

Кристаллография, 2019, T. 64, № 4, стр. 638-644
Применение эмпирического потенциала Si–O–C для моделирования методом переключения связей аморфных атомных структур и переходных слоев
1 Национальный исследовательский центр“Курчатовский институт”
Москва, Россия
* E-mail: ivanbelov1977@gmail.com
Поступила в редакцию 25.01.2018
После доработки 25.01.2018
Принята к публикации 27.04.2018
Аннотация
Представлены параметры эмпирического потенциала REBO2 для системы Si–O–C. Расчеты, полученные с использованием этого потенциала, показали хорошее совпадение с экспериментальными значениями длин и углов элементарных ячеек, энергии связей и поверхностной энергии для Si, SiO2 и SiC. Разработан и апробирован алгоритм переключения связей в многокомпонентной системе. Предложен метод моделирования структуры переходного слоя между кристаллическим Si и аморфным SiO2, а также между кристаллическим SiC и аморфным SiO2.
ВВЕДЕНИЕ
Компьютерный алгоритм для генерирования структур аморфных материалов под названием “моделирование атомной структуры методом переключения связей” был предложен в 1985 г. [1]. Он показал хорошее совпадение радиальной функции распределения атомов в aморфном Si и аморфном Ge с экспериментальными данными.
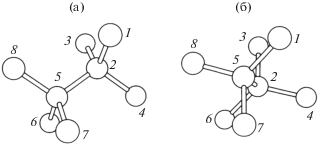
Основа этого метода – метод Монте-Карло с шагом, состоящим из поворота связи 5–2 (рис. 1) и небольшого изменения положений атомов 1, 3, 4, 7, 6, 8, чтобы две связи 1–2 и 5–6 исчезли, а две другие, 1–5 и 2–6, образовались. Координационное число атомов после переключения связей не изменяется.
Рис. 1.
Расположение связей в кристаллической кубической ячейке (а) и в новой структуре после переключения связей между атомами (б).
Каждый шаг рассматриваемого случайного процесса (переключения связей, диффузии, химических реакций) происходит с вероятностью exp(–ΔE/kT) при ΔE > 0 и равной единице при ΔE < 0, где ΔE – разница значений энергии начальной и конечной структуры в локальных минимумах, Т характеризует скорость процессов (переключения связей, диффузии и других). Это, по сути, параметр моделирования, и он не является температурой системы.
Если в случае T < Tm аморфная фаза при моделировании превращается в кристаллическую (в случае T > Tm наблюдается обратное), то при условии T = Tm система в итоге через большое количество шагов придет к канонически равновесной структуре.
В 1998 г. данный метод был применен в [2] для получения структуры аморфного SiO2. Были найдены энтропия, энергия и свободная энергия, приходящаяся на атом, что позволило найти параметр Т для генерирования данным методом структуры кварцевого стекла. Также было смоделировано влияние анизотропии на атомную структуру слоя, соединяющего кристаллическую фазу SiO2 и аморфный SiO2.
В 2000 г. данный метод был применен для моделирования атомной структуры, соединяющей кристаллическую фазу Si и аморфный SiO2 [3], и энергии атомов, а в 2002 г. – при моделировании окисления кремния [4].
Для ускорения моделирования и нахождения ΔE в настоящей работе использован тип эмпирического потенциала, известный как second-generation reactive empirical bond order potential (REBO2), в котором энергия взаимодействия описывается следующим образом:
Данный тип потенциала выбран благодаря тому, что он успешно применялся для моделирования атомной структуры в 3C-SiC в [5], где параметры для потенциала С–С взяты из [6, 7], а параметры потенциала Si–Si, Si–C – из [8]. Также он использовался для моделирования гетероэпитаксиального роста алмаза на кремнии [9] и карбиде кремния [10] как с параметрами из [8], так и с параметрами потенциала Si–Si, Si–C из [11]. Одним из его недостатков является то, что он не описывает взаимодействия с недоокисленными атомами как на далеком, так и на близком расстоянии.
ЭМПИРИЧЕСКИЙ ПОТЕНЦИАЛ Si–O–C
Нахождение неизвестных параметров эмпирического потенциала для системы Si–O–C может помочь в описании свойств керамики SiOC, а также в создании механизма производства силовой микроэлектроники на базе SiC, в котором в качестве диэлектрика используют оксид кремния, получаемый окислением SiC.
В настоящей работе параметры для взаимодействий C–C взяты из [12], а C–O – из [13]. Параметры потенциала для взаимодействий Si–Si, Si–C, Si–O получены самостоятельно и представлены в табл. 1.
Таблица 1.
Параметры потенциала для взаимодействий Si–Si, Si–C, Si–O, O–O
| Параметр | Единицы в системе СГС | |
|---|---|---|
| Взаимодействие | Si–Si | Si–O |
| A | 2.99 × 10–9 | 1.32 × 10–10 |
| Q | 0.0 | 2.13 × 10–9 |
| α | 2.47 × 108 | 1.78 × 107 |
| B1 | –8.72 × 10–10 | 1.7 × 10–10 |
| B2 | 0.0 | 9.56 × 10–12 |
| β1 | 1.75 × 108 | 2.33 × 107 |
| β2 | 0.0 | 2.25 × 107 |
| Dmin | 1.7 × 10–8 | 1.7 × 10–8 |
| Dmax | 1.8 × 10–8 | 1.8 × 10–8 |
| Взаимодействие | Si–C | O–O |
| A | 2.59 × 10–9 | 1.4 × 1011 |
| Q | 0.0 | 0.0 |
| α | 2.93 × 108 | 1.66 × 108 |
| B1 | –7.01 × 10–10 | 0.0 |
| β1 | 1.97 × 108 | 0.0 |
| Dmin | 1.7 × 10–8 | 1.75 × 10–8 |
| Dmax | 1.8 × 10–8 | 2.05 × 10–8 |
| Параметр | Значение |
|---|---|
| HC–C(NSi, NO > 0, NC), HC–O(NSi, NO, NC), HO–С(NSi, NO, NC), HO–O(NSi, NO, NC) | ${{H}_{{ij}}}(N_{{ij}}^{{{\text{Si}}}} + N_{{ij}}^{{\text{C}}},N_{{ij}}^{{\text{O}}})$ из [13] |
| HС–С(NSi,0,NC) | ${{H}_{{ij}}}(0,N_{{ij}}^{{{\text{Si}}}} + N_{{ij}}^{{\text{C}}})$ из [12] |
| HSi–C(NSi,0,NC), HSi–Si(NSi,0,NC), HC–Si(NSi,0,NC) | ${{H}_{{ij}}}(0,N_{{ij}}^{{{\text{Si}}}} + N_{{ij}}^{{\text{C}}})$ из [11] |
| HO–O(NSi + NC = 2, NO = 2), HO–O(NSi + NC = 1, NO = 1), HO–Si(NSi + NC = 0, NO = 2), HO–Si(NSi + NC = 1, NO = 1) | 0.4 |
| HSi–Si(NSi + NC = 0, NO = 5), HSi–Si(NSi + NC = 1, NO = 3), HSi–Si(NSi + NC = 1, NO = 4), HSi–Si(NSi + NC = 1, NO = 5), HSi–Si(NSi + NC = 2, NO = 2), HSi–Si(NSi + NC = 2, NO = 3), HSi–Si(NSi +NC =2, NO = 4) | 0.2 |
| HSi–Si(NSi + NC = 0, NO = 6), HSi–O(NSi + NC = 1, NO = 5) | 0.06 |
| HSi–Si(NSi + NC = 0, NO = 4) | 10.0 |
| Для не определенных выше HO–С(NSi, NO, NC), HOO(NSi, NO, NC), HO–Si(NSi, NO, NC) | 0.17(Ntot – 1)2, Ntot > 1, где ${{N}_{{{\text{tot}}}}} = N_{{ij}}^{{{\text{Si}}}} + N_{{ij}}^{{\text{O}}} + N_{{ij}}^{{\text{C}}}$ |
| Для не определенных выше Hij(NSi, NO, NC) | 0.2(Ntot – 3)2, Ntot > 3, где ${{N}_{{{\text{tot}}}}} = N_{{ij}}^{{{\text{Si}}}} + N_{{ij}}^{{\text{O}}} + N_{{ij}}^{{\text{C}}}$ |
Функция ${{\bar {B}}_{{ij}}}$ превращает потенциал 1REBO2 в многоатомный:
Слагаемое ${{F}_{{ij}}}(N_{{ij}}^{{{\text{tot}}}},N_{{ji}}^{{{\text{tot}}}},N_{{ij}}^{{{\text{conj}}}})$ учитывает зависимость энергии взаимодействия от количества связей. Однако в отличие от [11] в данном случае суммирование проходит не только по атомам С, но и по атомам Si как для $N_{{ij}}^{{{\text{tot}}}}$ = $\sum\nolimits_{k \ne i}^{{\text{Si}},\,{\text{C}}\_{\text{atom}}} {{{f}_{c}}({{r}_{{ik}}})} $, так и для $N_{{ij}}^{{{\text{conj}}}}$.
Рассмотрим функцию Bij в виде
Чтобы улучшить описание аморфного окcида кремния, вклад слагаемого g(Θijk) для связи Si–Si, взятого из [8], для ∠Si–Si–O увеличен за счет увеличения соответствующего параметра αSi–Si–O = 4. С этой же целью введена новая функция g(Θijk) связи Si–О для угла ∠Si–O–Si, найденная из квантово-механических расчетов, с параметром αO–Si–Si = 1 (табл. 2).
Таблица 2.
g(Θijk) для угла Si–O–Si
| ∠Si–O–Si, град | g(Θijk) | ∠Si–O–Si, град | g(Θijk) |
|---|---|---|---|
| 0 | 0.207 | 96 | 0.018 |
| 6 | 0.206 | 102 | 0.009 |
| 12 | 0.202 | 108 | 0.001 |
| 18 | 0.195 | 114 | –0.004 |
| 24 | 0.186 | 120 | –0.009 |
| 30 | 0.175 | 126 | –0.011 |
| 36 | 0.162 | 132 | –0.013 |
| 42 | 0.148 | 138 | –0.014 |
| 48 | 0.133 | 144 | –0.014 |
| 54 | 0.117 | 150 | –0.013 |
| 60 | 0.1 | 156 | –0.013 |
| 66 | 0.084 | 162 | –0.012 |
| 72 | 0.068 | 168 | –0.011 |
| 78 | 0.054 | 174 | –0.011 |
| 84 | 0.041 | 180 | –0.011 |
| 90 | 0.028 | – | – |
Данный потенциал протестирован на параметрах систем, таких как постоянные решетки a, b, c и ее углы α, β, γ, энергия всех связей Ep, поверхностная энергия на ячейку Es, равновесные расстояния r0, константы упругости С (табл. 3).
Таблица 3.
Сравнение экспериментальных и расчетных данных с результатами расчетов, полученными с использованием предложенного потенциала
| Фаза и ее параметры | Эксперименты и расчеты | Предложенный потенциал |
|---|---|---|
| Кубический кристаллический кремний, Ep, эВ [15] | 4.63 | 4.63 |
| Кубический кристаллический кремний, а, Å [15] | 5.43 | 5.43 |
| Si(001), Es, эВ [16] | 2.5 | 2.28 |
| Молекула Si2, Ep, эВ [11, 17] | 3.39 | 3.39 |
| Молекула Si2, r0, Å [18] | 2.24 | 2.2 |
| β-карбид кремния, Ep, эВ [19] | 6.34 | 6.37 |
| β-карбид кремния, а, Å [19] | 4.36 | 4.36 |
| Si-терминированный β-SiC(001), Es, эВ [20] | 2.74 | 2.76 |
| С-терминированный β-SiC(001), Es, эВ [20] | 2.70 | 2.74 |
| Молекула SiO, Ep, эВ [21] | 8.02 | 7.17 |
| α-кварц, Ep, эВ [22, 23] | 6.41 | 6.41 |
| α-кварц, a, b, c, Å [24] | 4.92, 4.92, 5.41 | 5.002, 5.002, 5.38 |
| α-кварц, α, β, γ, град [24] | 120, 90, 90 | 120, 90, 90 |
| α-кварц, C11, ГПа [25] | 78.0 | 75.6 |
| β-кварц, Ep, эВ [22, 23] | 6.39 | 6.38 |
| β-кварц, a, b, c, Å [26] | 5.0, 5.0, 5.46 | 5.14, 5.14, 5.64 |
| β-кварц, α, β, γ, град [26] | 120, 90, 90 | 120, 90, 90 |
| β-кристобалит, Ep, эВ [22, 23] | 6.39 | 6.32 |
| β-кристобалит, a, Å [23, 27, 28] | 7.16 | 7.59 |
МЕТОД ПЕРЕКЛЮЧЕНИЯ СВЯЗЕЙ ДЛЯ МОДЕЛИРОВАНИЯ АМОРФНЫХ СТРУКТУР
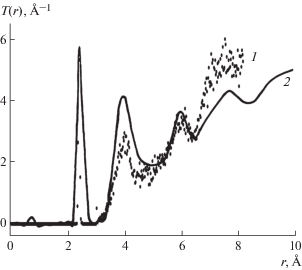
Для тестирования метода переключения связей смоделирована структура аморфного кремния. В качестве начальной структуры взята кубическая ячейка, состоящая из 216 атомов кремния. Выявлено, что при использовании предложенного потенциала минимальная температура переключения связей Tm, при которой происходит аморфизация кремния, составляет 2000 ± 200 К. Аморфная структура Si получена при 103 шагах, что соответствует 102 подтвержденным шагам метода Монте-Карло. График радиальной функции распределения атомов смоделированной структуры представлен на рис. 2.
Рис. 2.
Сравнение радиальной функции распределения атомов в полученном aморфном Si (1) с экспериментальными данными [31] (2).
Аналогично смоделирована структура аморфного SiO2, состоящая из 243 атомов. В качестве начальной взята структура α-кварца. В этом случае переключение связей осуществляется поворотом связи Si–O–Si как целого. То есть атомы на рис. 1 – это атомы Si. А кислород расположен на связях между атомами. При моделировании сделано 20 случайных шагов, а затем 102 шагов Монте-Карло, что соответствует 10 подтвержденным шагам Монте-Карло при температуре процесса 4000 К. Температура взята в 2 раза больше из-за того, что в данном случае при переключении связей меняются энергии сразу у двух связей – SiO и OSi. График радиальной функции распределения атомов смоделированной структуры продемонстрирован на рис. 3. Значения среднего угла 〈ΘSi–O–Si〉 и среднеквадратичного отклонения σSi–O–Si представлены в табл. 4.
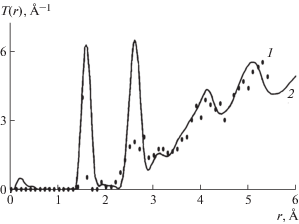
МЕТОД ПЕРЕКЛЮЧЕНИЯ СВЯЗЕЙ ДЛЯ МОДЕЛИРОВАНИЯ ПЕРЕХОДНЫХ СЛОЕВ
Для моделирования атомной структуры переходного слоя между кристаллическим Si и аморфным SiO2, состоящей из 440 атомов, добавлен процесс диффузии кислорода из связи Si–O–Si в связь Si–Si. В качестве начальной взята структура переходного слоя [3] между кристаллическим кремнием и сдавленным α-кристобалитом. Эта структура хороша тем, что в ней нет сильных искажений углов и длин связей, что должно минимизировать энергию взаимодействия атомов.
При моделировании выполнено 103 шагов, которые соответствуют 40 подтвержденным шагам Монте-Карло (32 переключения связей и восемь актов диффузии атома кислорода) при температуре процессов T = 4000 К. Заметим, что при этом моделировании переключали не только связи Si–O–Si, но и связи Si–Si с кислородом в ближайшем окружении.
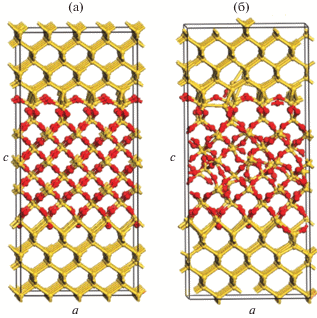
Такое маленькое количество шагов Монте-Карло не привело к каноническому равновесию, но показало несколько протекающих процессов. Как видно из рис. 4, структура α-кристобалита становится аморфной. В правом нижнем углу рисунка расположены два атома кислорода, которые окислили кристаллический кремний, а вверху рисунка наблюдается переключение связи Si–Si вблизи продиффундировавшего атома кислорода, что в дальнейшем должно помочь окислению кристаллического кремния. Малое количество диффундирующих атомов кислорода связано с тем, что ΔE в случае диффузии в среднем в 3 раза больше, чем ΔE при переключении связей.
Рис. 4.
Атомная структура переходного слоя: а – начальная между кристаллическим кремнием и сдавленным α-кристобалитом, б – смоделированная между кристаллическим Si и аморфным SiO2. Темным цветом обозначены атомы кислорода, светлым – кремния.
Изучение плотности энергии взаимодействия между материалами в структуре является ключом к пониманию свойств и определению стабильности структуры. Начальная структура состоит из 208 атомов Si с энергией взаимодействия 4.63 эВ в кристаллическом Si, 144 атомов O с энергией 7.67 эВ, 56 атомов Si с энергией взаимодействия 3.86 эВ в β-кристобалите и 32 атомов Si2+ с энергией взаимодействия 4.63 + 0.51 эВ. Добавка 0.51 эВ учитывает недоокисленние кремния Si2+, взята из [14]. Площадь поверхности соприкосновения материалов равна 2(2 × 7.56)2 Å2. При использовании потенциала Si–O–C была найдена полная энергия взаимодействия начальной структуры 2450.21 эВ. Поэтому плотность энергии взаимодействия между материалами в структуре равна: (2450.21 – 208 × 4.63 – 144 × 7.67 – ‒ 56 × 3.86 – 32(4.63 + 0.51)) эВ/(2 × 7.56)2/2 Å2 = = 4.48 мэВ/Å2.
Полная энергия смоделированной структуры переходного слоя между кристаллическим Si и аморфным SiO2 равна 2455.81 эВ. Модель включает в себя 202 атома Si с энергией взаимодействия 4.63 эВ в кристаллическом Si, 144 атома O с энергией 7.63 эВ, 56 атомов Si с энергией взаимодействия 3.59 эВ в аморфном кремнии, 26 атомов Si2+ с энергией взаимодействия 4.63 + 0.51 эВ и 12 атомов Si+ с энергией взаимодействия 4.63 + + 0.47 эВ. Добавка 0.47 эВ учитывает недоокисленние кремния Si+, взята из [14]. Плотность энергии взаимодействия между материалами в структуре в этом случае равна (2455.81 – 202 × 4.63 – 144 × × 7.63 – 56 × 3.59 – 26(4.63 + 0.51) – 12(4.63 + + 0.+ 47))эВ/ (2 × 7.56)2/2 Å2 = 5.68 мэВ/Å2. То есть смоделированная переходная структура с точки зрения энергии взаимодействия более выгодна, чем начальная, что также подтверждается графиками энергии взаимодействия атомов (рис. 5, 6), из которых видно, что энергия атомов O меняется незначительно, а атомы Si в переходном слое в смоделированной структуре значительно стабильнее, чем в начальной структуре.
Рис. 5.
Энергия взаимодействия атома Si в зависимости от координаты z: а – в структуре кристаллического кремния и сдавленного α-кристобалита, б – в смоделированной структуре переходного слоя между кристаллическим Si и аморфным SiO2.
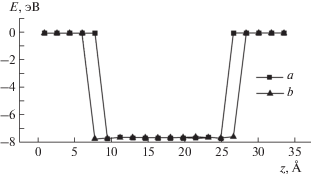
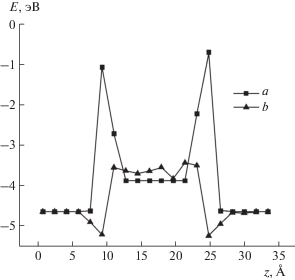
Рис. 6.
Энергия взаимодействия атома O в зависимости от координаты z: а – в структуре кристаллического кремния и сдавленного α-кристобалита, б – в смоделированной структуре переходного слоя между кристаллическим Si и аморфным SiO2.
Для тестирования метода переключения связей в многокомпонентной системе в качестве начальной структуры, состоящей из 1296 атомов, взята структура кристаллического SiC и кристобалита SiO2. Эта структура хороша тем, что в ней нет сильных искажений углов и длин связей и сохранено координационное число для всех атомов, что должно минимизировать энергию их взаимодействия. Осуществляется переключение окисленных связей и связей с атомом кислорода в ближайшем окружении.
Разработанный алгоритм переключения связей в многокомпонентной системе действует следующим образом. Предположим, что в первом приближении переключение связей – это в основном поворот связи 5–2, остальные сдвиги атомов могут быть получены в результате релаксации структуры в локальный минимум энергии. Поэтому достаточно знать положение атомов 2 и 5 после переключения связей. Атом 2 должен находиться рядом с атомами 3, 6, 4. То есть если известны характерные длины связей 2–3, 2–6, 2–4, то атом 2 должен находиться на пересечении трех сфер с радиусами 2–3, 2–6, 2–4 с центрами в атомах 3, 6, 4. Для окисленных связей 2–3, 2–6, 2–4 берут другие характерные длины. Аналогично можно найти расположение атома 5. Понятно, что пересечение трех сфер может дать два возможных варианта расположения атомов 2 и 5. Из этих четырех возможных вариантов выбирают те, которые ближе всего к характерной длине связи 2–5 и которые не приводят к изменению координационного числа атомов.
При моделировании выполнены 4 × 102 шагов, которые соответствуют 23 подтвержденным шагам Монте-Карло (19 переключений связей и четыре процесса диффузии атома кислорода) при температуре T = 4000 К. Малое количество продиффундировавших атомов кислорода связано с тем, что ΔE диффузии кислорода из связи Si–O–Si в связь Si–C в среднем в 5 раз больше, чем ΔE переключения связей. Выявлено, что предложенный алгоритм достаточен для моделирования методом переключения связей структур переходного слоя между кристаллическим SiC и аморфным SiO2.
ЗАКЛЮЧЕНИЕ
Представлены параметры эмпирического потенциала REBO2 для системы Si–O–C. Расчеты, полученные с использованием этого потенциала, показали хорошее совпадение с экспериментальными значениями параметров и углов элементарных ячеек, энергии связей и поверхностной энергии для Si, SiO2 и SiC. Разработан и апробирован алгоритм переключения связей в многокомпонентной системе. Радиальная функция распределения атомов при моделировании методом переключения связей aморфного Si и SiO2 с применением предложенного потенциала показала качественное совпадение с экспериментальными данными. Смоделированная структура переходного слоя между кристаллическим Si и аморфным SiO2 с точки зрения энергии взаимодействия более выгодна, чем структура переходного слоя между кристаллическим кремнием и сдавленным α-кристобалитом.
Работа выполнена при частичной поддержке Российского фонда фундаментальных исследований (проект № НК 15-29-01291).
Список литературы
Wooten F., Winer K., Weaire D. // Phys. Rev. Lett. 1985. V. 54. № 13. P. 1392.
Tu Y., Tersoff J., Grinstein G., Vanderbild D. // Phys. Rev. Lett. 1998. V. 81. № 22. P. 4899.
Tu Y., Tersoff J. // Phys. Rev. Lett. 2000. V. 84. P. 4393.
Tu Y., Tersoff J. // Phys. Rev. Lett. 2002. V. 89. P. 086102.
Gao F., Weber W.J. // Nucl. Instrum. Method Phys. Res. B. 2002. V. 191. P. 504.
Brenner D.W. // Phys. Rev. B. 1990. V. 42. P. 9458.
Brenner D.W. // Phys. Rev. B. 1992. V. 46. P. 1948.
Dyson A.J. // Surf. Sci. 1996. V. 355. P. 140.
Dyson A.J. // Surf. Sci. 1997. V. 45. P. 375.
Dyson A.J. // Surf. Sci. 1998. V. 24. P. 396.
Dyson A.J. // Molec. Phys. 1999. V. 96. P. 1491.
Brenner D.W. // J. Phys.: Condens. Matter. 2002. V. 14. P. 783.
Ni B., Lee K.-H., Sinnott S.B. // J. Phys.: Condens. Matter. 2004. V. 16. P. 7261.
Hamann D.R. // Phys. Rev. B. 2000. V. 61. P. 9899.
Properties of Silicon. Emis Datareviews Ser. № 4. / Ed. Harriss G.L. London: INSPEC, 1988.
Yin M.T., Cohen M.L. // Phys. Rev. B. 1981. V. 24. P. 2303.
Lide D.R. Handbook of Chemistry and Physics. Boca Raton: CRC Press, 1991.
Peyerimhoff S.D., Buenker R.J. // Chem. Phys. 1982. V. 72. P. 111.
Lambrecht W.R.L., Segall B., Methfessel M., van Schilfgaarde M. // Phys. Rev. B. 1991. V. 44. P. 3685.
Yan H., Smith A.P., Joínsson H. // Surf. Sci. 1995. V. 330. P. 265.
Lambrecht W.R.L., Segall B., Methfessel M., van Schilfgaarde M. // Phys. Rev. B. 1991. V. 44. P. 3685.
Sanderson R.T. // J. Inorg. Nucl. Chem. 1968. V. 30. P. 375.
CRC Handbook of Chemistry and Physics. Boca Raton: CRC, 1983.
Yu J., Phillpot S.R., Sinnott S.B. // Phys. Rev. B. 2007. V. 75. P. 233203.
Levien L., Prewitt C.T., Weidner D.J. // Am. Mineral. 1980. V. 65. P. 920.
Herrmann F. // Ultrasonics. 1999. V. 37. P. 335.
Wright A.F., Lehmann M.S. // J. Solid State Chem. 1981. V. 36. P. 371.
Wyckoff R.W.G. Crystal Structures. New York: Interscience, 1974.
Wright A.F., Leadbetter A.J. // Philos. Mag. 1975. V. 31. P. 1391.
Mauri F., Pasquarello A., Pfrommer B.G. et al. // Phys. Rev. B. 2000. V. 62. P. 4786.
Mozzi R.L., Warren B.E. // J. Appl. Cryst. 1969. V. 2. P. 164.
Kugler S., Molnár G., Petö G. et al. // Phys. Rev. B. 1989. V. 40. P. 8030.
Susman S., Volin K.J., Price D.L. et al. // Phys. Rev. B. 1991. V. 43. P. 1194.
Susman S., Volin K.J., Montague D.G. et al. // Phys. Rev. B. 1991. V. 43. P. 11076.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография