

Кристаллография, 2020, T. 65, № 4, стр. 624-629
Выращивание кристаллов флюоритовых твердых растворов системы CdF2–PbF2–MnF2 и их спектроскопические свойства
И. И. Бучинская 1, *, С. Х. Батыгов 2, А. Г. Иванова 1
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Институт общей физики им. А.М. Прохорова РАН
Москва, Россия
* E-mail: buchinskayaii@gmail.com
Поступила в редакцию 10.01.2020
После доработки 07.02.2020
Принята к публикации 10.02.2020
Аннотация
Концентрационная серия кристаллов твердого раствора (Pb0.67Cd0.33)1 – xMnxF2 + х (0 ≤ x ≤ 0.7) и кристалл Cd0.93Mn0.07F2 выращены из расплава методом вертикальной направленной кристаллизации (Бриджмена) во фторирующей атмосфере. Однофазные кристаллы флюоритовой структуры получены при легировании Pb0.67Cd0.33F2 фторидом марганца вплоть до 5 мол. %. При более высоком содержании MnF2 в кристаллах выпадает примесная тетрагональная фаза, изоструктурная группе соединений Ba2MF6 (M = Mn, Co, Zn). Проведены исследования оптического поглощения и люминесценции кристаллов Cd0.93Mn0.07F2 и (Pb0.67Cd0.33)0.95Mn0.05F2. Спектры поглощения указывают на октаэдрическую симметрию окружения ионов Mn2+ в трехкомпонентном кристалле (Pb0.67Cd0.33)0.95Mn0.05F2. Широкая полоса люминесценции Mn2+ в (Pb0.67Cd0.33)0.95Mn0.05F2 с максимумом 605 нм наблюдается только при 77 K.
ВВЕДЕНИЕ
Дифторид кадмия и высокотемпературная модификация дифторида свинца кристаллизуются в структурном типе флюорита с параметрами кристаллической решетки а = 5.388 и 5.940 Å соответственно [1, 2]. В системе PbF2–CdF2 образуется непрерывный флюоритовый твердый раствор Pb1 – xCdxF2 (0 ≤ x ≤ 1) с минимумом на кривых плавкости при х = 0.33 ± 0.02 и температуре Тпл = = 745 ± 5°С [2, 3]. Структурные исследования и исследования методом ядерного магнитного резонанса твердого раствора состава Pb0.67Cd0.33F2 свидетельствуют о существовании тетрагональных упорядоченных кластеров Pb2CdF6 при сохранении кубической макросимметрии [4–7]. Кристаллы Pb0.67Cd0.33F2 выращивались неоднократно [8–11]. Высокие плотность и радиационная стойкость в сочетании с относительно низкой температурой плавления делают их перспективными конструкционными и функциональными оптическими материалами [12, 13].
Некоторые оптические и радиационные характеристики кристаллов Pb0.67Cd0.33F2 и влияние на них ряда примесей изучались в [14]. Кристаллический материал Pb0.67Cd0.33F2 имеет низкий уровень световыхода (<0.1% от NaI : Tl) и является радиатором черенковского излучения. Из всех исследованных примесей только MnF2 изменяет люминесцентные характеристики Pb0.67Cd0.33F2 (увеличивает световыход до уровня ∼0.5% от NaI : Tl, при этом значительно увеличивает время высвечивания >300 нс).
Для кристаллов твердого раствора Cd1 – xMnxF2 при концентрациях 0.05 < x < 0.07 обнаружен максимальный световыход (∼2000 фотонов/МэВ) на длине волны λ ≈ 550 нм [14] (при х > 0.02 эти кристаллы достигают радиационной стойкости 10 Мрад). Люминесценция CdF2 и кубического PbF2, слаболегированных Mn2+, изучалась в [15] и [16] соответственно.
Представляется интересным продолжить исследование оптических свойств смешанных кристаллов (Pb0.67Cd0.33)1 – xMnxF2.
Фазовые диаграммы систем PbF2–MnF2 [17, 18] и CdF2–MnF2 [19] известны. В первой образуется тетрагональное соединение Pb2MnF6, которое отнесено к пр. гр. P42/nbc с параметрами элементарной ячейки a = 7.98, c = 16.92 Å [17, 18, 20]. Есть данные о существовании PbMnF4 [21]. Твердых растворов на основе компонентов в этой системе не обнаружено. Во второй системе существует обширная область флюоритового твердого раствора Cd1 – xMnxF2 вплоть до х ≤ 0.79. На основании этого в тройной системе CdF2–PbF2–MnF2 можно ожидать образования флюоритового твердого раствора (Pb0.67Cd0.33)1 – xMnxF2.
Цель данной работы – выращивание кристаллов твердого раствора (Pb0.67Cd0.33)1 – xMnxF2, определение предела вхождения в него ионов Mn2+ (x) и изучение спектроскопических характеристик образца с максимальной концентрацией Mn2+.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Выращивание кристаллов (Pb0.67Cd0.33)1 ‒ xMnxF2 с х = 0, 0.01, 0.03, 0.05, 0.07 проводили методом вертикальной направленной кристаллизации (методом Бриджмена) в открытом многоячеистом графитовом тигле в смешанной атмосфере Не и продуктов пиролиза тетрафторэтилена. В качестве исходных веществ использовали коммерческие реактивы марки “х. ч.”, предварительно высушенные в вакууме и проплавленные во фторирующей атмосфере для очистки от кислородсодержащих примесей. Температурный градиент в ростовой зоне составлял ∼50 K/см, скорость опускания тигля – 3.5 мм/ч. Потери вещества в процессе выращивания не превышали 2% от массы исходной загрузки. Для сравнительных исследований в аналогичных условиях выращен кристалл состава по шихте Cd0.93Mn0.07F2.
Фазовый состав и параметры элементарной ячейки полученных кристаллов определяли методом порошкового рентгенофазового анализа (РФА). Съемку рентгенограмм проводили на порошковом рентгеновском дифрактометре RigakuMiniFlex 600 (Rigaku, Япония) с использованием излучения CuKα в диапазоне углов 2θ от 10° до 100°. Идентификацию фаз выполняли в программе PXDRL (Rigaku, Япония) по базе данных ICDD PDF-2 (версия 2017). Параметры элементарных ячеек определяли по программе DICVOL [22] и уточняли методом Ле Бейла [23] в программе Jana2006 [24].
Спектры поглощения измеряли на спектрофотометре Cary 5000 в диапазоне длин волн 300–600 нм.
Спектры люминесценции возбуждали светодиодами с разной длиной волны излучения и измеряли на миниспектрометре FSD-10 при T = 77 и 300 K.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Выращенные кристаллы (Pb0.67Cd0.33)1 – xMnxF2 имели розоватую окраску (рис. 1), интенсивность которой нарастала с увеличением содержания марганца. Окраска свидетельствовала о присутствии ионов Mn2+, хотя нельзя исключать наличия других валентных состояний. В кристаллах, содержащих по шихте 3, 5 и 7 мол. % MnF2, наблюдалась ячеистая субструктура, вызванная концентрационным переохлаждением расплава при направленной кристаллизации. В кристалле с 7 мол. % MnF2 присутствовали видимые глазом мелкодисперсные включения, количество которых увеличивалось к его верхней части.
Рис. 1.
Кристаллы (Pb0.67Cd0.33)1 – xMnxF (0.01 ≤ x ≤ ≤ 0.07), выращенные (а) и просветленные для оптического просмотра (б).

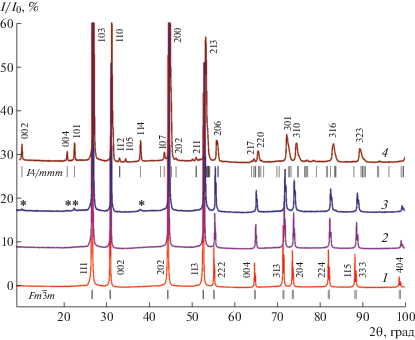
Рентгенофазовый анализ показал, что кристаллы, выращенные из шихты состава Pb0.67Cd0.33F2 с добавлением MnF2 1 и 3 мол. %, полностью однофазны, имеют флюоритовую структуру (пр. гр. $Fm\bar {3}m$) (рис. 2, спектр 1). При содержании 5 мол. % MnF2 по шихте (рис. 2, спектры 2, 3) в самой верхней части кристалла фиксируются следы второй фазы, содержание которой увеличивается с ростом концентрации MnF2 в шихте (рис. 2, спектр 4). Рефлексы примесной фазы проиндицированы в тетрагональной сингонии (пр. гр. I4/mmm) с параметрами решетки а = 4.0127(1) и с = = 16.8983(1) Å. Эта флюоритоподобная фаза изоструктурна группе соединений Ba2MF6 (M = Mn, Co, Zn) [25, 26] с близкими значениями параметров решетки. Вероятно, тетрагональная флюоритоподобная фаза образуется на основе соединения Pb2MnF6, в котором один или оба катиона частично замещены Cd2+.
Рис. 2.
Рентгенограммы образцов в системе Pb0.67Cd0.33F2–MnF2: 1 – 1 мол. % MnF2; 2 – 5 мол. % MnF2, низ кристалла; 3 – 5 мол. % MnF2, верх кристалла (* – появление примесной фазы); 4 – 7 мол. % MnF2. Показаны положения рефлексов Брэгга для фаз пр. гр. $Fm\bar {3}m$ и I4/mmm.
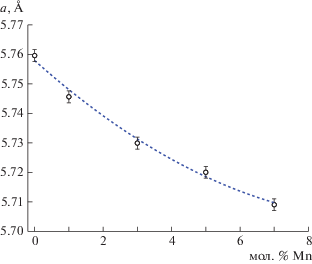
В результате кристаллизации из расплава флюорита смешанного состава (Pb0.67Cd0.33)1 – x MnxF2, содержащего ионы Mn2+ с меньшим ионным радиусом, чем у Pb2+ и Cd2+ [27], происходит закономерное уменьшение параметра элементарной ячейки. Параметр решетки флюоритовой фазы изменяется от 5.75(9) Å для Pb0.67Cd0.33F2 (что полностью совпадает с ранее полученным значением 5.75963 ± 0.00007 Å [28]) до 5.70(9) Å для кристалла с 7 мол. % MnF2 (рис. 3).
Рис. 3.
Зависимость параметра флюоритовой фазы в кристаллах (Pb0.67Cd0.33)1 – xMnxF2 от содержания Mn2+.
Кристалл с составом по шихте Cd0.93Mn0.07F2 однофазный, принадлежит структурному типу флюорита. Его параметр элементарной ячейки изменяется от 5.37(4) Å для нижней части до 5.36(4) Å для верхней. По зависимости параметра решетки от состава [19]: а = 5.3888 – 0.19х, проведена оценка изменения состава кристалла по длине в результате направленной кристаллизации. Выявлено, что содержание в нем MnF2 изменяется от 6 до 8 мол. %. Вероятно, центральная часть кристалла близка по составу к составу исходной шихты.
Образцы для спектроскопических исследований были вырезаны из средних по длине частей выращенных кристаллов. На рис. 4 показаны их спектры поглощения, а также спектр поглощения кристалла MnF2, полученного в [29]. Спектры содержат полосы, обусловленные электронными переходами в ионе Mn2+. Спектр поглощения исследуемого кристалла Cd0.93Mn0.07F2 отличается от спектра возбуждения, приведенного в [15], наличием сложной структуры в полосе 400 нм, связанной с переходом 6A1 → 4E, 4A1(G) в ионе Mn2+. В [16, 30–32] сложная структура этой полосы в кристаллах флюоритовой структуры объяснялась образованием центров Mn2+ с некубической симметрией, но для кристалла Cd0.93Mn0.07F2 более вероятным представляется перекрытие перехода 6A1 → 4T2(G) с переходом 6A1 → 4E, 4A1(G) (рис. 4, кривая 1). Спектр поглощения Mn2+ в матрице Cd0.67Pb0.33F2, также имеющей кубическую структуру, значительно отличается от спектра в Cd0.93Mn0.07F2, в то же время положения полос Mn2+ в Cd0.67Pb0.33F2 близки к положениям полос поглощения в MnF2, имеющем структуру рутила (кривая 3). Положения полос поглощения и соответствующие переходы в ионах марганца в разных матрицах приведены в табл. 1.
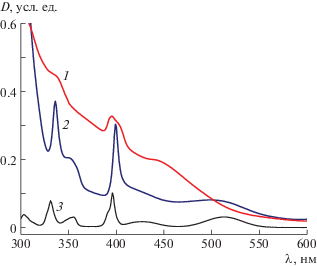
Таблица 1.
Положения полос поглощения Mn2+ в исследованных кристаллах
| Переход | Длина волны, нм | ||
|---|---|---|---|
| (Pb0.67Cd0.33)0.95Mn0.05F2 B = 771 см–1 Δ = 756 см–1 |
Cd0.93Mn0.07F2 B = 775 см–1 Δ = 480 см–1 |
MnF2 B = 778 см–1 Δ = 813 см–1 |
|
| 6A1 → 4T1(G) | 509 | 450 | 516 |
| 6A1 → 4T2(G) | 432 | 410 | 430 |
| 6A1 → 4A1, 4E(G) | 400 | 396 | 397 |
| 6A1 → 4T2(D) | 350 | 340 | 350 |
| 6A1 → 4E(D) | 330 | 330 | 330 |
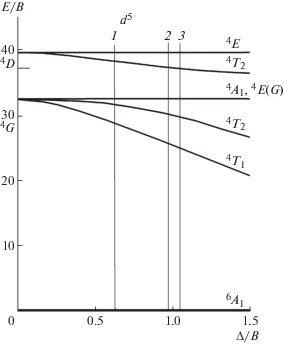
Для уточнения положений полос использовали диаграмму Танабе–Сугано (рис. 5). Параметры Рака B и кристаллического поля Δ рассчитывали с использованием перехода 6A1 → 4A1, 4E(G), соответствующего полосе поглощения в области 400 нм, положение которой слабо зависит от силы кристаллического поля, и перехода 6A1 → 4T1(G), соответствующего наиболее длинноволновой полосе поглощения. Разности энергий этих полос соответствуют значениям Δ/B, отмеченным вертикальными линиями на рис. 5. Значения B и Δ, рассчитанные с использованием значений Δ/B, приведены в табл. 1. Величина Δ для Cd0.67Pb0.33F2 (756 см–1) ближе к его величине в MnF2 (813 см–1), чем в CdF2 (480 см–1), что дает основание предполагать октаэдрическую симметрию окружения Mn2+ в Cd0.67Pb0.33F2.
Рис. 5.
Часть диаграммы Танабе–Сугано для Mn2+. Вертикальными линиями указаны значения Δ/B для Cd0.93Mn0.07F2 (1), (Pb0.67Cd0.33)0.95Mn0.05F2 (2) и MnF2 (3).
Спектры люминесценции образцов состава Cd0.93Mn0.07F2 и (Pb0.67Cd0.33)0.95Mn0.05F2 показаны на рис. 6. Спектр люминесценции образца Cd0.93Mn0.07F2 при комнатной температуре состоит из полосы с максимумом при 522 нм и полушириной 55 нм, приписанной переходу 4T1(G) → 6A1 в ионе Mn2+ [15]. Охлаждение до T = 77 K вызывает смещение максимума на 531 нм и уменьшение полуширины полосы до 34 нм.
Рис. 6.
Спектры люминесценции при возбуждении в области 400 нм: 1 – Cd0.93Mn0.07F2 (300 K), 2 – Cd0.93Mn0.07F2 (80 K), 3 – (Pb0.67Cd0.33)0.95Mn0.05F2 (77 K).
В образце (Cd0.67Pb0.33)0.95Mn0.05F2 при комнатной температуре люминесценция Mn2+ не возбуждается, но при охлаждении до 77 K появляется полоса люминесценции с максимумом при 605 нм и полушириной 59 нм. Наблюдаемая большая полуширина этой полосы в сравнении с кристаллами Cd0.93Mn0.07F2 может указывать на частичное разупорядочение в (Cd0.67Pb0.33)0.95Mn0.05F2.
ЗАКЛЮЧЕНИЕ
Выращены из расплава трехкомпонентные кристаллы (Pb0.67Cd0.33)1 – xMnxF2 (0 ≤ x ≤ 0.7). Выявлено, что однофазная область флюоритового твердого раствора (при его кристаллизации из расплава с последующим медленным охлаждением) существует вплоть до 5 мол. % MnF2. При более высоких концентрациях MnF2, предположительно на основе соединения Pb2MnF6 с частично замещенными катионами Cd2+, появляется примесная тетрагональная фаза. Для определения состава ее катионной подрешетки следует получить монокристаллические образцы.
Спектры поглощения кристаллов (Pb0.67Cd0.33)1 – xMnxF2 содержат слабые полосы поглощения, обусловленные запрещенными по спину переходами в ионе Mn2+. Положения полос указывают на образование Mn+2 центров с локальной октаэдрической симметрией окружения в кубической кристаллической матрице. Люминесценция кристалла (Pb0.67Cd0.33)0.95Mn0.05F2 наблюдается только при 77 К, ее спектр состоит из широкой полосы с максимумом 605 нм. По сравнению со спектром Cd0.93Mn0.07F2 полоса люминесценции значительно уширена и сдвинута в длинноволновую область.
Авторы выражают благодарность Д.Н. Каримову за помощь в обработке результатов эксперимента.
Работа выполнена при поддержке Министерства науки и высшего образования в рамках выполнения работ по Государственному заданию в части роста и структурного исследования кристаллов. Рентгенодифракционные исследования выполнены с использованием оборудования ЦКП ФНИЦ “Кристаллография и фотоника” при поддержке Минобрнауки (проект RFMEFI62119X0035).
Список литературы
Федоров П.П., Соболев Б.П. // Кристаллография. 1992. Т. 37. Вып. 5. С. 1210.
Бучинская И.И., Федоров П.П. // Успехи химии. 2004. Т. 73. № 4. С. 404.
Buchinskaya I.I., Fedorov P.P., Sobolev B.P. // SPIE Proc.: Solid State Crystals: Growth and Characterization. Zakopane, Poland. 1996. V. 3178. P. 59.
Бузник В.М., Суховской А.А., Вопилов В.А. и др. // Журн. неорган. химии. 1997. Т. 42. № 12. С. 2092.
Fedorov P.P., Buchinskaya I.I., Zhurova E.A. et al. // Conf: Structure and Properties of Crystalline Materials. Dubna. 4–6 March 1997. Abstrs. P. 42.
Мацулев А.И., Иванов Ю.Н., Лившиц А.И. и др. // Журн. неорган. химии. 2000. Т. 45. № 2. С. 296.
Trnovcova V., Fedorov P.P., Ozvoldova M. et al. // J. Optoelectron. Adv. Mater. 2003. V. 5. № 3. P. 627.
Kosacki I., Dynowska E. // J. Cryst. Growth. 1980. V. 50. № 2. P. 575.
Kosacki I. // Appl. Phys. A. 1989. V. 49. P. 413.
Buchinskaya I.I., Fedorov P.P., Sobolev B.P. // SPIE: Solid State Crystals: Growth and Characterization. Zakopane, Poland. 1996. V. 3178. P. 59.
Попов П.А., Матовников А.В., Моисеев Н.В. и др. // Кристаллография. 2015. Т. 60. № 1. С. 128.
Васильченко В.Г., Мотин Ю.Д., Кречко Ю.А. и др. // Неорган. материалы. 1993. Т. 29. № 6. С. 739.
Соболев Б.П., Быстрова А.А., Бучинская И.И. и др. // Патент RU 2061114, 27.05.1996.
Kuptsov S.I., Solov’ev A.S., Vasil’chenko V.G. et al. // Nucl. Instrum. Methods Phys. Res. B. 1995. V. 103. P. 323.
Alonso P.J., Alcala R. // J. Luminescence. 1981 V. 22. P. 321.
Shcherbakov V.D., Nizamutdinov A.S. // J. Luminescence. 2019. V. 205. P. 37.
Samouel M., Seances C.R. // Acad. Sci. C. 1968. V. 268. P. 409.
Samouel M. // Rev. Chem. Miner. 1971. V. 8. P. 537.
Федоров П.П., Саттарова М.А., Ольховая Л.А. и др. // Высокочистые вещества. 1991. Т. 3. С. 191.
Dorshow R., Hogg R.D., Jaccarino V. // Phys. Lett. A. 1979. V. 37. P. 250.
Demortian G., Tressaud A., Tanguy B. et al. // Rev. Chem. Miner. 1987. V. 24. P. 117.
Boultifand A., Louer D. // J. Appl. Cryst. 2004. V. 37. P. 724. https://doi.org/10.1107/S0021889804014876
Le Bail A. // Powder Diffr. 2005. V. 20. № 4. P. 316.
Petříček V., Dušek M., Palatinus L. // Z. Kristallogr. Cryst. Mater. 2014. B. 229. № 5. S. 345.
Chenavas J., Capponi J.J., Joubert J.C., Marezio M. // Mater. Res. Bull. 1974. V. 9. P. 13.
Von Schnering H.G. // Z. Anorg. Allg. Chem.1967. B. 353. S. 13
Shannon R.D. // Acta Cryst. A. 1976. V. 32. № 5. P. 751.
Попов П.А., Матовников А.В., Моисеев Н.В. и др. // Кристаллография. 2015. Т. 60. № 1. С. 111. https://doi.org/10.7868/S0023476115010178
Икрами Д.Д., Кузнецова Н.И., Сидоров В.С., Ручкин Е.Д. // Журн. неорган. химии. 1984. Т. 29. № 9. С. 2360.
Щербаков В.Д. // Учен. зап. Казан. ун-та. Сер. физ.-мат. науки. 2010. Т. 152. кн. 4. С. 21.
Щербаков В.Д. // Учен. зап. Казан. ун-та. Сер. физ.-мат. науки. 2015. Т. 157. кн. 4. С. 172.
Щербаков В.Д. // Кристаллография. 2017. Т. 62. № 3. С. 433.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография