 4 | Hm | 412 | 64 | 43 | 55 | 416 | 29 | 19 | 13 |
Рис. 1.
Рентгеновский фотоэлектронный спектр Ce 3d в Hm. Исходные кристаллы (а), после травления Ar+ (б).
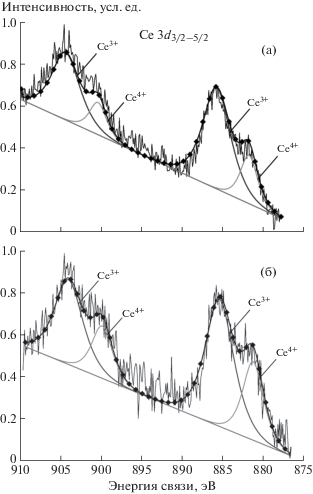
Таблица 4.
Данные РФЭС по валентному состоянию церия в кристаллах синтезированных Mt и Hm
| Образец* | Фотоэлектронный пик | Энергия связи, эВ | Полуширина пика, эВ** | Химическая форма | Ат. % |
|---|---|---|---|---|---|
| 2 | 3d5/2 3d5/2 |
882.0 885.8 |
2.8 3.6 |
Ce4+ Сe3+ |
28 72 |
| То же, Ar+ | 3d5/2 3d5/2 |
881.7 885.5 |
3.0 3.3 |
Ce4+ Ce3+ |
38 62 |
| 3 | 3d5/2 3d3/2 |
882.0 900.6 |
3.0 3.0 |
Ce4+ | 30 |
| 3d5/2 3d3/2 |
885.8 904.4 |
3.5 3.5 |
Ce3+ | 70 | |
| То же, Ar+ | 3d5/2 3d3/2 |
881.2 899.8 |
3.0 3.0 |
Ce4+ | 31 |
| 3d5/2 3d3/2 |
885.4 904.0 |
4.1 4.1 |
Ce3+ | 69 |
* Номер опыта по табл. 1. Ионное травление аргоновой плазмой в течение 10 мин (∼60 нм).
Таблица 5.
Характеристики спектров РФЭС Fe 2p3/2 (энергия связи, эВ) и соотношение валентных форм железа в поверхности кристаллов Mt (1, 2) и Hm (3) (после 5 мин травления Ar+)
| Образец | Высокоспиновый мультиплет Fe3+, номера пиков* | Высокоспиновый мультиплет Fe2+, номера пиков* | $\frac{{{\text{F}}{{{\text{e}}}^{{3 + }}}}}{{{\text{F}}{{{\text{e}}}^{{2 + }}}}}$ | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | ||
| 1 | 710.6 | 711.8 | 713.0 | 714.2 | 708.3 | 709.5 | 710.7 | 1.1 |
| 2 | 710.6 | 711.8 | 713.0 | 714.2 | 708.4 | 709.6 | 710.8 | 1.2 |
| 3 | 710.0 | 711.2 | 712.4 | 713.6 | 708.5 | 709.7 | 710.9 | 1.1 |
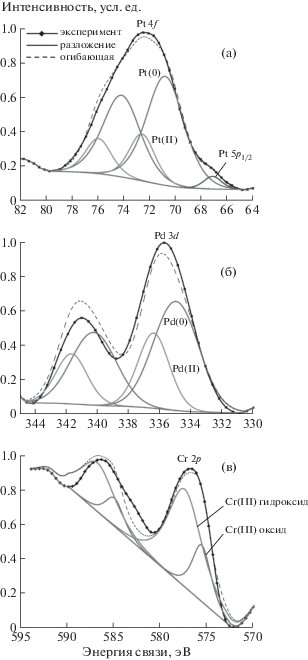
Аккумуляция БМ пиритом и Mt. Ранее было показано [24], что поверхность кристаллов гидротермального пирита химически модифицирована в пирротиноподобную неавтономную фазу с базовым составом Fe2+[S, S2, Sn]2–, способную поглощать как анионные (${\text{SO}}_{3}^{{2 - }}$, ${{{\text{S}}}_{2}}{\text{O}}_{3}^{{2 - }}$, ${\text{SO}}_{4}^{{2 - }}$), так и катионные примеси. Распространение ПНФ зависит от физико-химических условий роста: при высоких fS2 ее компоненты составляют менее 20% от Fe и S пирита, тогда как в полифазных системах в ассоциации с пирротином достигают 70%, и ПНФ образует слой толщиной более 500 нм. Имеются как косвенные, так и прямые свидетельства того, что ПНФ является причиной аккумуляции элементов-примесей в пирите, прежде всего несовместимых, к которым относятся БМ. На рис. 2 представлена зависимость среднего содержания равномерно распределенной Pt от удельной поверхности среднего кристалла в размерной фракции, полученная методом СВАДМ. Подобные зависимости с необычно высокими коэффициентами детерминации наблюдались и для Au. При расчете содержания элемента в разных формах поверхностные избытки относили к массе всего кристалла, иначе эти вклады трудно сравнивать между собой. На рис. 2 видно, что содержание поверхностной формы на порядок превосходит содержание структурной. Если относить концентрацию только к поверхностному слою ПНФ, то содержания Pt и Au возрастают, но при этом они все же, как правило, не достигают значений, достаточных для уверенного определения химических форм металлов методом РФЭС. Такие данные удалось получить для Pt и Pd на кристаллах Mt (рис. 3), синтезированных при 500°С в присутствии небольшой добавки хрома, который играл роль индикатора состава ПНФ. Формы Cr(III) подтверждают ее оксигидроксидный характер, как и в опытах с РЗЭ. Таким образом, в отличие от общепринятого мнения о восстановлении БМ на минеральных поверхностях до нейтрального состояния [25, 26] в гидротермальных ростовых системах фиксируется наличие двух форм каждого из элементов – Me0 и MeII. Это показывает, что имеет место не только восстановительная адсорбция, как при низких, близких к нормальным условиям Р, Т-параметрах, но и поглощение этих элементов ПНФ в химически связанной форме. В отношении Mt следует обратить внимание на одно важное обстоятельство. Добавляя в систему Mn и вызывая кристаллизацию твердых растворов (Fe, Mn)[Fe2O4], можно препятствовать образованию ПНФ, приводя к пологости кривые СБМ–Sуд. Причина этого в обогащении поверхности марганцем, что подтвердил послойный анализ методом ЛА-ИСП-МС [17]. То есть на поверхности остается слишком мало железа для формирования Fe-оксигидроксидной (подобной гидромагнетиту) поверхностной фазы. Таким образом, развитие ПНФ сильно зависит от физико-химических условий и химического состава среды, что позволяет говорить о типоморфизме и типохимизме минеральных поверхностей [24].
Рис. 2.
Зависимость среднего содержания равномерно распределенной Pt в пирите от удельной поверхности среднего кристалла в размерной фракции. Синтез при 500°С и 100 МПа. Указано содержание (ppm) структурной и поверхностно-связанной форм элемента.
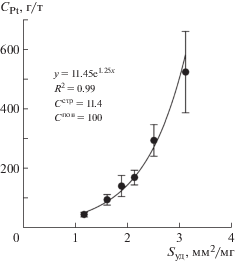
Рис. 3.
Рентгеновские фотоэлектронные спектры Pt 4f (a), Pd 3d (б) и Сr 2p (в) кристаллов Mt, полученных при 500°С и 100 МПа. Фиксируются две валентные формы Pt и Pd. Cr(III) служит индикатором оксигидроксидного состава ПНФ.
В свете представленных выше экспериментальных данных следует обратить внимание на одно важное обстоятельство. Спонтанное осаждение вещества на кристаллах при закалке, безусловно, имеет место в интервале от температуры опыта до ∼380°C – критической точки, ниже которой кристаллы перестают контактировать с раствором. Растворимость фаз в таких многокомпонентых системах в растворах NH4Cl не изучалась, есть только оценочные данные, полученные методом физико-химического моделирования с использованием программного комплекса “Селектор” [27]. Расчеты выполнены при участии В.А. Бычинского (ИГХ СО РАН) для реальных условий проведенных экспериментов на примере пирита. Они показывают, что такой механизм формирования поверхностного слоя и его обогащения микроэлементами теоретически (в приближении временного равновесия) возможен. Однако признание такой возможности вступает в противоречие с фактами. Простые балансовые расчеты показывают, что закалочное осаждение вещества не может обеспечить наблюдаемые концентрации микроэлементов в поверхностном слое. Возьмем для примера Pt в пирите (рис. 2). В этом опыте, описанном в [28] под номером D23-4, в растворе из ловушки установлено 0.42 мг/кг Pt. Содержание Pt в поверхностной форме, в расчете на массу среднего кристалла в пробе, составило 100 мг/кг (рис. 2). Масса полученных кристаллов около 900 мг, откуда находим количество вещества Pt – 0.09 мг. Если даже допустить, что на поверхность выросших кристаллов (и только на нее) при закалке разгружается весь флюид с содержанием 0.42 мг/кг Pt, находившийся во вкладыше объемом ∼50 мл, то получим только 0.02 мг Pt, т.е. в 4.5 раза меньше. Если учесть реальное соотношение площадей поверхности кристаллов и стенок вкладыша и ловушки, также вполне доступных для осаждения Pt, различие составит уже несколько порядков величины. Другие аргументы, опровергающие закалочную природу рассматриваемого явления, приведены в разделе Обсуждение результатов.
Эволюция ПНФ в процессе роста. Наблюдения в СЭМ и АСМ указывают на два возможных варианта развития событий в процессе преобразования поверхности и перехода внешних, контактирующих с флюидом слоев ПНФ внутрь кристалла. Нанофазы могут агрегироваться на участках поверхности, содержащих различные несовершенства, например выходы дислокаций или дефектов упаковки. Такой вариант представлен на рис. 4. Здесь показана поверхность кристаллов пирита, полученных в трехфазной ассоциации пирит–пирротин–Mt при 450°С. Образующиеся путем агрегации наноразмерных частиц мезокристаллы микронных размеров имеют переменный состав и содержат повышенные концентрации примесных элементов, включая Au. С другой стороны, имеются свидетельства того, что вещество примеси может поступать из-под поверхности по путям облегченной диффузии. Это можно видеть на рис. 5 на примере пирита с примесью Ag. Форма кристаллов фазы примеси примерного состава Ag1.7S вблизи кристаллографических ямок травления отражает их симметрию. В большинстве случаев ямки полностью или частично закрыты образующейся подобным образом фазой. Часто видны “шнуры” из вещества микрофазы вследствие его транспорта по дислокационному каналу. Детализация формы ямок методами СЭМ и АСМ важна для интерпретации условий их образования. В данном случае имеем дело с небольшими и сравнительно крутыми ямками с плоским дном, что может свидетельствовать о том, что их зарождение определяется дефектами именно в поверхностном слое [29].
Рис. 4.
Примеры агрегации наноразмерных фаз на поверхности кристаллов пирита (СЭМ). Пирамидки фазы, близкой по составу к моносульфиду (Fe0.71Mn0.07K0.04)S, образуются путем агрегации наноразмерных прекурсоров (а, б), нанофазы декорируют параллельные штрихи на грани куба, предположительно связанные с дефектом упаковки (в, г).

Рис. 5.
Мезокристаллические фазы на основе нестехиометрического сульфида AgI отражают симметрию дислокационных ямок травления (СЭМ). Стрелкой показан жгут, соединяющий кристалл с ямкой.

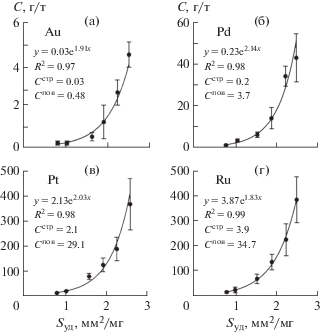
Изучение природного материала. На рис. 6, 7 приведены зависимости СБМ–Sуд для Au и платиноидов (Pt, Pd, Ru), полученные по методу СВАДМ для сосуществующих пирита и арсенопирита золоторудного месторождения Дегдекан. Сопоставление с рис. 2 указывает на идентичность ситуации для природных и синтезированных кристаллов. В количественном отношении поверхностно-связанная форма примерно на порядок превосходит структурную по содержанию всех изученных элементов в обоих минералах, подтверждая, что в основном именно она определяет валовое содержание равномерно распределенного элемента. Пирит по сравнению с арсенопиритом лучше накапливает все эти элементы как в объеме, так и особенно в поверхности. Причина этого состоит, по-видимому, в разной степени развития ПНФ на поверхности сосуществующих минералов: если на кристаллах рудных пиритов толщина слоя достигает 500 нм, то в случае арсенопирита она существенно меньше, на уровне 100 нм [30, 31].
Рис. 6.
Зависимости средних содержаний равномерно распределенных БМ в пирите от удельной поверхности среднего кристалла в размерной фракции. Месторождение Дегдекан.
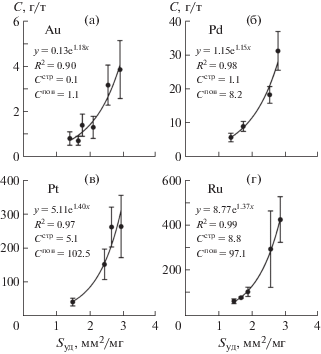
Рис. 7.
Зависимость средних содержаний равномерно распределенных БМ в арсенопирите от удельной поверхности среднего кристалла в размерной фракции. Месторождение Дегдекан.
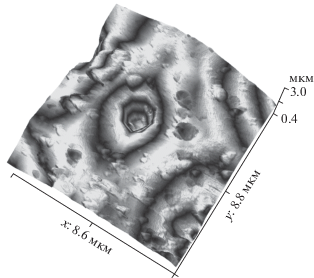
На поверхности кристаллов пирита обнаруживаются микроскопические ямки травления, подобные наблюдаемым в эксперименте. На рис. 8 представлено изображение характерной ямки в АСМ. Как и в предыдущем случае (рис. 5), это неглубокие ямки (обычно до 1 мкм) с крутыми стенками и плоским дном, иногда с микрокристалликом на дне. Вероятно, выделявшееся вблизи ямки вещество ПНФ не сохранилось впоследствии, хотя и обеспечило повышенные содержания микроэлементов (Au и платиноидов) в поверхности [32]. Полученные данные обосновывают геохимическую роль ПНФ в распределении микроэлементов как для экспериментальных, так и для природных систем. Это необходимо учитывать на практике при анализе образцов руд и разработке технологии извлечения из них полезных компонентов.
Рис. 8.
Изображение в атомно-силовом микроскопе естественной ямки травления на поверхности пирита. Проба Др-15/09, месторождение Дегдекан [32].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Рассмотренные во Введении особенности можно согласовать с полученными экспериментальными результатами, если предположить, что поверхностный слой кристалла, содержащий ПНФ, принимает участие в ростовом процессе. Определяя характер этого участия, исходим из следующих соображений. При кристаллизации в равновесии с раствором находится не объем кристалла, а его поверхностный слой. Равновесие понимается в смысле равенства химических потенциалов с учетом ∆µ, необходимого для роста, т.е. как вынужденное равновесие под действием внешнего вынуждающего фактора [33]. Этот слой структурно реконструирован и химически модифицирован в ПНФ, не отделимую от матричного кристалла. Измененное состояние слоя объясняется тем, что твердая фаза в равновесии с пересыщенным раствором характеризуется более высоким значением химического потенциала по сравнению твердой фазой в насыщенном растворе [34]. Избыток μ может реализоваться как в изменении структуры поверхностной фазы (например, степени упорядоченности или типа полиморфной модификации), так и в изменении ее химического состава (например, поглощении примеси, несовместимой с объемной структурой). Если в системе совместно образуются несколько фаз, то именно ПНФ, а не объемные их части будут находиться в локальном равновесии. Относительная устойчивость подобных систем диктуется принципом непрерывности фазообразования на сосуществующих минеральных поверхностях [35], согласно которому ПНФ на разных объемных фазах способны приспосабливаться друг к другу при изменении физико-химических условий роста. В этом возможная причина сохранения в парагенезисах фаз с более высокой и более низкой растворимостью. Тяжелые металлы переносятся в гидротермальных растворах в форме комплексных соединений. На поверхности растущего кристалла эти комплексы могут либо распадаться, либо реагировать с веществом поверхности. При распаде вещество будет хаотически выпадать на поверхности всех совместно образующихся фаз. Но в случае, если комплекс реагирует с поверхностью, становится возможным отбор компонентов путем формирования ПНФ, имеющей общие структурные и химические элементы как с матрицей, так и с данным комплексом. Только определенный комплекс способен “доращивать” ПНФ до некоторого критического размера, что и является элементарным микроскопическим процессом роста кристалла в такой сложной ассоциации.
С увеличением толщины слоя ПНФ наступает момент, когда теряется диффузионная связь прилегающих к матричной поверхности участков слоя с пересыщенным раствором, и тогда часть слоя трансформируется по типу когерентного или полукогерентного твердофазного превращения в структуру матричного кристалла. При этом примеси, с ним структурно несовместимые, обосабливаются в форме нано- и микровключений на дефектах, порах и других несовершенствах переходной зоны. ПНФ имеет дополнительные возможности по аккомодации несовместимых элементов, поэтому примесь частично остается в ее составе, но “избыточная” ее часть выводится из граничного слоя на поверхность, образуя собственные фазы. Показанная на рис. 5 сульфидная фаза AgI образуется за счет диффузии вещества из переходной зоны к поверхности по облегченным путям – дислокационным скоплениям, выходящим на поверхность и проявляющимся в виде ограненных ямок травления. Такую же роль проводников играют приповерхностные трещины и поры. Но не всегда примесь достигает поверхности, и тогда на границе образуются субмикронные и наноразмерные частицы примесной фазы. В любом случае коэффициент распределения будет сильно завышен как для переходного слоя, так и для ПНФ в целом по сравнению с объемом кристалла. Подобный эффект в физической химии известен, он имеет место при разделении изотопов простых ионов хорошо растворимых солей, когда, казалось бы, нет препятствий для диффузионных процессов выравнивания состава в поверхностном слое. Но максимально его действие сказывается на малорастворимых веществах и несовместимых примесях, как было показано выше на примере БМ и РЗЭ в сульфидах и оксидах железа. Важно понимать, что данное явление имеет термодинамическую, а не кинетическую природу [36], и поверхностное обогащение примесью нельзя трактовать как зону диффузии, даже если примесный элемент образует широкую изоморфную смесь в матричном кристалле. Традиционная кинетическая трактовка сегрегации примеси и микрозональности (“осцилляторной зональности”) минеральных кристаллов, основанная на ростовом числе Пекле [10], непригодна в случае несовместимых элементов, таких как БМ. Дело в том, что БМ могут частично восстанавливаться (рис. 3) в процессе эволюции ПНФ при агрегации составляющих ее наноразмерных фрагментов или распаде под влиянием изменившихся условий. Несовместимые элементы склонны к эндокриптии, т.е. они используют для своего вхождения в минералы не регулярные позиции атомов в идеальной кристаллической структуре, а разного рода дефекты и их комплексы [37, 38]. Но тогда крайне сложно объяснить, как формируются высоко детерминированные зависимости, показанные на рис. 2, 6, 7. Ведь количество хаотически возникающих дефектов, на которых восстанавливаются БМ, зависит от площади поверхности, а не от удельной поверхности кристалла. Столь четкие зависимости от удельной поверхности с R2 на уровне 90–99% будут иметь место только в том случае, если поглощающая элемент ПНФ гомотетична кристаллу (т.е. геометрически повторяет его форму) и покрывает значительную часть его поверхности. По этой же причине эффект обогащения микроэлементами не может быть следствием осаждения вещества при закалке и нуклеации фаз по гетерогенному механизму; в этом случае содержание элемента было бы пропорционально поверхности контакта кристалла с флюидной фазой, а не его удельной поверхности. Действительно, разные участки в различной степени доступны для осаждаемого вещества при закалке, часть поверхности кристаллов внутри сростков вообще закрыта от флюида в момент закалки. Другие отличия ПНФ и продуктов их эволюции от автономных закалочных фаз состоят в наличии упорядоченных поверхностных структур, закономерном изменении состава с глубиной (например, снижении содержания кислорода в пирите в глубь кристалла), различии морфологии и состава сегрегаций на гранях, относящихся к разным простым формам [28, 39].
Агрегация практически двумерных ПНФ, как и их переход в объем кристалла, сопровождается сбросом “мешающих” такому переходу (несовместимых с матрицей) примесей. Они в значительной мере остаются в составе промежуточных мезо-кристаллических фаз субмикронного и микронного размера. Вероятно, эффект обогащения поверхности многими несовместимыми элементами обязан главным образом этим фазам. С другой стороны, это означает, что микровключения в минералах, особенно на их поверхности, могут не отражать адекватно состав маточного раствора из-за более высоких коэффициентов распределения и сокристаллизации для ПНФ.
ВЫВОДЫ
Поверхностные неавтономные фазы, по-видимому, являются теми субстанциями, которые регулируют монокристальный рост и распределение элементов в сложных поликомпонентных системах в условиях кристаллизации многофазных кристаллических сообществ.
Существование ПНФ имеет серьезные последствия для теории рудообразования и практики оценки экономического потенциала рудных месторождений в плане определения “истинной” и “скрытой” металлоносности и выработки рациональной технологии переработки рудного сырья.
Рассмотренный механизм концентрирования микроэлементов в условиях эндогенного рудообразования путем их вхождения в состав ПНФ важен для правильной интерпретации геохимических параметров, таких как коэффициенты распределения и коэффициенты сокристаллизации элементов, а также для понимания генезиса микровключений в минералах.
Авторы выражают благодарность за помощь в аналитической части работы Ю.Л. Михлину, Т.М. Пастушковой, И.Ю. Вороновой и А.П. Чебыкину, в экспериментальной части – Д.Н. Бабкину, в геохимической – Р.Г. Кравцовой. Авторы признательны рецензенту за ценные замечания.
Исследование выполнено в рамках государственного задания № 0350-2016-0025 и при поддержке Российского фонда фундаментальных исследований (проекты № 16-35-00102, 15-05-05767, 15-05-00612, 16-05-00104, 18-05-00077), Комплексной программы Сибирского отделения РАН (проект № II.2П/IX.130-5), Интеграционного проекта ИНЦ СО РАН № 1.3.
Список литературы
Русанов А.И. Фазовые равновесия и поверхностные явления. Л.: Химия, 1967. 388 с.
Аптекарь Л.И. // Докл. АН СССР. 1978. Т. 240. № 5. С. 1135.
De Yoreo J.J., Vekilov P.G. // Rev. Mineral. Geochem. 2003. V. 54. P. 57.
Vekilov P.G. // Cryst. Growth Des. 2010. V. 10. P. 5007.
Гликин А.Э. // Зап. Рос. минерал о-ва. 2007. Т. 136. С. 1.
Pals D.W., Spry P.G., Chryssoulis S. // Econ. Geol. 2003. V. 98. P. 479.
Barra F., Reich M., Campos E. et al. // 11-th SGA Biennial Meeting on Let’s Talk Ore Deposits. Antofagasta, Chile. 2011. V. 1–2. P. 749.
Tauson V.L., Akimov V.V., Spiridonov A.M. et al. // Exp. Geosci. 2016. V. 22. P. 63.
Burton J.A., Prim R.C., Slichter W.P. // J. Chem. Phys. 1953. V. 21. P. 1987.
Watson E.B. // Geochim. Cosmochim. Acta. 1996. V. 24. P. 5013.
Derby J.J., Chelikowsky J.R., Sinno T. et al. // Advances in Crystal Growth. Eds. Skowronski M. et al. AIP Conference Proc. 916. N.Y.: Melville, 2007. P. 139.
Fougerouse D., Reddy S.M., Saxey D.W. et al. // Am. Mineral. 2016. V. 101. P. 1916.
Смагунов Н.В., Таусон В.Л., Овчинникова О.В. // Кристаллография. 2004. Т. 49. № 2. С. 356.
Tauson V.L. // Eur. J. Mineral. 1999. V. 11. P. 937.
Таусон В.Л., Кравцова Р.Г. // Записки Всерос. минерал. о-ва. 2002. Ч. 131. № 4. С. 1.
Таусон В.Л., Кравцова Р.Г., Смагунов Н.В. и др. // Геология и геофизика. 2014. Т. 55. № 2. С. 350.
Таусон В.Л., Бабкин Д.Н., Пастушкова Т.М. и др. // Геохимия. 2016. № 2. С. 165.
Химический анализ в геологии и геохимии / Под ред. Г.Н. Аношина. Новосибирск: Гео, 2016. 622 с.
Moulder J.E., Sticle W.F., Sobol P.E. et al. Handbook of X-ray photoelectron spectroscopy. Eden Prairie MN: Perkin-Elmer Corp., 1992. 261 p.
Таусон В.Л., Бабкин Д.Н., Пастушкова Т.М. и др. // Геохимия. 2012. № 3. С. 251.
Grosvenor A.P., Kobe B.A., Biesinger M.C. et al. // Surf. Interface Anal. 2004. V. 36. P. 1564.
Smythe D.J., Brenan J.M. // Earth Planet. Sci. Lett. 2016. V. 453. P. 260.
Smythe D.J., Brenan J.M. // Geochim. Cosmochim. Acta. 2015. V. 170. P. 173.
Таусон В.Л., Бабкин Д.Н., Лустенберг Э.Е. и др. // Геохимия. 2008. № 6. С. 615.
Bancroft G.M., Jean G. // Nature. 1982. V. 298. P. 730.
Jean G.E., Bancroft G.M. // Geochim. Cosmochim. Acta. 1995. V. 59. P. 3351.
Чудненко К.В. Термодинамическое моделирование в геохимии: теория, алгоритмы, программное обеспечение, приложения. Новосибирск: Гео, 2010. 287 с.
Таусон В.Л., Липко С.В., Арсентьев К.Ю. и др. // Геохимия. 2017. № 9. С. 759.
Сангвал К. Травление кристаллов: теория, эксперимент, применение. М.: Мир, 1990. 492 с.
Таусон В.Л., Кравцова Р.Г., Гребенщикова В.И. и др. // Геохимия. 2009. № 3. С. 245.
Кравцова Р.Г., Таусон В.Л., Никитенко Е.М. // Геохимия. 2015. № 11. С. 991.
Таусон В.Л., Кравцова Р.Г., Акимов В.В. и др. // Докл. РАН. 2018. Т. 478. № 2. С. 221.
Tauson V.L., Akimov V.V. // Geochim. Cosmochim. Acta. 1997. V. 61. P. 4935.
Ахумов Е.И. // Журн. неорган. химии. 1987. Т. 32. С. 248.
Таусон В.Л. // Докл. РАН. 2009. Т. 425. № 5. С. 668.
Бочкарев А.В., Трефилова А.Н., Бобров М.Ф. и др. // Журн. физ. химии. 2003. Т. 77. С. 2075.
Урусов В.С., Таусон В.Л., Акимов В.В. Геохимия твердого тела. М.: ГЕОС, 1997. 500 с.
Таусон В.Л. // Геохимия. 1999. № 6. С. 665.
Таусон В.Л., Липко С.В., Смагунов Н.В. и др. // Докл. РАН. 2014. Т. 455. № 2. С. 210.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография