

Кристаллография, 2019, T. 64, № 4, стр. 583-585
Изучение связывания производных триазола в активном центре имидазолглицерофосфатдегидратазы из Mycobacterium tuberculosis методом молекулярной динамики
Ю. К. Агапова 1, *, В. И. Тимофеев 1, 2, А. С. Комолов 1
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
* E-mail: agapova.jk@gmail.com
Поступила в редакцию 06.02.2019
После доработки 06.02.2019
Принята к публикации 14.02.2019
Аннотация
Построены модели комплексов имидазолглицерофосфатдегидратазы из Mycobacterium tuberculosis с некоторыми производными триазола, для которых известно, что они являются ингибиторами гомологичного фермента растительного происхождения. Методом молекулярной динамики показано, что данные соединения стабильно связываются в активном центре имидазолглицерофосфатдегидратазы из M. tuberculosis. Описаны их положение и окружение в активном центре фермента. Данная работа может быть использована для модификации исследуемых производных триазола при создании на их основе селективных противотуберкулезных лекарств.
ВВЕДЕНИЕ
Имидазолглицерофосфатдегидратаза (IGPD) катализирует шестой этап биосинтеза важнейшей аминокислоты – гистидина. IGPD отвечает за превращение имидазолглицерофосфата (IGP) в имидазолацетолфосфат (IAP) [1]. Гистидин является одной из 20 аминокислот, входящих в состав белков, и необходим бактерии для ее роста и развития. Ферментативный путь синтеза гистидина из 5-фосфорибозил-1-пирофосфата протекает в 10 стадий и задействует в общей сложности 11 ферментов. При этом биосинтез гистидина осуществляется только у бактерий, растений, грибов и полностью отсутствует у млекопитающих, которые получают гистидин вместе с пищей [2, 3]. Так как блокирование данного фермента в патогенной бактерии не нарушит метаболические процессы в организме хозяина, IGPD является привлекательной мишенью для поиска потенциальных противотуберкулезных лекарств.
Пространственная структура свободного фермента и его комплекса с природным субстратом (IGP) установлена при разрешении 2.02 и 2.1 Å соответственно [4]. Биологический ансамбль IGPD включает в себя 24 субъединицы, а в состав каждого из 24 его активных центров входят аминокислотные остатки трех соседних субъединиц. IGPD является металлозависимым ферментом, в каждом из 24 активных центров которого содержатся два иона марганца [4, 5]. Положение активного центра и природа входящих в него аминокислотных остатков установлены при анализе расположения субстрата в комплексе IGPD/IPG. В комплексе IGPD/IPG имидазольная группа субстрата IGP расположена в активном центре между двумя ионами марганца (Mn1 и Mn2) приблизительно на расстоянии 6 Å от каждого иона марганца; N- и C-атомы имидазольного кольца IGP участвуют в ван-дер-ваальсовых взаимодействиях с атомом CZ Met107А, а N1- и N2-атомы Arg99C формируют водородные связи с атомами OP5 и OP6 IGP соответственно.
Субстрат находится вблизи поверхности фермента и окружен преимущественно полярными остатками. Глицеролфосфатный фрагмент молекулы субстрата посредством водородных связей контактирует с остатками Glu21A, Arg99A, Glu180A, Lys184A. Ион Mn1 взаимодействует с атомами NE2 остатков His47A, His176A, His74B, атомом OE1 остатка Glu180A, атомами N2, O3 субстрата IGP, а Mn2 связан с атомами NE2 остатков His73B, His152B, His177A, атомом OE1 остатка Glu77B, боковыми радикалами остатков Met107A, Arg99C, Lys174A, Glu21B. Показано, что растительные IGPD оказались эффективными мишенями для ингибиторов – производных триазола при конструировании на их основе гербицидов [6].
В настоящей работе проведено моделирование комплексов аналогов триазола с имидазолглицерофосфатдегидратазой из M. tuberculosis для оценки возможности использования исследуемых лигандов в качестве прототипов высокоселективных противотуберкулезных лекарств.
МАТЕРИАЛЫ И МЕТОДЫ
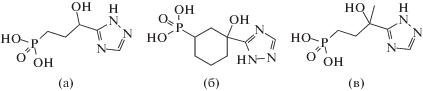
Построение трехмерных моделей комплексов. Для построения комплексов IGPD с тремя аналогами триазола (IRL 1695, IRL 1803, IRL 856, рис. 1) использовали метод молекулярного докинга (интернет-сервис Mcule [https://mcule.com]).
Рис. 1.
Структурные формулы аналогов триазола, ингибиторов IGPD: IRL 1695 (а), IRL 1803 (б), IRL 856 (в).
В качестве мишени использовали структуру IGPD из M. tuberculosis (PDB_ID: 4gqu). Для каждого из трех ингибиторов были построены четыре модели лиганд–рецептор. Для дальнейшего анализа были выбраны лучшие согласно оценочной функции докинга модели комплексов.
Молекулярно-днамическая симуляция моделей комплексов PSP с пептидами. Поведение комплексов IGPD из M. tuberculosis изучали в молекулярно-динамическом (МД) эксперименте. Для проведения МД-симуляции выбрано силовое поле OPLS-AA/L [7]. Область моделирования представляла собой кубическую ячейку 35 × 35 × 35 нм. Белок помещали в центр ячейки, весь объем которой заполняли водой с использованием модели воды spc216. Расчеты проводили с использованием программного пакета GROMACS [8].
На первом этапе МД-эксперимента провели минимизацию энергии комплексов для релаксации структуры, чтобы избежать стерических столкновений при дальнейшей симуляции. Затем на подвижность атомов системы были наложены ограничения, и при помощи симуляции системы в термостате и баростате установлены температура 300 К и давление 1 атм. Использовали алгоритмы поддержания температуры и давления – термостат Berendsen [9] и баростат Parrinello-Rahman [10]. После этого все ограничения были сняты и проведен МД-эксперимент, параметры которого представлены в табл. 1. В результате для каждого из комплексов получены траектории движения всех атомов продолжительностью 10 нс. Все вычисления проводили на суперкомпьютере НИЦ “Курчатовский институт”.
Таблица 1.
Параметры молекулярно-динамического эксперимента
| Система в термостате | |
| Термостат | Berendsen |
| Температура, К | 300 |
| Система в баростате | |
| Баростат | Parrinello-Rahman |
| Устанавливаемое давление, атм | 1 |
| Температура, К | 300 |
| Продолжительность NVT и NPT динамики, пс | 100 |
| Шаг временной сетки, пс | 0.002 |
| Молекулярная динамика | |
| Шаг временной сетки dt, пс | 0.002 |
| Количество шагов | 10 000 000 |
| Длина траектории, нс | 10 |
| Количество записанных фреймов | 100 |
| Интегратор | Leap-frog |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Полученные методом молекулярного докинга модели комплексов IGPD с тремя аналогами триазола (IRL 1695, IRL 1803, IRL 856) были ранжированы согласно оценочной функции докинга (табл. 2). В отобранных четырех вариантах положения ингибиторов в активных центрах каждого комплекса различались не более чем на 0.3 Å. Для моделей трех комплексов, выбранных согласно лучшей оценочной функции докинга, был проведен расчет методом молекулярной динамики. Рассчитываемое время для каждой из трех моделей составило 10 нс. Сравнение полученных моделей после 10 нс МД-симуляции с начальным положением ингибиторов в активном центре фермента показало, что все существенные взаимодействия ингибиторов с белком сохраняются в конечной точке МД-траектории. С помощью программы LigPlot [11] был проведен анализ аминокислотного окружения каждого из ингибиторов на 10 нс МД-траектории (рис. 2).
Таблица 2.
Оценочная функция докинга для комплексов IGPD с IRL 1695, IRL 1803 и IRL 856
| Модель | Значение оценочной функции докинга | ||
|---|---|---|---|
| IRL 1695 | IRL 1803 | IRL 856 | |
| 1 | –4.9 | –5.4 | –5.4 |
| 2 | –4.8 | –5.4 | –5.2 |
| 3 | –4.7 | –5.3 | –5.0 |
| 4 | –4.4 | –5.0 | –4.5 |
Рис. 2.
Окружение аналогов триазола, ингибиторов IGPD, в активном центре фермента IGPD после 10 нс МД-симуляции: IRL 1695 (а), IRL 1803 (б), IRL 856 (в).
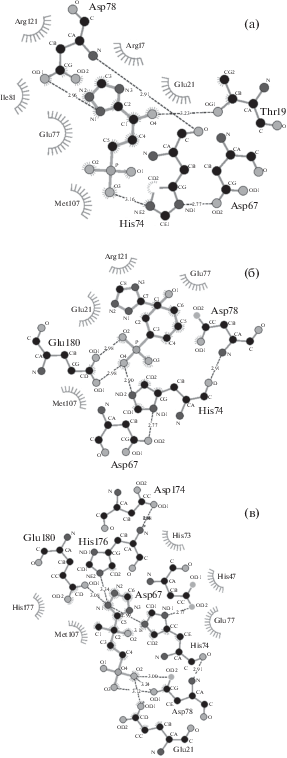
Из рисунка видно, что как фосфатная (IRL 1695/O3-HIS74/NE2), так и триазольная (IRL 1695/N1-ASP178/OD1) группы ингибитора IRL 1695 образуют с молекулой белка водородные связи, тогда как только фосфатная группа ингибитора IRL 1803 имеет водородные связи с белком (IRL 1803/O3-GLU180/OE2, IRL 1803/O4-GLU180/OE1, IRL 1803/O4-HIS74/NE2). Ингибитор IRL 856 образует значительно больше водородных связей с молекулой белка, причем в отличие от IRL 1695 и IRL 1803 к гистидинам активного центра обращена не фосфатная, а триазольная группа ингибитора. Таким образом, несмотря на большую степень сходства исследуемых ингибиторов (IRL 1695 отличается от IRL 856 только отсутствием CH3-группы, рис. 1), имеется существенная разница в связывании этих ингибиторов в активном центре фермента. Полученные структурные данные будут использованы как основа для разработки высокоселективных противотуберкулезных препаратов.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (грант № 18-34-00876) в части расчетов молекулярного докинга и молекулярной динамики и при поддержке Министерства науки и высшего образования в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН в части анализа результатов молекулярного моделирования.
Список литературы
Ames B.N., Garry B., Herzenberg L.A. // J. Mol. Biol. 1960. V. 33. P. 533.
Alifano P., Fani R., Lio P. et al. // Microbiol. Rev. 1996. V. 60. P. 44.
Stepansky A., Leustek T. // Amino Acids. 2006. V. 30. P. 127.
Ahangar M.S., Khandokar Y., Nasir N. at al. // Acta Cryst. F. 2011. V. 67. P. 1451.
Подшивалов Д.Д., Манджиева Ю.Б., Сидоров-Бирюков Д.Д. и др. // Кристаллография. 2018. Т. 63. № 1. С. 82.
Mori I., Fonne-Pfister R., Matsunaga S. et al. // Planta Physiol. 1995. V. 107. P. 719.
Jorgensen W.L., Maxwell D.S., Tirado-Rives J. // J. Am. Chem. Soc. 1996. V. 118. P. 11225.
Abraham Murtola T., Schulz R. et al. // Software X. 2015. V. 1–2. P. 19.
Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F. et al. // J. Chem. Phys. 1984. V. 81. P. 3684.
Parrinello M., Rahman A. // J. Chem. Phys. 1982. V. 76. № 5. P. 2662.
Swindells M.B., Laskowski R.A. // J. Chem. Inf. Model. 2011. V. 51. P. 2778.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография