

Кристаллография, 2019, T. 64, № 5, стр. 747-753
Изучение связывания субстратов с двумя положительно заряженными аминокислотными остатками олигопептидазой В из Serratia proteamaculans методом молекулярной динамики
Ю. К. Агапова 1, *, А. А. Талызина 2, Ю. С. Зейфман 1, Т. В. Фатеева 1, В. И. Тимофеев 1, 3, А. Г. Михайлова 4, Т. В. Ракитина 1, 4, **
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 Московский физико-технический институт
Москва, Россия
3 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
4 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
* E-mail: agapova.jk@gmail.com
** E-mail: taniarakitina@yahoo.com
Поступила в редакцию 07.11.2018
После доработки 07.11.2018
Принята к публикации 25.12.2018
Аннотация
Методом молекулярной динамики изучено поведение пептидных субстратов GlyArgArgGly и GlyLysArgGly, содержащих аргинин или лизин в Р2-положении, в активном центре олигопептидазы В из гамма-протеобактерии Serratia proteamaculans. Показано, что природа остатка в Р2-положении определяет не только его собственную позицию и контакты с аминокислотным окружением в субстратсвязывающем кармане, но и конформационную динамику всего пептида. При этом только остаток в Р1-положении имеет фиксированные контакты с S1-субстратсвязывающим центром фермента, тогда как контакты остатка в Р2-положении могут варьировать в зависимости от его природы. Полученные результаты демонстрируют, что молекулярно-динамический эксперимент дополняет и корректирует знания, полученные при рентгенокристаллографических исследованиях, позволяющих определить пространственную структуру молекулы, находящейся в одной из многочисленных альтернативных конформаций.
ВВЕДЕНИЕ
Олигопептидаза В из психрофильной бактерии Serratia proteamaculans proteamaculans (PSP) [1–6] является трипсиноподобной сериновой пептидазой [КФ 3.4.21.83], принадлежащей семейству пролилолигопептидаз (POP), характерной особенностью которого является наличие С-концевого α/β-гидролазного каталитического домена, содержащего каталитическую триаду Ser532, Asp617 и His652 (нумерация дана по аминокислотной последовательности PSP), и N-концевого регуляторного β-пропеллерного домена, препятствующего проникновению высокомолекулярных субстратов к активному центру, расположенному в полости между доменами [7]. Олигопептидазы В (OpdBs) или кодирующие их гены обнаружены в прокариотах, одноклеточных эукариотах и в некоторых высших растениях [8]. Установлено, что эти ферменты относятся к факторам патогенеза трипаносомозов, лейшманиозов и других паразитарных инфекций и поэтому являются потенциальными мишенями для разработки терапевтических противоинфекционных средств [9].
Наиболее хорошо изучены OpdBs из простейших и только для них получены пространственные структуры. Кристаллические структуры OpdBs из Leishmania major и Trypanosoma brucei в комплексе с ингибитором, аналогом переходного состояния – антипаином (PDB_ID: 2XE4 [10], 4BP9 [11]), а также пространственные структуры близкородственных POP из млекопитающих и бактерий в комплексе с ковалентно связанными ингибиторами (PDB_ID: 1QFS [7], 2BKL [12], 3IVM [13]) или пептидными субстратами в случае каталитически неактивных мутантов POP из мозга Sus scrofa и археи Aeropyrum pernix (PDB_ID: 1UOO, 1UOQ, 1UOP [14] и 2HU8 [15]) создали основу для поиска конкурентных ингибиторов методом молекулярного докинга и изучения субстратной специфичности OpdBs методами молекулярной динамики (МД).
Известно, что протозойные и бактериальные OpdBs, включая PSP, гидролизуют субстраты с парой положительно заряженных остатков с более высокой эффективностью, чем субстраты с одиночными остатками Arg или Lys [7]. Первоначально на основании анализа трехмерной модели OpdB из Escherichia coli, а также данных направленного мутагенеза OpdB из Salmonella enterica предполагалось, что S2-центр, взаимодействующий с аминокислотным остатком в P2-положении субстрата, включает в себя аминокислотные остатки Asp460 и Asp462 [16, 17], из которых только первый консервативен среди OpdBs [18]. Последующие структурные исследования протозойных OpdBs в комплексе с антипаином опровергли данные предположения [10, 11]. В протозойных ферментах остатки Asp или Glu, занимающие позицию Asp460, участвовали в образовании междоменного солевого мостика SB3 (Arg333-Asp460, нумерация по аминокислотной последовательности PSP), сохраняющегося в бактериальных OpdBs, тогда как в предполагаемом S2-субстратсвязывающем кармане не было обнаружено кислых аминокислотных остатков. Встраивание остатков Arg или Lys в P2-положение антипаина, ковалентно связанного с OpdB из L. major, показало, что оба остатка одинаково взаимодействуют с белком путем образования водородной связи и стэкинг-взаимодействия с Tyr499 (Tyr455 в PSP) [10]. Однако общий механизм взаимодействия противоречит данным о том, что PSP гидролизует субстраты с аргинином в P2-позиции в 3 раза эффективнее, чем с лизином, а ее точечный мутант Asp649Ala, наоборот, предпочитает субстраты с лизином в P2-позиции [18]. Таким образом, вопрос о том, как регулируется вторичная субстратная специфичность у OpdBs оставался открытым.
Целью данной работы было изучение структурных факторов, определяющих вторичную субстратную специфичность PSP. Для решения этой задачи были получены трехмерные модели PSP в комплексе с пептидными субстратами GlyArgArgGly и GlyLysArgGly, проведена МД-симуляция полученных моделей и проанализированы возможные контакты между аминокислотными остатками PSP и остатками аргинина и лизина в P2-положениях субстрата.
МАТЕРИАЛЫ И МЕТОДЫ
Построение трехмерных моделей комплексов. Построение модели закрытой формы PSP с использованием web-сервиса I-TASSER [19] описано в [18]. Для построения трехмерных моделей комплексов PSP с пептидами GlyArgArgGly и GlyLysArgGly использовали пространственную структуру каталитически неактивного мутанта Ser554Ala пролилолигопептидазы из мозга кабана (Sus scrofa) в комплексе с тетрапептидом GlyPheArgPro, содержащим положительно заряженный остаток в P2-положении (PDB_ID: 1UOO) [14]. Структуру 1UOO совмещали с моделью PSP, после чего координаты пептида GlyPheArgPro переносили в модель PSP. Далее с использованием программы PyMol аминокислотные остатки пептида GlyPheArgPro в модели PSP заменяли на Gly(Arg/Lys)ArgGly, при этом положительные аминокислотные остатки в P1- и Р2-положениях субстрата получили нумерацию Arg726 и Arg(Lys)725 соответственно.
МД-симуляция моделей комплексов PSP с пептидами. Поведение комплексов PSP c пептидами GlyArgArgGly и GlyLysArgGly изучали в МД-эксперименте. Для проведения МД-симуляции было выбрано силовое поле OPLS-AA/L [20]. Область моделирования представляла собой кубическую ячейку 10.403 × 10.403 × 10.403 нм. Белок помещали в центр ячейки, весь объем которой заполняли водой с использованием модели воды spc216. Расчеты проводили с использованием программного пакета GROMACS [21]. Общий заряд системы нейтрализовали путем добавления 37 ионов хлора.
На первом этапе МД-эксперимента провели минимизацию энергии комплексов для релаксации структуры, чтобы избежать стерических столкновений при дальнейшей симуляции. Затем на подвижность атомов системы были наложены ограничения, и при помощи симуляции системы в термостате и баростате установлены температура 300 К и давление 1 атм. Использовали алгоритмы поддержания температуры и давления – “термостат Berendsen” [22] и “баростат Parrinello-Rahman” [23]. После этого все ограничения были сняты и проведен МД-эксперимент, параметры которого представлены в табл. 1. В результате для каждого из комплексов получили траектории движения всех атомов продолжительностью 40 нс. Все вычисления проводили на суперкомпьютере НИЦ “Курчатовский институт”.
Таблица 1.
Параметры молекулярно-динамического эксперимента
| Система в термостате Berendsen | |
| Температура, К | 300 |
| Система в баростате Parrinello-Rahman | |
| Устанавливаемое давление, атм | 1 |
| Температура, К | 300 |
| Продолжительность NVT- и NPT- динамики, пс | 100 |
| Шаг временной сетки, пс | 0.002 |
| Молекулярная динамика | |
| Шаг временной сетки dt, пс | 0.002 |
| Количество шагов | 50 000 000 |
| Длина траектории, нс | 50 |
| Количество записанных фреймов | 500 |
| Интегратор | Leap-frog |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Чтобы понять механизм взаимодействия PSP с субстратами, имеющими пару положительно заряженных остатков, была проведена 40-наносекундная МД-симуляция комплексов олигопептидазы В с пептидами GlyArgArgGly и GlyLysArgGly, полученных путем моделирования, и на протяжении симулируемой траектории проанализированы взаимодействия положительно заряженных остатков в Р1- и P2-положениях субстратов с их аминокислотным окружением.
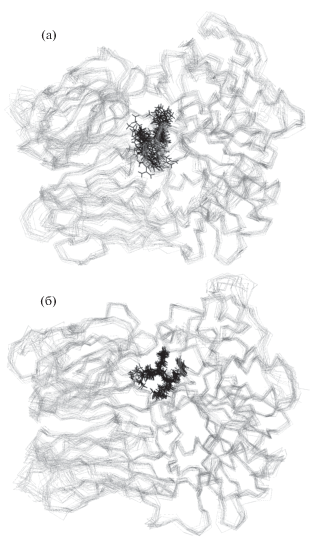
При анализе пространственных структур комплексов в разных точках траектории использовали программы PyMol. На рис. 1 представлены МД-траектории движения пептидов GlyArgArgGly и GlyLysArgGly в междоменном интерфейсе PSP. Отметим, что пептид GlyArgArgGly обладает гораздо большей подвижностью, чем GlyLysArgGly.
Рис. 1.
МД-траектории движения пептидов GlyArgArgGly (а) и GlyLysArgGly (б) в междоменном интерфейсе PSP.
С помощью программы LigPlot [24] был проведен анализ аминокислотного окружения каждого из пептидных субстратов на протяжении 15 нс МД-траектории и выявлены аминокислотные остатки PSP, способные формировать полярные (расстояние до 3.35 Å) контакты с атомами положительно заряженных аминокислотных остатков пептидов GlyArgArgGly и GlyLysArgGly, находящихся в P1- и P2-положениях. Полярные контакты, обнаруженные при анализе участка МД-траектории с 25 по 32 нс, представлены в табл. 2. Для анализа стабильности этих контактов были определены расстояния между соответствующими атомами на всем протяжении МД-траектории. Результаты представлены в виде графической зависимости изменения расстояния между атомами от времени МД-симуляции (рис. 2–4).
Таблица 2.
Контакты положительно заряженных остатков в Р1- и Р2-положениях пептидных субстратов GlyArgArgGly и GlyLysArgGly с аминокислотным окружением PSP, выявленные на участке МД-траектории (МД_Т) с 25 по 32 нс
| МД_Т | GlyArgArgGly | GlyLysArgGly | ||
|---|---|---|---|---|
| нс | P2 (Arg725) | P1 (Arg726)* | P1 (Arg726)** | P2 (Lys725) |
| 25 | Arg726NH1–Glu576OE2*** | Arg726N–Tyr455OH | Lys725N–Tyr225OH | |
| 25.5 | Arg726N–Tyr455OH | Lys725N–Tyr225OH | ||
| 26 | Arg726N–Tyr455OH | Lys725NZ–Ser242OG | ||
| 26.5 | Arg726O–Ser532OG | Arg726N–Tyr455OH | Lys725N–Tyr225OH Lys725NZ–Ser242OG | |
| 27 | Arg726N–Tyr455OH | Lys725N–Tyr225OH Lys725NZ–Ser242OG Lys725O–Gln619NE2 |
||
| 27.5 | Arg725NH2–Asp460OD2 | Arg726N–Ser457OG Arg726O–Tyr452OH | Arg726N–Tyr455OH Arg726O–Ser532OC |
Lys725N–Tyr225OH |
| 28 | Arg725NH1–Ser458O Arg725NH1–Asp460OD2 |
Arg726N–Ser457OG Arg726O–Tyr452OH |
Lys725N–Tyr225OH | |
| 28.5 | Arg725NH1–Ser458O | Arg726N–Ser457OG Arg726O–Tyr452OH |
Arg726N–Tyr455OH | Lys725N–Tyr225OH Lys725NZ–Ser242OG |
| 29 | Arg725NH1–Ser458O | Arg726N–Ser457OG Arg726O–Tyr452OH |
Arg726N–Tyr455OH | Lys725N–Tyr225OH Lys725NZ–Ser242OG |
| 29.5 | Arg725NH1–Asp460OD2 | Arg726N–Ser457OG Arg726NH1–Ser454OC | Arg726N–Tyr455OH | Lys725NZ–Ser242OG |
| 30 | Arg725NH1–Ser458O Arg725NH2–Tyr452OH |
Arg726N–Ser457OG Arg726NH1–Ser454OG |
Lys725N–Tyr225OH | |
| 30.5 | Arg725NH1–Ser458O Arg725NH2–Ser458O Arg725NH1–Asp460OD2 |
Arg726N–Ser457OG | Lys725N–Tyr225OH | |
| 31 | Arg725NH1–Asp460OD2 | Arg726NH1–Glu576OE2*** Arg726N–Ser457OG |
Lys725N–Tyr225OH | |
| 31.5 | Arg725NH1–Ser458O Arg725NH2–Tyr452OH |
Arg726N–Ser457OG Arg726O–Tyr452OH | Arg726N–Tyr455OH | Lys725N–Tyr225OH |
| 32 | Arg725NH1–Ser458O | Arg726N–Ser457OG Arg726O–Tyr452OH | Arg726N–Tyr455OH | Lys725N–Tyr225OH |
Рис. 2.
Графики изменения расстояния между атомами аргинина в P1 (Arg726) положениях пептидных субстратов GlyArgArgGly (а) и GlyLysArgGly (б) и атомами S1-субстратсвязывающего центра (Glu576) и серина каталитической триады (Ser532) PSP, образующими на протяжении 40 нс МД-симуляции полярные контакты: Glu576OE1-Arg726NH2 (черный цвет), Glu576OE2-Arg726NH1 (светло-серый цвет) (а); Glu576OE2-Arg726NH2 (черный цвет), Glu576OE1-Arg726NH1 (светло-серый цвет) (б); Ser532OG-Arg726O (темно-серый цвет) (а, б).
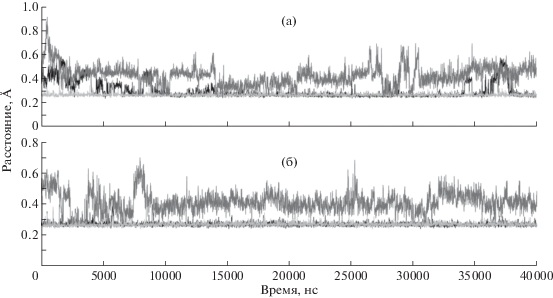
Рис. 3.
Графики изменения расстояния между атомами лизина и аргинина в P2 (Lys725) и P1 (Arg726) положениях пептидного субстрата GlyLysArgGly и атомами аминокислотных остатков PSP, образующими на протяжении 40 нс МД-симуляции полярные контакты: Tyr455OH-Arg726N (серый цвет), Tyr225OH-LYS725N (черный цвет) (а); Ser242OG-LYS725NZ (серый цвет), GLN619NE2-LYS725O (черный цвет) (б).
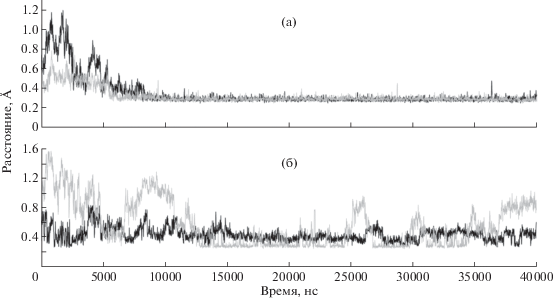
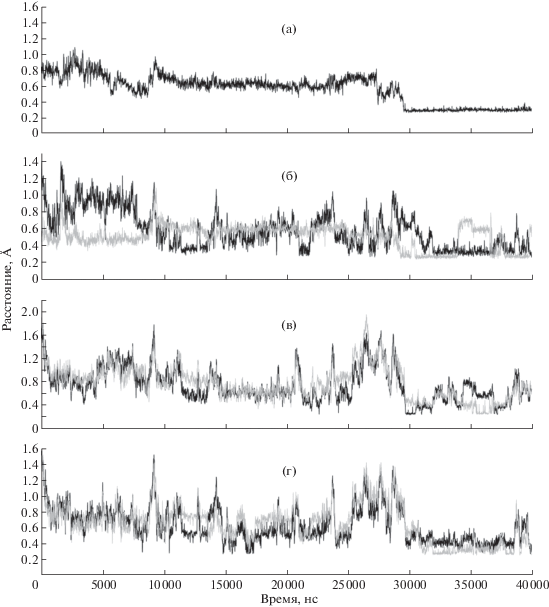
Рис. 4.
Графики изменения расстояния между атомами аргинина в P2 (Arg725) и P1 (Arg726) положениях пептидного субстрата GlyArgArgGly и атомами аминокислотных остатков PSP, образующими на протяжении 40 нс МД-симуляции полярные контакты: Ser457OG-Arg726N (a); Tyr452OH-Arg725NH2 (черный цвет), Tyr452OH-Arg726O (серый цвет) (б); Asp460OD2-Arg725NH2 (черный цвет), Asp460OD1-Arg725NH1 (серый цвет) (в); Ser458O-Arg725NH2 (черный цвет), Ser458O-Arg725NH1 (серый цвет) (г).
Установлено, что для обоих субстратов на всем протяжении МД-траектории наблюдается взаимодействие гуанидиновой группы аргинина в P1-положении (Arg726) с атомами кислорода карбоксильной группы боковой цепи остатка Glu576, который, как было установлено ранее, является структурной детерминантой первичной субстратной специфичности OpdBs [10, 11, 16, 17]. Анализ расстояний Glu576OE1-Arg726NH2 и Glu576OE2-Arg726NH1 (в случае пептида GlyArgArgGly) и Glu576OE1-Arg726NH1 и Glu576OE2-Arg726NH2 (в случае пептида GlyLysArgGly) показал, что хотя бы один из этих контактов сохраняется на протяжении всей МД-траектории (рис. 2). В ближайшем окружении аргинина в Р1-положении (Arg726) обоих субстратов также присутствует аминокислотный остаток Ser532 каталитической триады PSP, атом кислорода гидроксильной группы которого периодически приближается к карбонильной группе аргинина в Р1-положении (контакты Ser532OG-Arg726O на рис. 2), что обеспечивает возможность нуклеофильной атаки. Остальные контакты остатков в Р1 (Arg726) и Р2 (Arg725 или Lys725) положениях пептидов GlyArgArgGly и GlyLysArgGly различаются, хотя в обоих случаях помимо отрицательно заряженных аминокислотных остатков во взаимодействиях с субстратами участвуют преимущественно остатки тирозина и серина (табл. 2).
Пептид GlyLysArgGly удерживается в субстратсвязывающем кармане за счет водородных связей между атомами азота основной цепи остатков Lys725 и Arg726 и гидроксильными группами остатков Tyr225 и Tyr455 соответственно, о чем свидетельствует расстояние менее 3.35 Å на основной части МД-траектории (контакты Tyr225OH-Lys725N и Tyr455OH-Arg726N на рис. 3а). Кроме того, Lys725 может формировать менее стабильные (наблюдаемые на отдельных участках МД-траектории) полярные контакты: Ser242OG-Lys725NZ и Gln619NE2-Lys725О (рис. 3б, табл. 2).
В случае пептида GlyArgArgGly гуанидиновая группа аргинина в Р2-положении (Arg725), с одной стороны, препятствует стабилизации системы характерных водородных связей вплоть до 28–29 наносекунды симуляции, о чем свидетельствует незначительное количество контактов, наблюдаемых на участке МД-траектории с 25 по 27.5 нс, а с другой стороны, приводит к образованию более многочисленных альтернативных контактов (табл. 2). Графики зависимости расстояний между контактирующими атомами от времени МД-симуляции, приведенные на рис. 4, подтверждают, что формирование ряда стабильных контактов начинается на 28–29 наносекунде симуляции. Например, помимо стабильных контактов с атомами кислорода карбоксильной группы остатка Glu576 (рис. 2) на 29 наносекунде симуляции Arg726 образует дополнительный стабильный контакт Ser457OG-Arg726N и менее стабильный контакт Tyr452OH-Arg726O (рис. 4а, 4б). Стабилизации контакта Tyr452OH-Arg726O, вероятно, препятствует конкуренция между карбонильной группой Arg726 и аминогруппой Arg725 за гидроксильную группу Tyr452, о чем свидетельствуют меняющиеся в противофазе расстояния Tyr452OH-Arg726O и Tyr452OH-Arg725NH2 (рис. 4б). В то же время карбоксильная группа Asp460 может конкурировать с гидроксильной группой Tyr452 за гуанидиновую группу Arg725 (табл. 2, рис. 4б, 4в), которая может участвовать в образовании еще двух контактов: Ser458O-Arg725NH1 и Asp460OD1-Arg725NH1, первый из которых является более стабильным (рис. 4в, 4г, табл. 2).
Данные МД-симуляции показали, что в пептидных субстратах с двумя положительно заряженными остатками только остаток в Р1-положении имеет фиксированные контакты с S1-субстратсвязывающим центром PSP, тогда как контакты остатка в Р2-положении зависят от его природы и могут быть вариабельными. Ранее на основании сравнения структур комплексов OpdBs из L. major и T. brucei (PDB_ID: 2XE4 и 4BP9) с антипаином такая вариабельность была показана только для остатков в Р3- и Р4-положениях [11]. Полученные результаты демонстрируют, что МД-эксперимент дополняет и корректирует знания, полученные при рентгенокристаллографических исследованиях, позволяющих получать информацию о пространственной структуре молекулы, находящейся в одной из многочисленных альтернативных конформаций. В результате аргинин в Р2-положении субстрата в какие-то моменты взаимодействует с Asp460, как предполагали в [16, 17], а в другие моменты не имеет этого контакта, как показано в [10, 11].
Полученные результаты позволяют выдвинуть предположение, почему PSP гидролизует субстраты с аргинином в Р2-положении более эффективно, чем с лизином в этом положении [18]. Как было сказано выше, OpdBs и родственные РОРs состоят из С-концевого α/β-гидролазного каталитического домена и N-концевого регуляторного β-пропеллерного домена, соединенных гибким линкером. На основании серии структурных работ было установлено, что в отсутствие субстрата наблюдается постоянный переход молекулы из каталитически неактивной (открытой) формы, в которой как сами домены, так и аминокислотные остатки каталитической триады разобщены, в активную (закрытую) форму, в которой сами домены и остатки каталитической триады сближены друг с другом [10–16]. Активный центр, расположенный в полости между доменами, становится доступен для субстратов только в открытой форме фермента. В случае пептида GlyArgArgGly все наблюдаемые при МД-симуляции контакты аргининов в Р1- и Р2-положениях субстрата формируются за счет остатков каталитического домена, т.е. прочность связывания субстрата GlyArgArgGly не зависит от угла между расходящимися доменами. В случае пептида GlyLysArgGly аргинин в Р1-положении формирует стабильные контакты с остатками каталитического домена, а лизин в Р2-положении – преимущественно с остатками β-пропеллерного домена. При таком варианте связывания пептида с ферментом расстояние между доменами определяет возможность формирования и прочность контакта. Данный факт, а также меньшее суммарное количество контактов с аминокислотным окружением, наблюдаемое при МД-симуляции пептида GlyLysArgGly, вероятно, и является причиной более слабого связывания субстратов с лизином в Р2-положении по сравнению с субстратами с аргинином в Р2-положении.
Работа выполнена при финансовой поддержке Российского научного фонда (проект №17-14-01256) в части моделирования и молекулярно-динамической симуляции комплексов олигопептидазы В из S. proteamaculans, а также при поддержке Министерства науки и высшего образования в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН в части сравнительного анализа пространственных структур пролилолигопептидаз.
Список литературы
Михайлова А.Г., Лихарева В.В., Хайруллин Р.Ф. и др. // Биохимия. 2006. Т. 71. № 5. С. 697.
Хайруллин Р.Ф., Михайлова А.Г., Себякина Т.Ю. и др. // Биохимия. 2009. Т. 74. № 10. С. 1427.
Михайлова А.Г., Хайруллин Р.Ф., Демидюк И.В. и др. // Биохимия. 2011. Т. 76. № 4. С. 591.
Михайлова А.Г., Хайруллин Р.Ф., Коломийцева Г.Я. и др. // Биохимия. 2012. Т. 77. № 3. С. 384.
Mikhailova A.G., Khairullin R.F., Demidyuk I.V. et al. // Protein Exp. Purif. 2014. V. 93. P. 63.
Михайлова А.Г., Некрасов А.Н., Зинченко А.А. и др. // Биохимия. 2015. Т. 80. № 11. С. 1606.
Fulop V., Bocskei Z., Polgar L. // Cell. 1998. V. 94. P. 161.
Kaushik S., Sowdhamini R. // BMC Genomics. 2014. V. 15. P. 985.
Coetzer T.H., Goldring J.P., Huson L.E. // Biochimie. 2008. V. 90. P. 336.
McLuskey K., Paterson N.G., Bland N.D. et al. // J. Biol. Chem. 2010. V. 285. № 50. P. 39249.
Canning P., Rea D., Morty R.E. et al. // PloS One. 2013. V. 8. № 11. P. e79349.
Shan L., Mathews I.I., Khosla C. // Proc. Natl. Acad. Sci. USA. 2005. V. 102. P. 359.
Li M., Chen C., Davies D.R. et al. // J. Biol. Chem. 2010. V. 285. P. 21487.
Szeltner Z., Rea D., Renner V. et al. // J. Biol. Chem. 2003. V. 278. P. 48786.
Kiss A.L., Hornung B., Radi K. et al. // J. Mol. Biol. 2007. V. 368. P. 509.
Gerczei T., Keseru G.M., Naray-Szabo G. // J. Mol. Graph. Model. 2000. V. 18. P. 7.
Morty R.E., Fu P.V., Andrews N.W. // J. Bacteriol. 2002. V. 184. P. 3329.
Mikhailova A.G., Rakitina T.V., Timofeev V.I. et al. // Biochimie. 2017. V. 139. P. 125.
Yang J., Yan R., Roy A. et al. // Nature Methods. 2015. V. 12. P. 7.
Jorgensen W.L., Maxwell D.S., Tirado-Rives J. // J. Am. Chem. Soc. 1996. V. 118. P. 11225.
Abraham Murtola T., Schulz R. et al. // Software X. 2015. V. 1–2 . P. 19.
Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F. et al. // J. Chem. Phys. 1984. V. 81. № 8. P. 3684.
Parrinello M., Rahman A. // J. Chem. Phys. 1982. V. 76. № 5. P. 2662.
Swindells M.B., Laskowski R.A. // J. Chem. Inf. Model. 2011. V. 51. P. 2778.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография