

Кристаллография, 2020, T. 65, № 4, стр. 588-592
Рентгеноструктурное и термографическое исследование потенциально мезоморфного 4-[4-октилокси-2-гидроксибензилиден)]цианоанилина
Л. Г. Кузьмина 1, *, П. Калле 1, И. И. Константинов 2, **, Э. Х. Лермонтова 1
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
* E-mail: kuzmina@igic.ras.ru
** E-mail: konst@ipc.ac.ru
Поступила в редакцию 16.10.2019
После доработки 16.10.2019
Принята к публикации 11.11.2019
Аннотация
Методом рентгеноструктурного анализа и дифференциальной сканирующей калориметрии изучен потенциально мезоморфный 4-[4-октилокси-2-гидроксибензилиден)цианоанилин C8H17OC6H3(OH)–CH=N–C6H4CN (I). Кристаллическая упаковка соединения не обнаруживает особенностей, типичных для мезоморфных кристаллов, что согласуется с данными калориметрии об отсутствии мезоморфных свойств у соединения I.
ВВЕДЕНИЕ
Настоящая работа является продолжением комбинированных исследований, основанных на методах рентгеноструктурного анализа (РСА) и дифференциальной сканирующей калориметрии (ДСК), гомологических рядов органических соединений, способных образовывать мезофазу при плавлении и/или охлаждении изотропного расплава. Эти исследования нацелены на более глубокое понимание природы мезофазы и причин, приводящих к ее формированию. Мезофаза – это особое агрегатное состояние вещества, промежуточное между жидкостью и кристаллом. Превращения кристалл–мезофаза–жидкость происходят как фазовые переходы, т.е. сопровождаются определенными тепловыми эффектами.
Впервые мезофаза как новое агрегатное состояние была описана ∼130 лет назад [1], и за прошедшее время предпринимались многочисленные попытки создания ее теории [2–15]. Однако эти попытки до настоящего времени нельзя считать успешными, поскольку ни одна из существующих теорий не дает близких к экспериментальным значений энтальпии фазовых переходов. Более того, не удается предсказать число фазовых переходов с участием мезофазы. Следовательно, модели мезофазы, положенные в основу теорий, не отражают каких-то ключевых ее особенностей, что делает актуальными дальнейшие структурные и калориметрические исследования потенциально мезоморфных органических соединений.
Наиболее изучены из них алкил- и алкилоксибензоаты, а также алкил- и алкилоксисалицилиденанилины [16–21]. Многие из этих соединений являются мезоморфными и обнаруживают либо энантиоморфный, либо монотропный мезоморфизм, тогда как другие вообще не являются мезоморфными. Это относится даже к ближайшим представителям одного гомологического ряда. Для установления причин указанных различий в настоящей работе методами РСА и ДСК проведены исследования 4-[4-октилокси-2-гидроксибензи-лиден]цианоанилина C8H17OC6H3(OH)–CH=N–C6H4CN (I).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез. Соединение I синтезировали по реакции 3-гидрокси-4-алкилоксибензальдегида с соответствующим анилином в абсолютном этаноле по методике [22]. Реакционную смесь кипятили в течение 3 ч при перемешивании, а затем быстро отфильтровывали. Растворитель удаляли в процессе испарения. Продукт перекристаллизовывали из абсолютного этанола. Чистоту продукта проверяли методом тонкослойной хроматографии.
Дифференциальная сканирующая калориметрия. Образец, состоящий из небольшого числа кристаллов, помещали в оптическую систему ДСК (Linkam DSC600), оснащенную микроскопом Olympus BX43, где исследовали термическое поведение соединения. Скорость нагрева составила 5 град/мин.
Рентгеноструктурный анализ. Монокристалл I исследован на CCD-дифрактометре Smart Apex-II в потоке охлажденного азота (излучение MoKα, λ = 0.71073 Å, графитовый монохроматор, ω-сканирование). Экспериментальные отражения обработаны по программе SAINT [23]. Структура расшифрована прямыми методами и уточнена методом наименьших квадратов в анизотропном приближении для всех атомов, кроме атомов водорода. Положения атомов водорода рассчитаны геометрически и уточнены по модели наездника. Кристаллографические параметры, а также параметры расшифровки и уточнения структуры I приведены в табл. 1. Все расчеты выполнены по программам Olex2 [24].
Таблица 1.
Кристаллографические характеристики, данные эксперимента и результаты уточнения структуры I (CCDC № 1910751)
| Химическая формула | C22H26N2O2 |
|---|---|
| М, г/моль | 350.45 |
| Пр. гр., Z | P$\bar {1}$, 2 |
| a, b, c, Å | 5.8487(5), 11.1002(10), 15.0699(13) |
| α, β, γ, град | 92.1680(10), 98.5470(10), 101.342(2) |
| V, Å3 | 946.33(14) |
| Dx, г/см3 | 1.230 |
| μ, см–1 | 0.079 |
| Т, K | 150 |
| Размеры кристалла, мм | 0.42 × 0.26 × 0.12 |
| F(000) | 376.0 |
| Пределы h, k, l | –7 < h < 7, –15 < k < 15, –20 < l < 20 |
| (sin θ/λ)max | 1.193 |
| Число отражений: измеренных/независимых, Rint | 10 518/5003, 0.0340 |
| Число параметров | 237 |
| S | 1.025 |
| R1/wR2 (все отражения) | 0.0786/0.1332 |
| R1/wR2 (I > 2σ(I)) | 0.0468/0.1173 |
| Δρmin/Δρmax, э/A3 | –0.20/0.33 |
| Программы | SAINT [23], Olex2 [24] |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Согласно данным ДСК соединение I плавится при 85.3°C; плавление не сопровождается образованием мезофазы. Охлаждение изотропного расплава также не сопровождается образованием мезофазы. Сразу образуется кристаллическая фаза, причем при более низкой температуре (73.7°C), чем температура плавления. Таким образом, данное соединение не является мезоморфным.
Образование переохлажденного расплава типично для жидкокристаллических соединений, но природа этого явления, насколько известно, в литературе не обсуждалась. Различие температур плавления и кристаллизации расплава соединения I можно объяснить формированием из расплава менее плотной модификации, чем исходный кристалл, полученный из раствора при комнатной температуре. Действительно, в расплаве алкильная цепочка молекулы в отличие от ее ароматического ядра обладает существенной конформационной подвижностью, что фактически означает увеличение ее эффективных размеров. Поэтому очертания и размеры молекулы I в расплаве и растворе различны, а значит, различаются геометрические требования к формированию кристаллического окружения. Это может быть причиной образования разных модификаций при кристаллизации из раствора и расплава. Высокотемпературная кристаллическая модификация с неизбежностью будет менее плотной и будет характеризоваться более низкой температурой плавления.
Строение молекулы I показано на рис. 1. Основной скелет молекулы почти плоский. Двугранный угол между плоскостями бензольных колец близок к нулю; его поворотная и наклонная компоненты равны 3.20(4)° и 0.45(4)° соответственно. Углеродный каркас алифатической цепочки плоский и почти копланарен бензольному кольцу С9…С14; поворотная и наклонная компоненты этого двугранного угла соответственно равны 0.44(7)° и 2.26(3)°. Согласно Кэмбриджскому банку структурных данных [25] двугранный угол между двумя бензольными кольцами в молекулах салицилиден-анилинов (156 примеров) варьируется в широких пределах (0°–56°). Это свидетельствует о том, что молекулы данного класса проявляют конформационную подвижность в отношении взаимного поворота бензольных колец. Например, двугранный угол между бензольными кольцами в 4-этоксисалицилиден-4'-метиланилине (С2Н5O–C6H3(OН)–СН=N–C6H4–CH3) составляет 18.7° и 29.2° в двух кристаллографически независимых молекулах [20]. В других представителях того же гомологического ряда, а именно в 4-пропилоксисалицилиден-4'-бутилланилине (С3H7O–C6H3(OН)–СН=N–C6H4–C4H9 и 4-децилоксисалицилиден-4'-гексиланилине, эти углы соответственно равны 50.4° и 42.2° [21].
Рис. 1.
Молекулярная структура кристалла I, эллипсоиды атомных смещений приведены на уровне вероятности 50%.
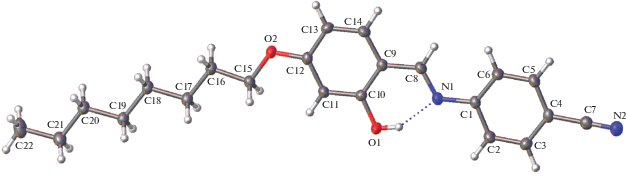
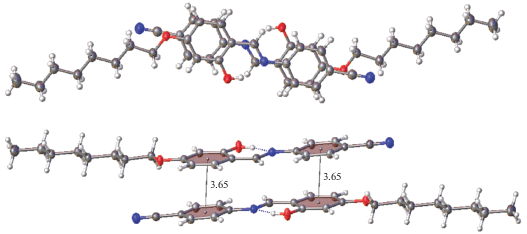
Величина рассматриваемого двугранного угла в конкретной структуре определяется кристаллической упаковкой и/или электронными эффектами заместителей в молекуле. Конформационная подвижность салицилиден-анилиновых молекул подавляет тенденцию формирования π–π-стэкинг-взаимодействующих элементов упаковки (димеров, стопок). Действительно, в кристаллических упаковках упомянутых ранее 156 структур не обнаружено ни единого случая образования таких элементов. Введение в положение 4 салицилиден-анилиновой молекулы π-заместителя, например нитрильной группы (молекула I), должно усилить π-сопряжение по всему салицилиден-анилиновому каркасу молекулы, а значит, понизить конформационную подвижность и сделать молекулу более склонной к π–π-стэкинг-взаимодействиям кристаллической упаковки в процессе роста кристалла. Действительно, центросимметрично связанный стэкинг-димер обнаружен в данном кристалле (рис. 2). В этом димере бензольные кольца эффективно проецируются друг на друга. Расстояние между центроидами бензольных колец соседних молекул (3.65 Å) удовлетворяет π–π-стэкинг-взаимодействию.
Введение в молекулу салицилал-анилина нитрильной группы может сильно повлиять на формирование кристаллической упаковки не только вследствие уплощения геометрии молекулы, но и благодаря склонности этой группы образовывать вторичные связи с аналогичными группами соседних молекул. Обычно для таких направленных взаимодействий требуется антипараллельное расположение соседних CN-групп. На рис. 3 показан слой кристаллической упаковки, состоящий из бесконечных лент, образованных вторичными связями CN···CN. Чередующиеся расстояния С…N между соседними антипараллельными фрагментами CN составляют 3.30 и 3.39 Å, что отвечает слабым направленным взаимодействиям. Еще одной особенностью рассматриваемого слоя является то, что почти все межмолекулярные расстояния между атомами алифатических цепочек (H…H, C…H, C…C) оказываются больше соответствующих сумм ван-дер-ваальсовых радиусов (∼2.4, ∼3.0, ∼3.8 Å соответственно). И только одно расстояние C…C между концевыми атомами C22 соседних лент соответствует ван-дер-ваальсовому контакту (3.50 Å).
Рис. 3.
Слой кристаллической упаковки, включающий в себя бесконечные ленты. Межмолекулярные расстояния приведены в ангстремах.
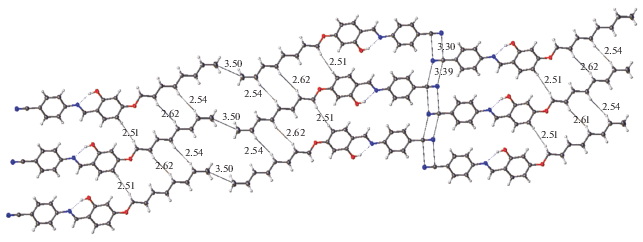
Рассматриваемый слой имеет все признаки кристаллической упаковки, типичной для мезоморфных соединений. Последняя построена из чередующихся плотно и рыхло упакованных областей. Рыхлые области образованы конформационно подвижными алифатическими цепочками, а плотные – жесткими ароматическими фрагментами молекул [16–21]. Только небольшое число контактов обнаруживается в “алифатических областях”, тогда как в плотно упакованных “ароматических областях” существует множество контактов, в том числе те, которые отвечают слабым направленным взаимодействиям (вторичным связям), а также ненаправленным дисперсионным контактам. Соединения, имеющие такую “разделенную” кристаллическую упаковку и множество слабых направленных взаимодействий, плавятся аномально с промежуточным образованием мезофазы, т.е. структурированной жидкости. Плавление начинается по рыхлым областям и приводит к разрушению дальнего порядка кристалла [26]. На начальном этапе плавления “ароматические” области сохраняют свою структурированность благодаря вторичным связям. Дальнейшее повышение температуры приводит к поэтапному разрушению всех типов вторичных связей, что в итоге завершается переходом к изотропной жидкости.
Однако рассмотренные особенности строения мезоморфных кристаллов не воспроизводятся во всем объеме кристалла I, чему препятствует способ наложения слоев. Он определяется соотношением двух тенденций – стремлением к возникновению π–π-стэкинг-взаимодействий между ароматическими кольцами соседних слоев и стремлением образовать типичную для мезоморфов “разделенную” кристаллическую упаковку. Эти две тенденции в данном случае оказались взаимоисключающими. Действительно, в геометрии рассмотренного слоя осуществившийся в кристалле I центросимметричный способ наложения слоев, необходимый для формирования π–π-стэкинг-взаимодействий между ароматическими кольцами соседних слоев, исключает возможность сближенного расположения алифатических областей соседних слоев. Таким образом, в процессе роста кристалла доминирует первая тенденция, что неизбежно приводит к возникновению кристаллической упаковки (рис. 4), не характерной для мезоморфов.
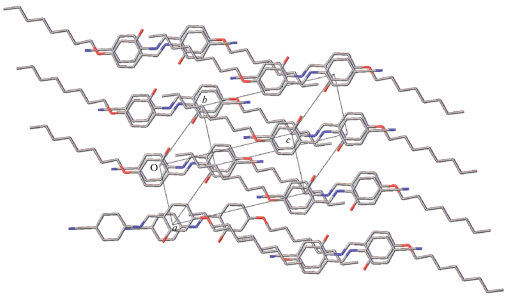
Усиление тенденции к π–π-стэкинг-взаимодействию в кристаллической упаковке благодаря включению в пара-положение молекулы π-заместителя – нитрильной группы – хотя и создает дополнительные элементы, необходимые для увеличения степени структурированности мезофазы, но негативно влияет на возможность реализовать “разделенную” кристаллическую упаковку. Это объясняет, почему плавление кристалла I не сопровождается формированием мезофазы.
Работа выполнена при финансовой поддержке Российского научного фонда (грант № 16-13-10273). Образцы синтезированы и охарактеризованы И.И. Константиновым в рамках государственной программы ИНХС РАН.
Список литературы
Lehman O. // Uber Fliessende Kristalle. Z. Phys. Chem. 1889. B. 4. S. 462.
Maier W., Saupe A. // Z. Naturforsch. 1960. B. 15a. S. 287.
Humphries R.L., James P.G., Luckhurst G.R.J. // Chem. Soc. Faraday Trans. II. 1972. V. 68. P. 1031.
De Gennes P.G. // Mol. Cryst. Liq. Cryst. 1973. V. 21. P. 49.
Mc Millan W.L. // Phys. Rev. A. 1973. V. 8. P. 1921.
Wulf A. // Phys. Rev. A. 1975. V. 1. P. 365.
Cotter M.A. // Mol. Cryst. Liq. Cryst. 1983. V. 97. P. 29.
Osipov M.A. // Handbook of Liquid Crystals / Eds. Demus D. et al. Weinheim: Wiley-VCH, 1998. P. 40.
Vertogen G., Jeu W.H. Thermotropic Liquid Crystals, Fundamentals. Berlin: Springer-Verlag, 1988. 324 p.
Gennes P.G., Prost J. The Physics of Liquid Crystals. New York: Oxford University Press, 1995. 616 p.
Velasco E., Mederos L., Sluckin T.J. // Liq. Cryst. 1996. V. 20. P. 399.
Singh S. // Phys. Rep. 2000. V. 324. № 2–4. P. 107.
Skoulios A., Guillon D. // Mol. Cryst. Liq. Cryst. 1988. V. 165. P. 317.
Tschiersk C. // J. Mater. Chem. 1998. V. 8. P. 1485.
Donnio B., Buathong S., Bury I., Guillon D. // Chem. Soc. Rev. 2007. V. 36. P. 1495.
Kuz’mina L.G., Konstantinov I.I., Lermontova E.K. // Mol. Cryst. Liq. Cryst. 2014. V. 588. P. 1.
Кузьмина Л.Г., Константинов И.И., Беззубов С.И. // Химия высоких энергий. 2016. Т. 50. № 6. С. 453.
Kuz’mina L.G., Konstantinov I.I., Churakov A.V., Navasardyan M.A. // Acta Cryst. E. 2017. V. 73. P. 1052.
Kuz’mina L.G., Konstantinov I.I., Churakov A.V. // Mol. Cryst. Liq. Cryst. 2018. V. 664. P. 95.
Кузьмина Л.Г., Навасардян М.А., Михайлов А.А. // Кристаллография. 2017. Т. 62. № 6. С. 889.
Кузьмина Л.Г., Навасардян М.А., Беззубов С.И. // Кристаллография. 2019. Т. 64. № 1. С. 72.
Yeap G-Y., Ha S-T., Lim Ph-L. et al. // Mol. Cryst. Liq. Cryst. 2006. V. 452. P. 63.
Bruker (2008). APEX2, SADABS and SAINT. Bruker AXS Inc. Madison, Wiskonsin, USA.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Cryst. 2009. V. 42. P. 339.
Groon C.R., Allen F.H. // Angew. Chem. 2014. V. 53. P. 662.
Kuz’mina L.G., Navasardyan M.A., Churakov A.V., Howard J.A.K. // Mol. Cryst. Liq. Cryst. 2016. V. 638. P. 60.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография