

Кристаллография, 2020, T. 65, № 2, стр. 190-196
О кристаллохимической “гибкости” структурного типа NiAs
Н. Н. Еремин 1, *, А. А. Артамонова 1, Н. Д. Гостищева 1, Е. М. Кочеткова 1, А. А. Межуева 1
1 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: neremin@geol.msu.ru
Поступила в редакцию 31.05.2019
После доработки 29.08.2019
Принята к публикации 29.08.2019
Аннотация
Доказана уникальность структурного типа NiAs. Исследования представительной выборки из более чем 300 кристаллических структур позволили определить поле устойчивости структурного типа NiAs на диаграмме Музера–Пирсона, а также показали, что средний атомный индекс упаковки для данного структурного типа находится в широком интервале, что связано с силой взаимодействий между соседними атомами металла в колонке октаэдров.
ВВЕДЕНИЕ
Толкование важнейшего в кристаллохимии термина “структурный тип” и в наши дни по-прежнему является достаточно неопределенным. Такая ситуация потребовала созыва специальной номенклатурной комиссии Международного союза кристаллографов [1]. Члены этой комиссии постулировали различные степени структурного подобия, причем некоторые из них не требуют полного геометрического равенства сравниваемых кристаллических структур. То есть понятие структурного типа сейчас не связано с жестким условием сохранения одной и той же пространственной группы симметрии, а оперирует более “мягким” условием сохранения близкого кристаллографического и кристаллохимического подобия с сохранением общей топологии (структуры должны иметь одинаковые строительные единицы, образованные координационными полиэдрами близкой формы, одинакового взаимного расположения и сочленения). Наглядным примером современного толкования этого термина является структурный тип перовскита, к которому принадлежат многочисленные неорганические соединения и минералы со стехиометрией АВХ3. Современные базы данных насчитывают в этом структурном типе несколько тысяч природных и синтетических соединений с кубическими, а чаще c псевдокубическими структурами, которые различаются небольшими атомными смещениями в различных направлениях относительно их позиций в кубическом прототипе с сохранением перовскитовой топологии (рис. 1а, 1б).
Рис. 1.
Координационные полиэдры в соединениях структурных типов перовскита и GdFeO3: а – неискаженный кубоктаэдр в кубическом прототипе перовскита, б – искаженный кубоктаэдр BaO12 в тетрагональном BaTiO3 (пр. гр. P4mm), в – 12 ближайших соседей Mg в бриджманите, г – выделяемый MgO8-полиэдр в бриджманите.
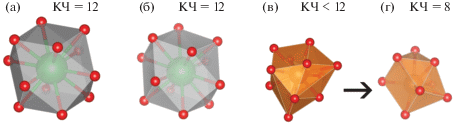
Значительное искажение структуры перовскита, приводящее к изменению ее топологии, характерно, например, для ромбической фазы высокого давления MgSiO3 (пр. гр. Pbnm; минералогическое название бриджманит), являющейся основной фазой нижней мантии Земли. В этом соединении разброс расстояний в первой координационной сфере магния велик: восемь расстояний Mg–О находятся в интервале 2.01–2.41 Å, а следующие четыре отстоят на расстояния 2.85–3.11 Å [2]. В цитируемой работе было продемонстрировано, что метод валентности связи и метод пересекающихся сфер указали, что четыре более удаленных атома кислорода не входят в первую координационную сферу магния; таким образом, координационное число (КЧ) магния равно: 12 – ‒ 4 = 8 (рис. 1в, 1г). Это означает, что бриджманит не может быть отнесен к структурному типу перовскита, а является представителем родственного структурного типа, который выделен в [3] под названием “структурный тип GdFeO3”. Как было отмечено в [4], к этому типу относится достаточно большое число кристаллических структур химических соединений из разных классов, в том числе CaGeO3, LuFeO3, SeCuO3, TeCoO3 и многих других.
Таким образом, к многочисленным статистическим обобщениям распространенности различных структурных типов следует подойти с определенной долей осторожности. С одной стороны, для высокосимметричных структур принадлежность к определенному типу может быть установлена однозначно (так, к структурному типу B1 (NaCl) по состоянию на 2013 г. относят 3555 неорганических соединений, а к структурному типу B2 (CsCl) – 1212). С другой стороны, для структур, содержащих варьируемые параметры (отношения параметров элементарных ячеек или координаты атомов в общих позициях), требуется индивидуальный анализ конкретной структуры на предмет ее принадлежности к тому или иному типу. Считается, что третьим по распространенности структурным типом со стехиометрией AX является B3 (кубический ZnS) и его псевдосимметричные производные (918 структур на 2013 г.). За четвертый ранг по распространенности конкурируют типы B8 (NiAs) (537 структур на тот же период) и B4 – гексагональный ZnS (518 структур), хотя более детальный анализ с учетом сказанного выше может внести в эти цифры определенные коррективы.
ТОПОЛОГИЯ СТРУКТУРНОГО ТИПА NiAs КАК УДОБНАЯ ОСНОВА ДЛЯ СОЗДАНИЯ РЯДОВ ВЫЧИТАНИЯ И ВНЕДРЕНИЯ И ПОЛИКАТИОННЫХ КРИСТАЛЛИЧЕСКИХ СТРУКТУР
При словесном описании известных кристаллических структур и определении новых структурных мотивов достаточно плодотворной оказалась концепция плотнейших анионных упаковок. В 2012 г. Лима Де Фариа [5] сделал подробный обзор базы данных кристаллических структур минералов (его выборка была равна 3248 структурам) и показал, что по крайней мере к 457 структурам (14% от общего количества) абсолютно законно применимо описание с позиций плотнейших упаковок либо плотных (например, ОЦК) упаковок и их производных.
Так, структурный тип галита NaCl описывается как плотнейшая кубическая упаковка (КПУ) анионов с заполнением всех октаэдрических пустот катионами. Тетраэдрические пустоты при этом остаются свободными. Структура B8 (NiAs) является гексагональным аналогом галита – она характеризуется плотнейшей гексагональной упаковкой (ГПУ) ионов мышьяка и заполнением всех октаэдрических пустот ионами Ni2+. В структуре сфалерита ZnS (B3) имеется КПУ ионов серы, в которой половина тетраэдрических пустот занята ионами Zn2+. Вторая модификация ZnS – вюрцит (B4) – характеризуется ГПУ ионов серы и также заполнением половины тетраэдрических пустот ионами Zn2+. Любопытно, что исключение из такого гексагонально-кубического “дуализма” – структурный тип Li2O (КПУ с заполнением всех тетраэдрических пустот), для которого отсутствует гексагональный аналог. В случае высокоионных соединений этот факт легко объяснить – топология ГПУ предусматривает соединение соседних тетраэдрических пустот гранями, что приводит к резкому возрастанию электростатического отталкивания между соседними мелкими катионами при попытке полного заселения пустот этого типа.
Однако если рассмотреть две соседние тетраэдрические пустоты в ГПУ как единый полиэдр (сдвоенный тетраэдр), то выясняется, что его объем достаточен для внедрения еще одного катиона, склонного к октаэдрической координации. Так, в структуре Ni2In (рис. 2) заняты все октаэдрические пустоты. Вторая половина атомов никеля (Ni2) располагается в центре оставшегося вакантным кластера из двух тетраэдрических пустот, соединенных в ГПУ общей гранью. Таким образом, КЧ(Ni2) = 5, а координационный полиэдр Ni2 – тригональная бипирамида. Такие особенности структурных соотношений приводят к тому, что в системах Ni–Те, Ni–Sb, Ni–Sn и Ni–In оказывается возможным непрерывный переход от структуры типа NiAs как к структуре типа Ni2In, так и к структуре типа CdI2 при переменном соотношении количеств компонентов. Если рассматривать структуру NiAs как основную (исходную), то структуру типа Ni2In можно считать “заполненной” по отношению к ней (фазой внедрения), а структуру типа CdI2 “дефектной” (фазой вычитания) (рис. 2).
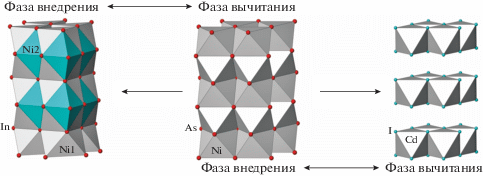
Вторым примером, иллюстрирующим образование такого гомологического ряда на основе структур типа B8, являются пирротины с общей формулой Fe1–xS (дефектные структуры троилита FeS; переход NiAs–CdI2 на рис. 2). Заметим, что собственно бездефектный троилит обнаружен в метеоритном веществе и в окислительно-восстановительных условиях земной поверхности нестабилен. Поэтому земной пирротин Fe1–xS всегда содержит железо в частично окисленном состоянии, его средняя валентность становится больше двух, что приводит к образованию катионных вакансий для соблюдения баланса зарядов и электронейтральности формулы. Очевидно, что чем выше окислительный потенциал среды, в которой происходит рост кристаллов пирротина, тем больше отклонение формулы пирротина от стехиометрического троилита. В этом гомологическом ряду можно выделить целый ряд промежуточных, относительно устойчивых фаз с постепенным понижением симметрии относительно исходного прототипа. Фазы Fe10S11 (91% октаэдров заполнен), Fe9S10 (90% октаэдров заполнено) гексагональные, а клинопирротин Fe7S8 (88% октаэдров заполнено) испытывает моноклинное искажение.
Незаполненные d-оболочки ионов переходных металлов оставляют возможности для валентных взаимодействий металл–металл, которые становятся более эффективными при сближении катионов друг с другом. Поэтому многие переходные металлы в разнообразных соединениях типа АХ (арсениды, антимониды, висмутиды, станниды, сульфиды, селениды, теллуриды) склонны кристаллизоваться именно в структурном типе NiAs, в котором октаэдры вокруг атомов металла имеют общие грани, что невозможно для структур типа NaCl. Более того, в этих случаях наблюдается не укорочение ребер, окаймляющих эти общие грани, а удлинение, что подчеркивает связывающий характер взаимодействий металл–металл. Таким образом, кристаллизация соединений в структурном типе NiAs дополнительно определяется так называемой концентрацией валентных электронов, излишек которых предписывает полиэдрам соединяться гранями для эффективного обеспечения взаимодействия металл–металл. Такие структуры были названы Партэ поликатионными [6]. Образование непрерывных стержней со связями М–М, проходящими через всю структуру вдоль оси c, объясняет магнитные (ферромагнетик) и электрические (проводник) свойства пирротинов. М–М-связи типичны для МХ-соединений переходных металлов с незаполненными электронными оболочками, поэтому они часто предпочитают именно структуру NiAs другим топологиям (например, джайпурит СоS). Кажущимся исключением является алабандин МnS со структурой NaCl, однако этот факт находит логичное объяснение тем, что у Мn2+ наполовину заполненная устойчивая d 5-конфигурация, поэтому соседние атомы Мn не стремятся обобществить свои d-электроны за счет образования связей металл–металл.
ВАРИАТИВНОСТЬ ПОЛОЖЕНИЯ СТРУКТУРНОГО ТИПА NiAs НА СОРТИРОВОЧНОЙ ДИАГРАММЕ МУЗЕРА–ПИРСОНА
С самого начала развития кристаллохимии стало ясно, что вопросом первостепенной важности является введение критериев для проведения границы между родственными структурными типами. Поэтому уже в ранних трудах классиков кристаллохимии появляются различные варианты так называемых структурных сортировочных диаграмм. Еще Гольдшмидт в 1934 г. [7] разделил различные структурные типы по отношению ионных радиусов (rA/rX) и поляризуемости (размеру) аниона. Очевидно, что такая диаграмма может результативно ограничить области стабильности лишь ионных соединений, но не кристаллов с существенно направленным либо металлическим характером химического взаимодействия. В качестве меры направленного характера химической связи между атомами Дехлингером в 1955 г. было предложено использовать среднее главное квантовое число $\tilde {n}$ наружных занятых электронных оболочек атомов, входящих в химическую формулу соединения.
Действительно, по мере увеличения $\tilde {n}$ атомные орбитали, участвующие в образовании связей, и, следовательно, сами связи постепенно теряют свою направленность. Направленность связи зависит и от других характеристик, например от разности электроотрицательностей атомов-партнеров Δχ. По мере возрастания Δχ связи становятся ионными, т.е. ненаправленными, вне зависимости от величины $\tilde {n}$. Поэтому в 1959 г. Музер и Пирсон [8] выделили области стабильности многих структурных типов для двойных соединений с различной стехиометрией в зависимости от среднего главного квантового числа $\tilde {n}$ и разности электроотрицательностей Δχ атомов M и Х. Разберем простейшую стехиометрию MХ. На диаграмме Музера–Пирсона, построенной в координатах $\tilde {n}$–Δχ (рис. 3а), важнейшие структурные типы МХ – СsСl (КЧ 8 : 8), NaCl (КЧ 6 : 6), сфалерита и вюрцита (КЧ 4 : 4) – занимают свои четко различимые поля cтабильности. Правее и выше (большие $\tilde {n}$ и Δχ) находится область стабильности структур типа СsС1с высокими КЧ и большой степенью ионности связи, левее и ниже других (малые $\tilde {n}$ и Δχ) – области стабильности тетраэдрических структур с низкими КЧ (4:4) и более высокой долей ковалентной связи. Широко распространенный структурный тип NaCl занимает промежуточное положение. Заметим, что на этой диаграмме авторы, очевидно, намеренно не привели сектор стабильности структурного типа NiAs, который, в принципе, должен располагаться в области малых Δχ и относительно больших $\tilde {n}$.
Рис. 3.
Диаграмма Музера–Пирсона для соединений МХ: а – поля устойчивости основных структурных типов согласно [8]: тип NaCl (кружки), тип СsСl (квадраты), тетраэдрические структуры типа сфалерита и вюрцита (треугольники); б – положение соединений структурного типа NiAs (заштрихованная область на диаграмме, данные настоящей работы).
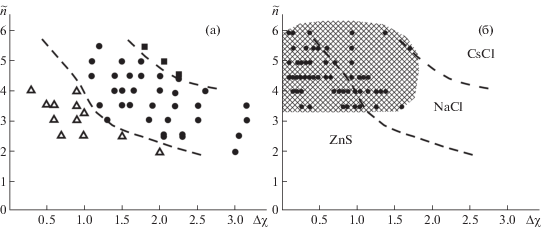
Однако, как отмечалось выше, многие переходные металлы в разнообразных соединениях типа АХ склонны кристаллизоваться именно в этом структурном типе. Важнейшим фактором становится концентрация валентных электронов, равная числу валентных электронов, деленному на число атомов в формуле соединения. При “избытке” электронов для заполнения анионной оболочки возможно образование связей катион–катион. Поэтому, как видно из проведенного анализа (рис. 3б), область стабильности этого структурного типа в координатах $\tilde {n}$–Δχ оказалась до статочно широкой и пересекающейся с другими. Этот факт вынудил авторов [8] выделить отдельную диаграмму МХ для переходных металлов. Такое специфическое поведение соединений структурного типа B8 необходимо учитывать при использовании подобной сортировочной диаграммы для предсказания возможной кристаллической структуры MX.
“ПОДВИЖНОСТЬ” СТРУКТУРНОГО ТИПА NiAs НА ДИАГРАММЕ ИНДЕКСА УПАКОВКИ
Для понимания основных структурных преобразований при изменении термодинамических параметров, в частности изменения давления, анализируют различные варианты диаграмм плотности упаковки. Напомним, что общей закономерностью полиморфных переходов при повышении давления является возрастание плотности, обусловленное увеличением КЧ. Однако кристаллохимический анализ с таких простых позиций не всегда возможен, так как первое КЧ, особенно для низкосимметричных структур, не всегда является корректной количественной характеристикой относительной плотности структуры, позволяющей правильно указать направление полиморфных переходов. Более правильной, чем первое КЧ, характеристикой оказался упаковочный коэффициент, который определяет долю занятого атомами кристаллического пространства. Эту долю легко найти, когда атомы одного размера (в случае металлов, ковалентных кристаллов простых веществ). Тогда отношение объема сферических атомов к полному объему ячейки есть не что иное, как коэффициент компактности структуры γ, равный 0.74 для всех плотнейших шаровых упаковок (КЧ = 12). Для ОЦК-структур (КЧ = 8) γ понижается до 0.68, для простой кубической структуры (КЧ = 6) γ = 0.52, а для структуры типа алмаза (КЧ = 4) γ равно лишь 0.34 (например, [9]).
В случае бинарных соединений величины γ могут варьироваться в достаточно широких пределах в зависимости от соотношения радиусов [10], а в случае более сложных стехиометрических соотношений (или низкосимметричных структур) определение величины γ затруднено либо вовсе невозможно. По этой причине в [11–13] был введен более объективный индекс упаковки α, называемый также критерием Лиу компактности гетероатомной структуры:
Здесь V – мольный объем, а R – ближайшее расстояние катион–анион. Удобно выражать мольный объем в см3/моль, а расстояние – в Å. Тогда значение α изменяется от 0.43 (в случае наиболее плотно упакованных металлов с ГЦК-решеткой или ГПУ) до 17.16 (в случае очень рыхлой молекулярной структуры “сухого льда” СО2) [13, 14]. Очевидно, что индекс упаковки α является функцией не только КЧ, но в общем случае также функцией соотношения периодов решетки, угла между осями и координат атомов в структуре. Систематический анализ индексов упаковки, проведенный в [13] для простых структурных типов неорганических веществ, показал, что на диаграммах в координатах V–R3 соединения, относящиеся к одному структурному типу, располагаются строго на одной линии.Однако непосредственное сравнение различных типов соединений с помощью индекса упаковки также затруднительно. Например, α для алмаза составляет 0.93, а для родственной ему структуры сфалерита – 1.85. Поэтому, как было предложено в [14], более правильно использовать для сравнения структурных типов средний атомный индекс упаковки $\bar {\alpha }$:
где т – число атомов в формуле соединения. Тогда для алмаза и сфалерита $\bar {\alpha }$ = 0.93. На рис. 4а в виде прямых линий приведены значения $\bar {\alpha }$ (определяемые углом наклона прямых в координатах V/m–R3) для некоторых распространенных структурных типов. Как видно из рисунка, в этих координатах все изоструктурные соединения действительно ложатся на равнонаклонную линию, соответствующую конкретному значению $\bar {\alpha }$ (табл. 1).Рис. 4.
Средний атомный индекс упаковки $\bar {\alpha }$, определяемый как угол наклона прямых в координатах V/m–R3, согласно [14] (а): A1 – структуры с ГЦК-ячейкой, А4 – алмаз, А7 – мышьяк, B1 – NaCl, B2 – CsCl, В3 – сфалерит, 1 – рутилоподобные структуры, 2 – α-РbО2, 3 – бадделиит, 4 – флюорит, 5 – коттунит, Q – кварц. Результаты ревизии диаграммы, проведенной в настоящей работе (б): 1 – кварц, 2 – сфалерит/вюрцит/алмаз, 3 – рутилоподобные структуры, 4 – мышьяк, 5 – флюорит, 6 – NaCl, 7 – α-РbО2, 8 – CsCl/α-вольфрам, 9 – магний, 10 – Ni2In, 11 – медь, 12 – MgCu2, пунктирная линия – NiAs.
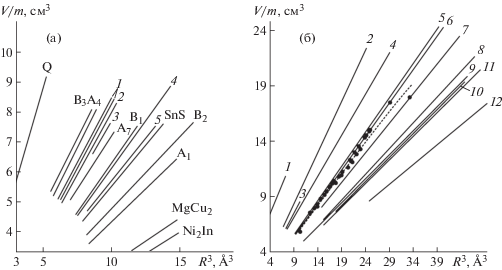
Таблица 1.
Средние индексы упаковки $\bar {\alpha }$ ряда структурных типов, представленных на рис. 4б
| Структурный тип | $\bar {\alpha }$ | Структуры в выборке |
|---|---|---|
| α-кварц SiO2 | 1.761 | 2 |
| Сфалерит–вюрцит–алмаз | 0.927 | 83 |
| TiO2 (рутил и его производные) | 0.829 | 14 |
| As | 0.753 | 4 |
| CaF2 | 0.619 | 33 |
| NaCl | 0.602 | 59 |
| NiAs | 0.602–0.532 | 57 |
| α-PbO2 | 0.536 | 4 |
| CsCl–α-W | 0.462 | 22 |
| Mg | 0.444 | 13 |
| Ni2In | 0.436 | 10 |
| Cu | 0.426 | 14 |
| Фазы Лавеса (на примере MgCu2) | 0.352 | 5 |
Проведенный статистический анализ более чем 300 кристаллических структур из представленных на рис. 4а типов показал следующее. В отличие от данных [14] структурный тип MgCu2 оказался более “плотноупакованным”, чем Ni2In. Это неудивительно, так как расположение атомов в очень плотных и компактных структурах фаз Лавеса характеризуется высокими КЧ – от 12 до 16. Такие высокие координации обеспечивает определенное соотношение радиусов атомов в структурах этих фаз (идеальное соотношение, при котором объем атомов различается ровно в 2 раза, равно 1.225 [15]). Таким образом, на графике из [14], очевидно, просто присутствует ошибка. Обратим также внимание на поведение структурного типа NiAs, который вообще отсутствует на оригинальном графике в [14]. В отличие от других структурных типов средний индекс упаковки в данном случае может меняться значительно: $\bar {\alpha }$(NiAs) охватывает весь диапазон значений от структурного типа NaCl (0.602) до структурного типа α-PbO2 (0.536). Изменение индекса упаковки в данном случае отражает усиление или ослабление взаимодействий металл–металл в колонке октаэдров. Как видно из рис. 5 и табл. 2, увеличение угла α1 приводит к укорочению расстояний металл–металл, увеличению площади общей грани октаэдров, уплощению самого октаэдра и поэтому к заметному уменьшению величины $\bar {\alpha }$.
Рис. 5.
Механизм повышения плотности упаковки в соединениях структурного типа NiAs путем уплощения октаэдров.
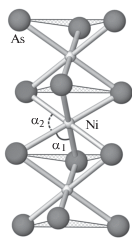
Таблица 2.
Изменение компактности соединений структурного типа NiAs в зависимости от сжатия октаэдров MX6
| Соеди-нение | V/m | R3 | $\bar {\alpha }$ | α1 | α2 |
|---|---|---|---|---|---|
| MgPo | 17.42 | 28.93 | 0.602 | 90.1 | 89.9 |
| MnTe | 15.04 | 24.97 | 0.602 | 90.08 | 89.92 |
| ScTe | 15.01 | 24.92 | 0.602 | 89.97 | 90.03 |
| HfPo | 14.42 | 23.96 | 0.602 | 89.98 | 90.52 |
| ZrPo | 14.63 | 24.34 | 0.601 | 91.87 | 88.13 |
| RhBi | 12.34 | 20.86 | 0.592 | 95.85 | 84.15 |
| NiSb | 10.32 | 17.69 | 0.583 | 97.9 | 82.1 |
| FeSn | 12.21 | 21.32 | 0.573 | 99.68 | 80.32 |
| BaPt | 18.07 | 33.35 | 0.542 | 103.54 | 76.45 |
ЗАКЛЮЧЕНИЕ
В результате проведенного исследования представительной выборки из более чем 300 кристаллических структур с различной стехиометрией можно сказать следующее. Уникальность топологии структурного типа B8 дает возможность образования многочисленных структурно-гомологических рядов, построенных по принципу структур вычитания (в предельном случае CdI2) и внедрения (в предельном случае Ni2In), что часто реализуется в системах Ni–Те, Ni–Sb и ряде других. Поле устойчивости структурного типа B8 на диаграмме Музера–Пирсона тяготеет к большим значениям $\tilde {n}$ и малым величинам Δχ, однако разнообразие возможных поликатионных взаимодействий среди его конкретных структурных представителей приводит к пересечению его поля устойчивости как с полем “тетраэдрических” структур типа сфалерита–вюрцита, так и с полем “октаэдрических” структур типа B1. Средний атомный индекс упаковки для структурного типа B8 может меняться в интервале 0.60–0.53, что регулируется силой взаимодействий между соседними атомами металлов в колонке октаэдров. Все это обеспечивает значительную уникальность данного кристаллохимически “гуттаперчевого” и многообразного структурного типа.
Список литературы
Lima-de-Faria J., Hellner E., Liebau F. et al. // Acta Cryst. A. 1990. V. 46. P. 1.
Урусов В.С. // Кристаллография. 2009. Т. 54. № 5. С. 795.
Ниггли П. Стереохимия / Ред. Бокий Г.Б. М.: Изд-во иностр. лит-ры, 1949. 359 с.
Sasaki S., Prewitt Ch.T., Liebermann R.C. // Am. Mineral. 1983. V. 68. P. 1189.
Lima-de-Faria J. // Eur. J. Mineral. 2012. V. 24. P. 163.
Parthé E. // Acta Cryst. V. 1973. V. 29. P. 2808.
Голдшмидт В.М. Кристаллохимия. Л.: ОНТИ, 1937. 60 с.
Mooser E., Pearson W.B. // Acta Cryst. 1959. V. 12. P. 1015.
Урусов В.С., Еремин Н.Н. Кристаллохимия. Краткий курс. М.: МГУ, 2010. 256 с.
Ворошилов Ю.В., Павлишин В.И. Основы кристаллографии и кристаллохимии. Рентгенография кристаллов Киев: КНТ, 2011. 568 с.
Альтшуллер Л.В., Подурец М.А., Симаков Г.В., Трунин Р.Ф. // ФТТ. 1973. Т. 15. № 5. С. 1430.
Liu L. // Phys. Earth Planet. Inter. 1974. V. 9. № 5. P. 338.
Liu L. // High Temperatures – High Pressures. 1981. V. 13. P. 387.
Урусов В.С., Пущаровский Д.Ю. // Минералогический журнал. 1984. Т. 6. № 3. С. 23.
Пирсон У. Кристаллохимия и физика металлов и сплавов. Т. 2. М.: Мир, 1977. 472 с.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография