

Кристаллография, 2020, T. 65, № 5, стр. 786-788
Расчет энергии взаимодействия имидазолглицерофосфатдегидратазы из Mycobacterium tuberculosis с производными триазола методом молекулярной динамики
Ю. К. Агапова 1, *, А. С. Комолов 1, В. И. Тимофеев 1, 2
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
* E-mail: agapova.jk@gmail.com
Поступила в редакцию 02.03.2020
После доработки 17.03.2020
Принята к публикации 18.03.2020
Аннотация
Методом компьютерного моделирования рассчитано изменение свободной энергии системы при связывании трех производных триазола в активном центре имидазолглицерофосфатдегидратазы из Arabidopsis thaliana и Mycobacterium tuberculosis. Проведено сопоставление с константами ингибирования, полученными ранее для данных ингибиторов для фермента из Arabidopsis thaliana. Показано, что для обоих ферментов изменение свободной энергии, рассчитанное методом MM–PBSA, коррелирует с константами ингибирования фермента растительного происхождения, полученными экспериментальным путем.
ВВЕДЕНИЕ
Имидазолглицерофосфатдегидратаза (ИГФД) катализирует шестой этап биосинтеза гистидина у бактерий, растений и грибов. Имидазолглицерофосфатдегидратаза катализирует превращение имидазолглицерофосфата в имидазолацетолфосфат [1]. В организмах млекопитающих данный фермент отсутствует (гистидин является так называемой незаменимой аминокислотой, которую млекопитающие должны употреблять с пищей). Этот факт делает ИГФД из Mycobacterium tuberculosis мишенью для разработки противотуберкулезных препаратов.
Ранее [2] было показано, что некоторые производные триазола способны ингибировать гомологичный фермент растительного происхождения из Arabidopsis thaliana. Для ИГФД из M. tuberculosis в настоящее время имеется большое количество структурных данных. Так, методом рентгеноструктурного анализа установлены пространственные структуры ИГФД из M. tuberculosis в свободной форме, в комплексе с имидазолглицерофосфатом, который является природным субстратом, а также в комплексе с различными ингибиторами – 3-амино-1,2,4-триазолом и (2S)-2-азанил-3-(4Н-1,2,4-триазол-3-ил)пропановой кислотой (рис. 1) при разрешении от 2.1 до 1.75 Å [3]. Биологически активная форма фермента состоит из 24 субъединиц, тогда как каждый из активных центров фермента включает в себя три соседние субъединицы. В каждом из активных центров фермента содержится по два иона марганца [3]. Для ИГФД из M. tuberculosis был проведен виртуальный скрининг потенциальных ингибиторов, а также исследование связывания ряда триазолов в активном центре фермента методами компьютерного моделирования [4, 5]. Для ИГФД из A. thaliana также имеется большое количество структурных данных. Так, известны пространственные структуры из A. thaliana в апо-форме, в комплексе с субстратом, а также в комплексе с двумя различными ингибиторами [6–8].
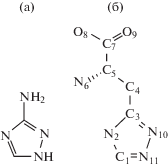
Рис. 1.
Структурные формулы 3-амино-1,2,4-триазола (а) и (2S)-2-азанил-3-(4Н-1,2,4-триазол-3-ил)пропановой кислоты (б).
В настоящей работе проведено исследование энергии взаимодействия имидазолглицерофосфатдегидратазы из M. tuberculosis и A. thaliana с тремя производными триазола методом компьютерного моделирования. Показана корреляция между экспериментально установленными константами ингибирования для растительного фермента и рассчитанными значениями изменения свободной энергии системы.
МАТЕРИАЛЫ И МЕТОДЫ
Построение трехмерных моделей комплексов ферментов и ингибиторов. Для построения комплексов обеих ИГФД с тремя аналогами триазола (IRL 1695, IRL 1803, IRL 856) [2, 5] был использован метод молекулярного докинга, реализованный в программе AutoDock Vina [9]. В качестве мишени использовали структуры ИГФД из Mycobacterium tuberculosis (PDB_ID: 5XDS) и Arabidopsis thaliana (PDB_ID: 5EKW). Обе структуры содержат ингибитор в активном центре, что позволило существенно уточнить результаты предыдущего исследования [5]. Были выбраны шесть моделей с наиболее низкой оценочной функцией докинга. Для достижения равновесного состояния комплексов было проведено моделирование молекулярной динамики полученных комплексов. Расчет проводили с использованием программного пакета AMBER [10] с CUDA-версией PMEMD [11]. Использованы силовое поле AMBER14SB [12] и модель воды TIP3P. Для релаксации структуры и во избежание стерических столкновений в последующих расчетах была проведена минимизация энергии. Температура и давление уравновешены до 1 атм. и 293 K путем расчетов в ансамблях NVT и NPT в течение 100 пс. Температуру и давление в системе контролировали с использованием термостата Ланжевена [13] и баростата Монте-Карло [14]. Шаг интегрирования составлял 2 фс.
Расчет изменения свободной энергии при связывании ингибиторов. Для расчета изменения свободной энергии при связывании ингибиторов применяли метод MM–PBSA [15]. На первом этапе с использованием уравновешенных моделей комплексов для каждой из шести моделей была расcчитана молекулярная динамика с шагом записи 10 пс. Полученный набор конфигураций моделируемой системы (снапшотов) был использован для применения программного скрипта, в котором реализован метод MM–PBSA из программного пакета AmberTools [10].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
С использованием метода MM–PBSA [15] рассчитано изменение свободной энергии при связывании трех производных триазола, которые являются ингибиторами ИГФД из A. thaliana и M. tuberculosis. Величины изменения свободной энергии для каждого из комплексов приведены в табл. 1. Также для сравнения с экспериментальными константами ингибирования в таблице приведены данные из [2]. Из таблицы видно, что для комплексов ингибиторов со структурой из Arabidopsis thaliana наблюдается хорошая корреляция с экспериментальными данными, из чего следует принципиальная применимость данного метода для разработки ингибиторов ИГФД методом компьютерного моделирования. Что касается комплексов из M. tuberculosis, также видно, что есть некая корреляция с данными о растительном ферменте, однако, наблюдаются большие погрешности измерения, что особенно заметно для комплексов M. tuberculosis ИГФД c IRL 1803 и IRL 1695. Данный эффект может быть связан как с точностью вычислений, так и с различиями в строении активного центра исследуемых ферментов. Однако можно утверждать, что, несмотря на данное обстоятельство, по меньшей мере один из трех исследуемых ингибиторов (IRL 856) может быть использован как прототип для разработки высокоэффективного и высокоселективного ингибитора ИГФД из Mycobacterium tuberculosis.
Таблица 1.
Расчетные значения изменения свободной энергии (ккал/моль) при связывании трех производных триазола в активном центре M. tuberculosis и A. thaliana и константа ингибирования (Ki) для фермента из A. thaliana [2]
| Ингибитор | M. tuberculosis | A. thaliana | Ki, нМ |
|---|---|---|---|
| IRL 1695 | –8.4 ± 4.0 | –16.8 ± 3.5 | 40 ± 6.5 |
| IRL 1803 | –17.4 ± 6.7 | –56.8 ± 4.1 | 10 ± 1.6 |
| IRL 856 | –63.9 ± 14.6 | –71.4 ± 8.1 | 8.5 ± 1.4 |
Работа выполнена при поддержке Российского фонда фундаментальных исследований (проект № 18-34-00876) в части расчета изменения свободной энергии при связывании ингибиторов и при поддержке Министерства науки и высшего образования РФ в рамках Государственного задания ФНИЦ “Кристаллография и фотоника” РАН в части анализа результатов молекулярного моделирования.
Список литературы
Ames B.N., Garry B., Herzenberg L.A. // J. Mol. Biol. 1960. V. 33. P. 533.
Mori I., Fonne-Pfister R., Matsunaga S. et al. // Plant. Physiol. 1995. V. 107. P. 719.
Ahangar M.S., Khandokar Y., Nasir N. et al. // Acta Cryst. F. 2011. V. 67. P. 1451.
Подшивалов Д.Д., Манджиева Ю.Б., Сидоров-Бирюков Д.Д. и др. // Кристаллография. 2018. Т. 63. № 1. С. 82.
Агапова Ю.К., Тимофеев В.И., Комолов А.С. // Кристаллография. 2019. Т. 64. № 4. С. 583.
Rawson S., Bisson C., Hurdiss D.L. // Proc. Natl. Acad. Sci. USA. 2018. V. 115. P. 1795.
Bisson C., Britton K.L., Sedelnikova S.E. et al. // Ang. Chem. Int. Ed. Engl. 2016. V. 55. P. 13485.
Bisson C., Britton K.L., Sedelnikova S.E. et al. // Structure. 2015. V. 23. P. 1236.
Trott A.J., Olson O. // J. Comput. Chem. 2010. V. 31. P. 455.
Case D.A., Cheatham T., Darden T. // J. Comput. Chem. 2005. V. 26. P. 1668.
Salomon-Ferrer R., Goetz A.W., Poole D. et al. // J. Chem. Theory Comput. 2013. V. 9. P. 3878.
Maier J.A., Martinez C., Kasavajhala K. et al. // J. Chem. Theory Comput. 2015. V. 11. P. 3696.
Allen M.P., Tildesley D.J. Computer Simulation of Liquids. New York: Oxford University Press, 1991. 385 p.
Steinbach P.J. // Proteins. 2004. V. 57. P. 665.
Genheden S., Ryde U. // Expert Opin. Drug Discov. 2015. V. 10. P. 449.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография