

Кристаллография, 2020, T. 65, № 2, стр. 234-243
Нитрило-трис-метиленфосфонато-манганат(ii) тетранатрия Na4[MnII{N(CH2PO3)3}] · 13H2O: синтез и некоторые структурные особенности
Н. В. Сомов 1, *, Ф. Ф. Чаусов 2, **, Н. В. Ломова 2, А. Н. Бельтюков 2, В. Г. Петров 2, М. А. Шумилова 2, Д. К. Жиров 2
1 Нижегородский государственный университет им. Н.Н. Лобачевского
Нижний, Новгород, Россия
2 Удмуртский федеральный исследовательский центр УрО РАН
Ижевск, Россия
* E-mail: somov@phys.unn.ru
** E-mail: chaus@yandex.ru
Поступила в редакцию 01.04.2019
После доработки 01.04.2019
Принята к публикации 17.04.2019
Аннотация
Синтезирована и исследована натриевая соль комплекса нитрило-трис-метиленфосфоновой кислоты (NTP) с марганцем (II); Na4[MnII{N(CH2PO3)3}] · 13H2O, пр. гр. P1$\bar {1}$, Z = 2, a = 11.2146(4), b = 11.2687(4), c = 12.4625(4) Å, α = 108.619(3)°, β = 97.149(3)°, γ = 116.991(4)°. Комплекс биядерный с хелатным строением; координационный полиэдр Mn – искаженная тригональная бипирамида, в основании которой лежат три атома O разных групп PO3 молекулы NTP, в одной вершине – атом азота NTP, в противоположной вершине – атом O соседней молекулы NTP. Кристаллическая упаковка островная, полиэдры Mn соединены попарно ребрами и окружены пространственной вязью полиэдров Na и молекул воды.
ВВЕДЕНИЕ
Строение и свойства координационных соединений марганца(II) имеют ряд характерных особенностей, обусловленных электронной конфигурацией 3d5. В высокоспиновом состоянии с атомным термом 6S энергия экстрастабилизации равна нулю при любой симметрии кристаллического поля, вследствие чего конфигурация координационного окружения в основном определяется геометрией кристаллической упаковки и стереохимическими свойствами лиганда. Преобладающий тип координационного полиэдра Mn(II) – октаэдр, что объясняется подходящим отношением кристаллохимических радиусов [1]: rcr(Mn2+) = 0.89, rcr(O2–) = 1.21 Å. Значительный ионный радиус ri(Mn2+) = 0.75 Å [1] и преимущественно ионный характер координационной связи обусловливают возможность координационного числа (КЧ) от 5 до 7 [2].
Одним из первых изученных координационных соединений Mn был комплекс с порфирином [3]. Впоследствии было описано большое количество аналогичных соединений [4], биологическая активность которых обусловлена окислительно-восстановительными свойствами системы Mn+2/Mn+3. Описаны марганецсодержащие ферменты с различной координацией атома Mn, например супероксид-дисмутаза (MnSOD) [5] с тригонально-бипирамидальной координацией и 3-дезокси-D-арабино-гептулозонат-7-фосфат-синтаза (DAHPS) [6], а также изопентенил-дифосфат-изомераза [7, 8] – с октаэдрической. Детально изучены летучие комплексы Mn(II) с макроциклическими лигандами, например 18-краун-6 [9, 10].
Тем не менее в классической координационной химии марганца остается немало “белых пятен”. Обзор [11] включает в себя большое разнообразие координационных соединений марганца с различной координацией, однако в поздней монографии [12] отмечено, что “бóльшая часть этого пребывает в затишье по отношению к химии, которая появилась с тех пор”.
Интересны кристаллохимические аспекты координации Mn(II) триподальными лигандами. Например, в комплексах с трис(2-бензимидазолилметил)амином (ntb) реализуются все три возможности для КЧ от 5 до 7 [13]: КЧ = 5 (координационный полиэдр – искаженная тригональная бипирамида) в комплексе [MnII(ntb)Cl]Cl · · 2.5MeOH, КЧ = 6 (координационный полиэдр – сильно искаженная тригональная бипирамида с дополнительной “шапочной” вершиной) в комплексе [MnII(ntb)(OAc)]Cl · 0.5MeOH · 3.25H2O и КЧ = 7 (координационный полиэдр – сильно искаженный октаэдр с дополнительной “шапочной” вершиной) в комплексе [MnII(ntb)(NO2)2] · · 4MeOH.
Распространенным триподальным лигандом является нитрило-трис-метиленфосфоновая кислота N(CH2PO3)3H6 (NTP), присутствие в которой PO3-групп обусловливает разнообразие мотивов связывания с металлами [14]. Ее комплексы с металлами являются объектами пристального исследования как за рубежом [15–17], так и в России [18–30]. Cоли и комплексы NTP с металлами широко применяются в технике в качестве ингибиторов отложений солей [31] и коррозии [32–34]. Поэтому синтез и исследование производных NTP представляет значительный прикладной интерес для расширения ассортимента потенциальных ингибиторов.
Ранее при взаимодействии ацетата марганца(II) с NTP при 200°С [35] были получены два комплекса NTP с Mn(II) – [Mn{NH(CH2PO3H)3}(H2O)3] и [Mn{NH(CH2PO3H)3}], представляющие собой нерастворимые в воде бесцветные кристаллы.
В настоящей работе описан синтез и некоторые особенности кристаллической, молекулярной и электронной структуры нитрило-трис-метиленфосфонато-манганата(II) тетранатрия Na4[MnII{N(CH2PO3)3}] · 13H2O (MnNaNTP).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез Na4[MnII{N(CH2PO3)3}] · 13H2O. К раствору 3.00 г (0.01 моль) NTP (дважды перекристаллизованной, менее 0.3% PO$_{4}^{{3 - }}$) в 400 см3 дистиллированной воды при 70–80°С при постоянном интенсивном перемешивании очень медленно, по каплям, добавили раствор 2.45 г (0.01 моль) MnSO4 · 5H2O (ЧДА, ГОСТ 435-77) в 100 см3 дистиллированной воды. Выпавший осадок отделили фильтрованием, промыли дистиллированной водой до нейтральной реакции, затем 95%-ным этанолом, диэтиловым эфиром и высушили на воздухе. Продукт идентифицирован методом рентгеноструктурного анализа (РСА) как ранее описанный [35] комплекс [Mn{NH(CH2PO3H)3}(H2O)3] (CCDC MOYBOR.cif). Выход 3.61 г (89%).
К полученному MnNTP добавили раствор 1.44 г (0.036 моль) NaOH (ХЧ, ГОСТ 4328-77) в 50 см3 дистиллированной воды и нагрели при постоянном перемешивании до 40–50°С. После растворения осадка жидкость профильтровали. При медленном испарении воды при комнатной температуре образовались прозрачные триклинные кристаллы MnNaNTP, которые отделили, промыли водой с добавлением 45%-ного этанола, затем 95%-ным этанолом, диэтиловым эфиром и высушили на воздухе. Выход 4.92 г (82%).
Рентгеноструктурный анализ. Кристаллографические характеристики, параметры рентгеноструктурных экспериментов и результаты уточнения структуры приведены в табл. 1. Первичный фрагмент структуры MnNaNTP был найден прямыми методами. Положения остальных атомов, включая атомы водорода, определены из разностного синтеза электронной плотности. Параметры всех атомов, кроме атомов водорода, уточняли в анизотропном приближении тепловых смещений методом наименьших квадратов (МНК) по |F|2. Уточнение параметров атомов водорода проходило в изотропном приближении в общем цикле МНК без ограничений. Результаты РСА депонированы в Кембриджский банк структурных данных (CCDC № 1896773).
Таблица 1.
Кристаллографические характеристики, данные эксперимента и результаты уточнения структуры
| Формула | Na4[MnII{N(CH2PO3)3}] · · 13H2O |
|---|---|
| M | 674.1 |
| Сингония, пр. гр., Z | Триклинная, P$\bar {1}$, 2 |
| a, b, c, Å | 11.2146(4), 11.2687(4), 12.4625(4) |
| α, β, γ, град | 108.619(3), 97.149(3), 116.991(4) |
| V, Å3 | 1260.47(9) |
| Dx, г/см3 | 1.776 |
| Излучение; λ, Å | MoKα; 0.71073 |
| μ, мм–1 | 0.873 |
| T, K | 293(2) |
| Размеры образца, мм | 0.163 × 0.13 × 0.075 |
| Дифрактометр | Oxford Diffraction Gemini S, Sapphire III |
| Тип сканирования | ω |
| Учет поглощения, Tmin/Tmax | Аналитически [36], 0.965/0.984 |
| θmin, θmax, град | 3.578, 31.468 |
| Пределы h, k, l | –16 ≤ h ≤ 16, –16 ≤ k ≤ 16 –17 ≤ l ≤ 17 |
| Число отражений: измеренных/независимых (N1)/RInt/с I > 2σ(I) (N2) | 24 974/7665/0.0329/6565 |
| Метод уточнения | Полноматричный МНК по F 2 |
| Число параметров | 435 |
| S | 1.036 |
| R1/wR1 по N1 | 0.0368/0.0747 |
| R1/wR1 по N2 | 0.0288/0.0703 |
| Δρmin/Δρmax, э/Å3 | –0.348/0.427 |
| Программы | CrysAlisPro [37], SHELX2014 [38], WinGX [39], VESTA3.0 [40] |
Рентгеновские фотоэлектронные спектры MnNaNTP получали на отечественном рентгеновском фотоэлектронном спектрометре ЭМС-3 (УдмФИЦ УрО РАН) [41] с магнитным энергоанализатором при возбуждении AlKα-излучением (hν = 1486.6 эВ). Раствор MnNaNTP (около 10%) в дистиллированной воде наносили на подложку из пиролитического графита, которую закрепляли в молибденовом держателе образца. Остаточное давление в рабочей камере составляло 10–5 Па. Калибровку энергоанализатора проводили по центру тяжести линии C1s (энергия связи Eсв = = 284.5 эВ). Коррекцию фона и неупругого рассеяния проводили методом Ширли [42], для статистической обработки полученных спектров использовали программу Fityk 0.9.8 [43].
Электронные спектры поглощения монокристалла MnNaNTP получали при помощи спектрометрического комплекса на базе монохроматора МДР-41, используя источник излучения на базе галогеновой лампы, в интервале 350–1000 нм.
ИК-спектры MnNaNTP и продуктов его термического разложения получали на ИК-фурье-спектрометре ФСМ-1201 в интервале 450–5000 см–1, прессуя таблетки, содержащие 1 мг вещества в 250 мг KBr.
Термогравиметрический анализ MnNaNTP проводили в атмосфере аргона на автоматизированном дериватографе Shimadzu DTG-60H в интервале 30–500°С при скорости нагрева 3 град/мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Структура кристалла MnNaNTP изображена на рис. 1. Межатомные расстояния и валентные углы приведены в табл. 2. Молекула NTP полностью депротонирована, включая атом N, который образует координационную связь с Mn. Координационный полиэдр Mn представляет собой искаженную тригональную бипирамиду, в основании которой находятся атомы O1, O6 и O7 молекулы лиганда. В одной вершине тригональной бипирамиды расположен атом азота молекулы NTP, в противоположной вершине – атом O1i. Геометрический параметр тригональности τ [44] для координационного полиэдра Mn составляет 0.56; степени подобия эталонным многогранникам [22, 45] приведены в табл. 3.
Рис. 1.
Кристаллическая структура MnNaNTP: координационные полиэдры марганца темные, натрия – светлые. Симметрично-эквивалентные позиции: (i) –x, –y, –z; (ii) –x, –y + 1, –z; (iii) –x + 1, –y, –z + 1.
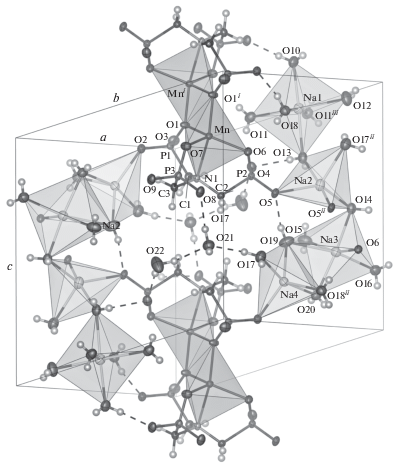
Таблица 2.
Межатомные расстояния d и валентные углы ω в структуре Na4[MnII{N(CH2PO3)3}] · 13H2O
| Связь | d, Å | Связь | d, Å | Связь | d, Å |
|---|---|---|---|---|---|
| Mn1–N1 | 2.3405(11) | N1–C3 | 1.4853(17) | P2–O4 | 1.5200(11) |
| Mn1–O1 | 2.1625(10) | C1–P1 | 1.8179(14) | P2–O5 | 1.5112(10) |
| Mn1–O6 | 2.0983(10) | C2–P2 | 1.8228(14) | P2–O6M | 1.5448(10) |
| Mn1–O7 | 2.0709(10) | C3–P3 | 1.8260(14) | P3–O7M | 1.5409(11) |
| Mn1–O1i | 2.0760(10) | P1–O1M | 1.5538(10) | P3–O8 | 1.5151(11) |
| N1–C1 | 1.4840(16) | P1–O2 | 1.5115(11) | P3–O9 | 1.5124(10) |
| N1–C2 | 1.4879(17) | P1–O3 | 1.5133(11) | Na1–O | 2.3404(15)–2.5092(15) |
| Na2–O | 2.3493(12)–2.4356(14) | Na3–O | 2.3470(14)–2.5941(17) | Na4–O | 2.2909(12)–2.4260(15) |
| Угол | ω, град | Угол | ω, град | Угол | ω, град |
| N1–Mn1–O1i | 161.65(4) | O1–P1–C1 | 102.06(6) | O1–P1–O2 | 111.05(6) |
| N1–Mn1–O1 | 79.67(4) | O2–P1–C1 | 110.38(6) | O1–P1–O3 | 112.20(6) |
| N1–Mn1–O6 | 84.65(4) | O3–P1–C1 | 107.15(6) | O2–P1–O3 | 113.33(7) |
| N1–Mn1–O7 | 84.55(4) | O4–P2–C2 | 107.97(6) | O4–P2–O5 | 113.60(6) |
| O1–Mn1–O6 | 127.83(4) | O5–P2–C2 | 108.01(6) | O4–P2–O6 | 110.26(6) |
| O1–Mn1–O7 | 113.01(8) | O6–P2–C2 | 104.24(6) | O5–P2–O6 | 112.21(6) |
| O6–Mn1–O7 | 114.56(4) | O7–P3–C3 | 104.30(6) | O7–P3–O8 | 111.24(6) |
| O1–Mn1–O1i | 82.02(4) | O8–P3–C3 | 108.86(7) | O7–P3–O9 | 111.87(6) |
| O–Na1–O | 169.87(6)–175.11(6); 84.37(6)–99.62(6) |
O9–P3–C3 | 107.45(6) | O8–P3–O9 | 112.66(6) |
| O–Na2–O | 170.16(5); 102.04(5)–135.83(5); 77.51(5)–97.13(5) |
O–Na3–O | 160.35(5)–171.00(6); 63.19(5)–102.80(5) |
O–Na4–O | 102.40(5)–162.17(5); 80.73(5)–95.41(5) |
Таблица 3.
Степень подобия Ф [22, 45] координационных полиэдров атомов металла в структуре Na4[MnII{N(CH2PO3)3}] · 13H2O эталонным многогранникам
| Атом | КЧ | Кубический тетраэдр | Тригональная бипирамида | Тетрагональная пирамида | Кубический октаэдр |
|---|---|---|---|---|---|
| Mn1 | 4 | 0.35660(9) | |||
| Mn1* | 5 | 0.25052(7) | 0.16946(5) | ||
| Na1 | 6 | 0.4662(2) | |||
| Na2 | 5 | 0.3681(4) | 0.2450(3) | ||
| Na3 | 6 | 0.2539(2) | |||
| Na4 | 5 | 0.38768(17) | 0.26924(12) |
Координационные полиэдры атомов Mn соединены друг с другом ребрами O1–O1i и образуют четырехчленный цикл Mn1–O1–Mn1i–O1i. Этот цикл плоский вследствие наличия центра симметрии. Различие между расстояниями Mn1–O1 и Mn1–O1i составляет 4.2%, так что цикл представляет собой слегка искаженный ромб.
При образовании комплекса замыкаются три пятичленных хелатных цикла N–Mn–O–P–C с общей связью N–Mn. Торсионный угол ψ(N1–Mn1–O1–P1) = 3.57(3)°, следовательно, расположение указанных атомов приближается к копланарному – к плоскости цикла Mn1–O1–Mn1i–O1i. Напротив, торсионные углы ψ(N1–Mn1–O6–P2) = 21.37(3)° и ψ(N1–Mn1–O7–P3) = = 13.88(3)° указывают на существенное отклонение двух других циклов N–Mn–O–P–C от плоскости.
Валентные углы при атоме азота (109.4(25)°) близки к углу в тетраэдре. Валентные углы при атомах фосфора (109.4(33)°) также близки к углу в тетраэдре; заметное отклонение (102.06(11)°–104.30(11)°) наблюдается лишь для атомов кислорода, участвующих в координации Mn. Расстояния P–O (среднее 1.525(11) Å) существенно различаются для атомов кислорода, участвующих в координации Mn (среднее 1.514(3) Å) и не связанных с Mn (среднее 1.547(3) Å). Наблюдаемая разница (средняя 0.033(6) Å) объясняется заметным участием электронной плотности кислорода в связи с Mn, она максимальна (0.039(4) Å) для атома O1, связанного с двумя атомами Mn.
Во внешней координационной сфере находятся ионы натрия (четыре иона Na+ на каждую структурную единицу, или восемь ионов Na+ на элементарную ячейку) и молекулы кристаллизационной воды (13 молекул H2O на каждую структурную единицу, или 26 молекул H2O на элементарную ячейку). Координационные полиэдры ионов Na1 и Na3 представляют собой искаженные октаэдры, ионы Na2 и Na4 координированы в конфигурации искаженных тригональных бипирамид (табл. 3). Полиэдры Na1 соседних структурных единиц комплекса соединены общим ребром O11–O11iii; полиэдры Na1–Na2–Na3 – общими вершинами O13 и O14, а полиэдры Na3 и Na4 – общим ребром O19–O20. Из 13 молекул кристаллизационной воды 11 координируют ионы Na+, а две являются сольватными. В кристаллической упаковке комплекса MnNaNTP присутствуют водородные связи (табл. 4).
Таблица 4.
Водородные связи в структуре Na4[MnII{N(CH2PO3)3}] · 13H2O (D – донор, А – акцептор)
| Связь | D–H, Å | H···A, Å | D···A, Å | D–H···A, град |
|---|---|---|---|---|
| O13–H7···O4 | 0.67(2) | 2.04(2) | 2.707(2) | 176(3) |
| O13–H8···O9 | 0.83(2) | 1.97(2) | 2.798(2) | 172(2) |
| O14–H9···O8 | 0.89(2) | 1.87(2) | 2.741(2) | 168(2) |
| O14–H10···O8 | 0.70(2) | 2.13(2) | 2.812(2) | 165(2) |
| O16–H11···O7 | 0.77(3) | 2.12(3) | 2.861(2) | 161(3) |
| O16–H12···O3 | 0.80(3) | 1.91(3) | 2.695(2) | 171(2) |
| O11–H13···O4 | 0.80(3) | 2.11(3) | 2.869(9) | 158(3) |
| O11–H14vO7 | 0.75(3) | 2.21(3) | 2.950(2) | 170(3) |
| O18–H15···O6 | 0.67(3) | 2.27(3) | 2.921(2) | 167(3) |
| O18–H16···O2 | 0.83(3) | 2.10(3) | 2.867(2) | 153(2) |
| O20–H17···O2 | 0.67(2) | 2.20(2) | 2.848(2) | 165(3) |
| O20–H18···O10 | 0.80(3) | 1.96(3) | 2.755(2) | 172(2) |
| O15–H19···O9 | 0.79(3) | 2.01(3) | 2.792(2) | 173(3) |
| O15–H20···O22 | 0.76(3) | 2.12(3) | 2.844(2) | 157(3) |
| O10–H21···O9 | 0.81(3) | 1.90(3) | 2.705(2) | 170(2) |
| O10–H22···O22 | 0.76(3) | 2.06(3) | 2.786(2) | 159(3) |
| O12–H23···O3 | 0.72(3) | 2.36(3) | 3.036(2) | 156(3) |
| O12–H24···O3 | 0.84(3) | 2.00(3) | 2.842(2) | 173(2) |
| O22–H25···O21 | 0.87(3) | 1.93(3) | 2.786(2) | 167(3) |
| O22–H26···O4 | 0.72(3) | 2.03(3) | 2.745(2) | 173(3) |
| O17–H27···O12 | 0.69(3) | 2.32(3) | 2.958(2) | 156(3) |
| O17–H28···O21 | 0.85(3) | 1.98(3) | 2.786(2) | 158(2) |
| O19–H29···O5 | 0.79(3) | 1.97(3) | 2.745(2) | 166(3) |
| O19–H30···O15 | 0.70(3) | 2.06(3) | 2.707(2) | 153(3) |
| O21–H31···O8 | 0.84(3) | 1.90(3) | 2.739(2) | 175(3) |
| O21–H32···O16 | 0.77(3) | 2.02(3) | 2.778(2) | 166(2) |
В целом структура комплекса MnNaNTP близка к структурам ранее описанных комплексов, содержащих в качестве комплексообразователя атом Zn [18], Cu [19] и Co [21]. Однако рассматриваемая структура резко отличается от структуры комплекса Ni [20], в которой координационный полиэдр атома Ni достроен до октаэдра присоединенной молекулой воды. Как было показано в [46], отличие структуры комплекса Ni от других членов ряда Na4[MII{N(CH2PO3)3}] · nH2O (MII = = Co, Ni, Cu, Zn) объясняется в рамках теории кристаллического поля бóльшей энергией экстрастабилизации иона Ni2+ в октаэдрическом поле по сравнению с тригонально-бипирамидальным. С точки зрения теории кристаллического поля ион Mn2+ с конфигурацией 3d5 не имеет выраженных стереохимических предпочтений; сходство структуры MnNaNTP со структурой комплекса Zn2+ (3d10) Na4[Zn{N(CH2PO3)3}] · 13H2O [18] вполне закономерно.
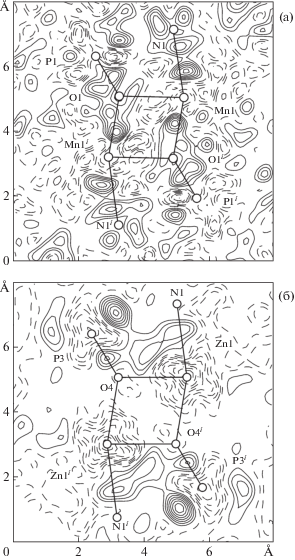
Однако распределение электронной плотности в структуре MnNaNTP (рис. 2а) резко отличается от аналогичного комплекса Zn (рис. 2б). Рельеф разностной электронной плотности в окрестности атомов Mn характеризуется глубоким минимумом вблизи центра атома Mn1, а также резко выраженными локальными максимумами на связях с атомами азота Mn1–N1 и Mn1i–N1i и атомами кислорода Mn1–O1i и Mn1i–O1. Атом марганца связан с атомом кислорода соседней структурной единицы прочнее (расстояние Mn1–O1i = 2.0760(10) Å), чем с атомом кислорода “своей” молекулы лиганда (Mn1–O1 = 2.1625(10) Å). В комплексе с цинком, напротив, образуются широкие области повышенной электронной плотности, охватывающие связи Zn1–N1, Zn1–O4 и O4–P3 (и соответственно Zn1i–N1i, Zn1i–O4i, O4i–P3i).
Рис. 2.
Карты разностной электронной плотности ρexp–ρcalc в плоскости цикла M–O–M–O комплексов MnNaNTP (а) и аналогичного соединения с Zn [18] (б). Шаг изолиний 0.05 э/Å3; изолинии отрицательной плотности – штриховые, положительной – сплошные.
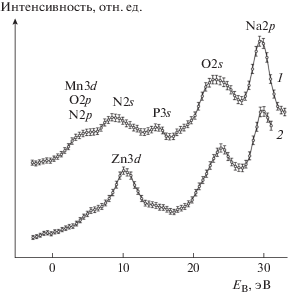
Различия в природе координационной связи объясняются существенными различиями в энергетическом спектре атомов Mn и Zn, которые проявляются в рентгеновском фотоэлектронном спектре валентной полосы комплексов (рис. 3). Для комплекса Mn характерно положение Mn3d-состояний вблизи уровня Ферми, что создает благоприятные условия для перекрывания Mn3d-орбиталей с O2p- и N2p-орбиталями. Вместе с тем валентные орбитали атома Mn незначительно перекрываются с локализованными N2s- и P3s-орбиталями. В комплексе с Zn максимум интенсивности Zn3d-полосы спектра соответствует энергии связи EB = 10.2 эВ. Вследствие этого возможно перекрывание Zn3d-орбиталей с N2s- и в меньшей степени с P3s-орбиталями.
Рис. 3.
Рентгеновские фотоэлектронные спектры валентной полосы комплексов MnNaNTP (1) и аналогичного соединения с Zn [18] (2). Засечками показаны 95%-ные доверительные интервалы для измеренных значений интенсивности.
Спектр Mn3s представлен дублетом с EB = 82.4 и 87.1 эВ (расщепление ΔMn3s = 4.7 эВ), что согласуется с данными [47] о мультиплетном расщеплении линии Mn3s в комплексах Mn(II). Соотношение интегральных интенсивностей линий дублета I2/I1 составляет 0.63. Оценка спинового магнитного момента атома Mn в магнетонах Бора μB по формуле μMn = 2μB[S(S + 1)]1/2 (где S – спиновое число неспаренных электронов, которое удовлетворяет соотношению S/(S + 1) = I2/I1) [48–51], приводит к значению μMn = 4.29μB. Положение дублета Mn2p3/2–Mn2p1/2, возникающего вследствие спин-орбитального расщепления, при EB = 641.8 и 652.9 эВ (ΔMn2p = 11.1 эВ) также характерно для координационных соединений Mn(II) [47].
Спектр P2p-состояний включает в себя одну неразрешенную составляющую с максимумом при EB = 132.8 эВ с шириной на половине высоты 1.5 эВ, что свидетельствует об эквивалентности положений атомов фосфора в структуре MnNaNTP и согласуется с результатами РСА. Спектр N1s имеет максимум при EB = 398.9 эВ, что свидетельствует об образовании координационной связи Mn–N; значения EB = 398.7–399.2 эВ характерны для N1s-состояний в N-координированных металлокомплексах [52–54].
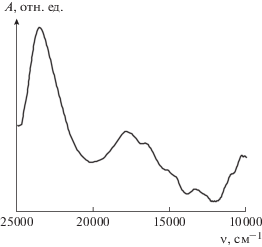
Описанная структура MnNaNTP подтверждается оптическими спектрами. Электронные спектры поглощения (рис. 4) содержат полосы с максимумами при 23 500 и 17 900 см–1, отнесение которых представляет трудности из-за недостатка литературных данных об электронных спектрах поглощения координационных соединений марганца(II) с тригонально-бипирамидальной координацией. По аналогии с [55] можно предположить, что полосы отвечают запрещенным по спину переходам из основного состояния 6A'1(6S) в возбужденные 4A'1(4G) (23 500 см–1) и 4E '(4G) (17 900 см–1).
ИК-спектр комплекса MnNaNTP приведен на рис. 5 (кривая 1). Колебаниям ν(MnII–O) внутренней координационной сферы комплекса соответствует дублет 495–575 см–1 (s–as) (по литературным данным в комплексах Mn(II) с октаэдрической координацией атомами кислорода [56] колебаниям ν(MnII–O) соответствуют полосы 465–525 см–1 (s–as); для комплексов с тригонально-бипирамидальной координацией [55] частота ν(MnII–O) возрастает до 520–570 см–1 (s–as)). Плечо около 660 см–1 относится, вероятно, к колебаниям мостика MnII–O–MnII [57]. Полосы в интервале 700–900 см–1 соответствуют деформационным колебаниям N–C–P–O скелета молекулы лиганда. Интенсивные полосы в области 900–1200 см–1 соответствуют валентным колебаниям групп PO3; полоса валентных колебаний локализованной связи P=O (1220 см–1) ослаблена и смещена в низкочастотную сторону, что свидетельствует о делокализации связи P=O при депротонировании лиганда. При 1320 и 1380 см–1 проявляются колебания δ(M–O–H). Колебаниям δ(CH2) соответствует слабо расщепленный дублет 1425–1430 см–1 (s–as).
Рис. 5.
ИК-спектры MnNaNTP (1) и продуктов его термического разложения (2) при 500°С в атмосфере Ar.
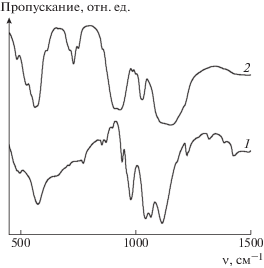
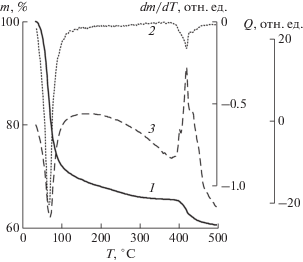
Отщепление молекул воды при термическом разложении MnNaNTP протекает в широком интервале температур (рис. 6): 30–150°С (–11.5 H2O) и 180–360°С (–1.5 H2O). Распад молекулы лиганда с разрывом связей N–C–P происходит при 380–480°С; потеря массы соответствует отщеплению CH3NH2. ИК-спектр продуктов термического разложения (рис. 5, кривая 2) – 480, 550, 720–750, 900, 920, 1050, 1100–1170 см–1 – близок к спектру натрий-марганец-фосфатного стекла с составом 40Na2O · 20MnO · 40P2O5 [58].
ВЫВОДЫ
Впервые синтезирован, выделен и исследован растворимый нитрилотрисметиленфосфонатный комплекс Mn(II). Его атомно-молекулярная и кристаллическая структура близка к ранее изученным комплексам Zn, Cu(II), Co(II) и резко отличается от структуры комплекса Ni(II).
Изучено распределение электронной плотности во внутренней координационной сфере комплекса Mn(II), которое существенно отличается от комплекса Zn. Показано, что эти отличия обусловлены особенностями электронной структуры комплекса. Оценена величина атомного магнитного момента Mn(II) в структуре полученного комплекса.
Методами ИК-спектроскопии и термогравиметрического анализа изучен механизм термического разложения комплекса.
Работа выполнена в рамках базовой части государственного задания высшим учебным заведениям и научным организациям в сфере научной деятельности (проект № 3.6502.2017/БЧ).
Список литературы
Shannon R.D. // Acta Cryst. A. 1976. V. 32. P. 751–767.https://doi.org/10.1107/S0567739476001551
Порай-Кошиц М.А., Полынова Т.Н., Школьникова Л.М. // Журн. Всесоюз. хим. о-ва им. Д.И. Менделеева. 1984. Т. 29. № 3. С. 43.
Zalesky J. // Z. Physiologische Chemie. 1904. B. 43. S. 11.
Boucher L.J. // Coord. Chem. Rev. 1972. V. 7. № 3. P. 289.https://doi.org/10.1016/s0010-8545(00)80024-7
Edwards R.A., Whittaker M.M., Whittaker J.W. et al. // Biochemistry. 2001. V. 40. P. 4622. https://doi.org/10.1021/bi002403h
Wagner T., Shumilin I.A., Bauerle R., Kretsinger R.H. // J. Mol. Biol. 2000. V. 301. P. 389. https://doi.org/10.1006/jmbi.2000.3957
Durbecq V., Sainz G., Oudjama Y. et al. // EMBO J. 2001. V. 20. P. 1530. https://doi.org/10.1093/emboj/20.7.1530
Bonanno J.B., Edo C., Eswar N. et al. // Proc. Natl. Acad. Sci. USA 2001. V. 98. P. 12896. https://doi.org/10.1073/pnas.181466998
Colton R., Mitchell S., Traeger J.C. // Inorg. Chim. Acta. 1995. V. 231. P. 87. https://doi.org/10.1016/0020-1693(94)04353-w
Zagorevskii D., Holmes J., Watson C., Eyler J. // Eur. J. Mass Spectrometry. 1997. V. 3. № 1. P. 27. https://doi.org/10.1255/ejms.22
Comprehensive Coordination Chemistry / Eds. Wilkinson G. et al. V. 4. Oxford: Pergamon Press, 1987. 1405 p.
Comprehensive Coordination Chemistry II / Eds. McCleverty J.A. et al. V. 4. Amsterdam: Elsevier, 2003. 847 p.
Lah M.S., Chun H. // Inorg. Chem. 1997. V. 36. P. 1782. https://doi.org/10.1021/ic960923o
Дятлова Н.М., Темкина В.Я., Попов К.И. Комплексоны и комплексонаты металлов. М.: Химия, 1988. 544 с.
Demadis K.D., Katarachia S.D., Koutmos M. // Inorg. Chem. Commun. 2005. V. 8. P. 254. https://doi.org/10.1016/j.inoche.2004.12.019
Cunha-Silva L., Mafra L., Ananias D. et al. // Chem. Mater. 2007. V. 19. P. 3527. https://doi.org/10.1021/cm070596q
Bazaga-García M., Angeli G.K., Papathanasiou K.E. et al. // Inorg. Chem. 2016.V. 55. P. 7414. https://doi.org/10.1021/acs.inorgchem.6b00570
Сомов Н.В., Чаусов Ф.Ф. // Кристаллография. 2014. Т. 59. № 1. С. 71. https://doi.org/10.1134/S1063774513050118
Сомов Н.В., Чаусов Ф.Ф. // Кристаллография. 2015. Т. 60. № 2. С. 233. https://doi.org/10.1134/S1063774515010228
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М., Федотова И.В. // Кристаллография. 2016. Т. 61. № 2. С. 238. https://doi.org/10.1134/S1063774516020243
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М., Федотова И.В. // Координац. химия. 2015. Т. 41. № 12. С. 729. https://doi.org/10.1134/S1070328415110081
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М. и др. // Кристаллография. 2017. Т. 62. № 6. С. 896. https://doi.org/10.1134/S1063774517050224
Сомов Н.В., Чаусов Ф.Ф., Ломова Н.В. и др. // Координац. химия. 2017. Т. 43. № 9. С. 545. https://doi.org/10.1134/S1070328417090093
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М. и др. // Координац. химия. 2017. Т. 43. № 12. С. 765. https://doi.org/10.1134/S1070328417120090
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М. // Кристаллография. 2016. Т. 61. № 3. С. 400. https://doi.org/10.1134/S1063774516030263
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М. // Кристаллография. 2016. Т. 61. № 4. С. 583. https://doi.org/10.1134/S1063774516040209
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М. и др. // Координац. химия. 2017. Т. 43. С. 369. https://doi.org/10.7868/S0132344X1706010X
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М. и др. // Кристаллография. 2018. Т. 63. № 6. С. 894. https://doi.org/10.1134/S1063774518050280
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М. и др. // Кристаллография. 2018. Т. 63. № 3. С. 415. https://doi.org/10.1134/S1063774518030276
Сомов Н.В., Чаусов Ф.Ф., Ломова Н.В. и др. // Журн. структур. химии. 2019. Т. 60. № 1. С. 87. https://doi.org/10.26902/JSC_id38758
Сомов Н.В., Чаусов Ф.Ф., Закирова Р.М. и др. // Журн. неорган. химии. 2018. Т. 63. № 1. С. 46. https://doi.org/10.1134/S0036023618010199
Кузнецов Ю.И. // Физикохимия поверхности и защита материалов. 2002. Т. 38. № 2. С. 122. https://doi.org/10.1023/A:1014904830050
Кузнецов Ю.И. // Успехи химии. 2004. Т. 73. С. 79. https://doi.org/10.1070/RC2004v073n01ABEH000864
Ломова Н.В., Чаусов Ф.Ф., Шабанова И.Н. // Изв. РАН. Сер. физ. 2018. Т. 82. № 7. С. 975. https://doi.org/10.3103/S1062873818070298
Cabeza A., Ouyang X., Sharma C.V.K. et al. // Inorg. Chem. 2002. V. 41. P. 2325. https://doi.org/10.1021/ic0110373
Clark R.C., Reid J.S. // Acta Cryst. A. 1995. V. 51. P. 887. https://doi.org/10.1107/S0108767395007367
Rigaku (2016). CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, Oxfordshire, England.
Sheldrick G.M. // Acta Cryst. A. 2008. V. 64. P. 112. https://doi.org/10.1107/S0108767307043930
Farrugia L.J. // J. Appl. Cryst. 1999. V. 32. P. 837. https://doi.org/10.1107/S0021889899006020
Momma K., Izumi F. // J. Appl. Cryst. 2011. V. 44. P. 1272. https://doi.org/10.1107/S0021889811038970
Trapeznikov V.A., Shabanova I.N., Kholzakov A.V., Ponomaryov A.G. // J. Electron Spectrosc. Related Phenomena. 2004. V. 137–140. P. 383. https://doi.org/10.1016/j.elspec.2004.02.115
Shirley D.A. // Phys. Rev. 1972. V. 55. P. 4709. https://doi.org/10.1103/PhysRevB.5.4709
Wojdyr M. // J. Appl. Cryst. 2010. V. 43. P. 1126. https://doi.org/10.1107/S0021889810030499
Addison A.W., Rao T.N., Reedijk J. et al. // J. Chem. Soc. Dalton Trans. 1984. № 7. P. 1349. https://doi.org/10.1039/DT9840001349
Сомов Н.В., Андреев П.В. // Кристаллография. 2018. Т. 63. № 1. С. 38. https://doi.org/10.1134/S1063774518010170
Чаусов Ф.Ф., Сомов Н.В., Наймушина Е.А., Шабанова И.Н. // Изв. РАН. Сер. физ. 2017. Т. 81. №3 . С. 312. https://doi.org/10.3103/S1062873817030078
Fujiwara M., Matsushita T., Ikeda S. // J. Electron Spectroscopy Related Phenomena. 1995. V. 74. № 3. P. 201. https://doi.org/10.1016/0368-2048(94)02375-1
Fadley C.S., Shirley D.A. // Phys. Rev. A. 1970. V. 2. P. 1109. https://doi.org/10.1103/PhysRevA.2.1109
Немошкаленко В.В., Алешин В.Г. Электронная спектроскопия кристаллов. Киев: Наукова думка, 1976. 336 с.
Маратканова А.Н., Соснов В.А., Шабанова И.Н., Келлер Н.В. // Журн. структур. хим. 1998. Т. 39. № 6. С. 1093. https://doi.org/10.1007/BF02903601
Шабанова И.Н., Ломова Н.В., Меньшиков А.З. // Журнал структур. хим. 2002. Т. 43. № 1. С. 85. https://doi.org/10.1023/A:1016073816481
Zanoni R., Boschi T., Licoccia S. et al. // Inorg. Chim. Acta. 1988. V. 145. № 2. P. 175. https://doi.org/10.1016/s0020-1693(00)83952-3
Huang J.-W., Liu Z.-L., Gao X.-R. et al. // J. Molecular Catalysis A: Chem. 1996. V. 111. № 3. P. 261. https://doi.org/10.1016/1381-1169(96)00202-6
Ferrer E.G., Baran E.J. // J. Electron Spectroscopy Related Phenomena. 1991. V. 57. № 2. P. 189. https://doi.org/10.1016/0368-2048(91)85022-l
Delaunay J., Hugel R.P. // Inorg. Chem. 1986. V. 25. № 22. P. 3957. https://doi.org/10.1021/ic00242a026
White W.B., Vito C., Scheetz B.E. // J. Cave Karst Studies. 2009. V. 71. № 2. P. 136.
Chu H.-A. // Frontiers Plant Sci. 2013. V. 4. P. @.https://doi.org/10.3389/fpls.2013.00146
Omrani R.O., Krimi S., Videau J.J. et al. // J. Non-Cryst. Solids. 2014. V. 389. P. 66. https://doi.org/10.1016/j.jnoncrysol.2014.02.006
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография