

Кристаллография, 2020, T. 65, № 5, стр. 780-785
Изменения четвертичной структуры нуклеотид-регулируемой пирофосфатазы из Desulfitobacterium hafniense при взаимодействии с лигандами, выявленные методом малоуглового рентгеновского рассеяния в растворе
Л. А. Дадинова 1, *, В. А. Анашкин 2, Э. В. Штыкова 1
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Научно-исследовательский институт физико-химической биологии им. А.Н. Белозерского Московского государственного университета им. М.В. Ломоносова
Москва, Россия
* E-mail: lubovmsu@mail.ru
Поступила в редакцию 28.02.2020
После доработки 28.02.2020
Принята к публикации 03.03.2020
Аннотация
Для установления механизма передачи сигнала между активным и регуляторным центрами регулируемой нуклеотидами неорганической пирофосфатазы интенсивно исследуется регуляция этих ферментов на молекулярном уровне. Однако отсутствие достоверных структурных данных о полноразмерном белке и его взаимодействии с лигандами не позволяет однозначно решить проблему. Методом малоуглового рентгеновского рассеяния впервые определена структура низкого разрешения регулируемой нуклеотидами пирофосфатазы из Desulfitobacterium hafniense, а также выявлены пространственные изменения полноразмерного фермента при его взаимодействии с аденозинмонофосфатом и диаденозинтетрафосфатом. Показано, что белок в разбавленных растворах существует как стабильный гомотетрамер, структура которого зависит от природы присоединенного лиганда. Полученные структурные результаты важны для понимания молекулярных основ регуляции ферментов этого семейства.
ВВЕДЕНИЕ
Неорганические пирофосфатазы (PPазы) – ферменты, присутствующие во всех живых организмах. Они катализируют гидролиз неорганического пирофосфата, превращая его в две молекулы фосфата и обеспечивая необходимые условия для таких реакций, как синтез белка, РНК и ДНК, делая таким образом эти ферменты незаменимыми для жизни [1]. Растворимые PPазы можно разделить на три структурно-независимых семейства (I, II, III). PPазы семейства II были открыты в 1998 г. [2, 3], и их до сих пор интенсивно исследуют [4, 5]. Эти ферменты существуют, как правило, в бактериях и архебактериях, включая патогены человека. Известно лишь несколько трехмерных структур высокого разрешения PPase семейства II [6–8]. Канонические РРазы семейства II состоят из двух каталитических доменов, связанных гибким линкером между N-концевым DHH- и C-концевым DHHA2-доменом и относятся к DHH-гидролазам, названным так по консервативному мотиву Asp–His–His [9]. Ферменты являются гомодимерами, в которых контакт осуществляется через DHH-домены (за счет формирования объединенного β-листа). Около четверти ферментов PPаз семейства II содержит вставку из 250 остатков в DHH-домене [5]. Вставка состоит из двух CBS-доменов (примерно по 60 аминокислотных остатков), названных в честь фермента цистатион-β-синтазы, в которой они были идентифицированы впервые, и DRTGG-домена (около 120 аминокислотных остатков). Эти PPазы обозначаются как CBS-PPазы. Известна кристаллическая структура регуляторной вставки из Clostridium perfringens, в которой пара CBS- и DRTGG-домен образуют гомодимерную структуру. CBS-домены довольно широко распространены в природе и обнаружены, например, в 75 белках человека и восьми белках Escherichia coli. Мутации в CBS-доменах ферментов и мембранных каналов у человека связаны с рядом наследственных заболеваний, включая гомоцист-инурию, пигментозный ретинит, синдром Бартера и остеопетроз [10]. CBS-домены всегда встречаются парами и образуют функциональный псевдодимер (модуль Бейтмана), способный связывать различные лиганды (производные аденозина, гуанозина, ионы магния и другие) [11]. Кроме того, CBS-домены участвуют в образовании структур более высокого порядка, формируя из двух модулей Бейтмана гомодимерные структуры – CBS-модули, состоящие уже из четырех CBS-доменов. В качестве лигандов CBS-доменов в CBS-PPазах выступают различные фосфатные производные аденозина [12–14]. Аденозинмонофосфат (AMФ) и аденозиндифосфат (AДФ) ингибируют CBS-PPазы, тогда как аденозинтрифосфат (АТФ) и диаденозинтетрафосфат (Ap4A) активируют его. Хотя каждая пара CBS-доменов содержит две потенциальные полости для связывания лигандов (CBS-модуль – четыре), регуляторная вставка CBS-PPазы связывает только две молекулы AMФ или одну молекулу Ap4A через оба аденозиновых фрагмента [15].
Несмотря на физиологическую важность регуляции пирофосфатаз CBS-доменами, детали базовых молекулярных механизмов функционирования этих ферментов начали выяснять недавно [16, 17]. Для полного понимания путей передачи сигнала между их активным и регуляторным центрами необходима структурная информация на атомном уровне, которая, к сожалению, на данный момент недоступна из-за трудностей кристаллизации полноразмерных CBS-PPаз. Таким образом, их структурное исследование остается актуальной задачей. Метод малоуглового рентгеновского рассеяния (МУРР) был выбран в качестве основного исследовательского подхода, поскольку его можно использовать для изучения структуры биологических объектов непосредственно в растворе, т.е. в условиях, наиболее близких к физиологическим [18, 19]. Целью настоящей работы был структурный анализ CBS-PPаз из D. hafniense (dh-PPаза) с помощью МУРР для получения информации о четвертичной структуре и олигомерном состоянии белка при его взаимодействии с лигандами. Результаты работы могут стать шагом к пониманию принципиально важных функциональных особенностей CBS-PPаз.
МАТЕРИАЛЫ И МЕТОДЫ
Выделение и очистка CBS-PPазы. Dh-PPаза выделена из клеток E. coli штамма BL21, трансформированных вектором pET-42b (Novagen), несущим соответствующий ген, как описано ранее [20], при использовании буфера Mops-KOH концентрацией 0.1 M, pH 7.2, 2 мM MgCl2, 0.1 мM CoCl2 и 150 мM KCl на стадии гель-фильтрации. Полученный белковый препарат был электрофоретически гомогенным.
Концентрацию белка определяли спектрофотометрически, используя значение $А_{{280}}^{{0.1{\text{\% }}}}$ = 0.478, рассчитанное из аминокислотного состава dh-PPазы по программе ProtParam [21]. Все эксперименты проводили в буфере Mops-KOH концентрацией 0.1 M, pH 7.2, 2 мM MgCl2, 0.1 мM CoCl2 и 150 мM KCl.
Эксперимент и анализ данных МУРР. Измерения проводили с помощью метода МУРР на станции EMBL-P12 BioSAXS накопительного кольца PETRAIII синхротрона DESY Европейской Лаборатории молекулярной биологии (EMBL) в Гамбурге, Германия. Станция оборудована роботизированной системой подачи и смены образца, 2D-детектором Pilatus 2M (DECTRIS, Switzerland). Интенсивность рассеяния регистрировали в диапазоне векторов 0.027 < s < 5.0 нм–1, длина волны рентгеновского излучения λ = 0.124 нм [22]. Измерения проводили в том же буфере, в котором был приготовлен образец, при температуре 10°C и постоянном контроле радиационного повреждения образца. Диапазон концентраций растворов белков варьировался от 1 до 3 мг/мл. Первичную обработку полученных данных проводили по стандартным методикам с использованием программы PRIMUS [23].
Радиус инерции Rg рассчитывали в приближении Гинье, которое справедливо в области sRg < 1.3:
где интенсивность рассеяния в нулевой угол I(0) определяется из экспериментальных данных Iexp(s) [24] и характеризует общее количество рассеивающей материи, пропорциональное квадрату молекулярной массы рассеивающей частицы.Молекулярные массы рассчитывали по данным МУРР двумя методами: с помощью подхода Байеса (MMBayesian) [24], в котором молекулярные массы определяли, в том числе, по интенсивности рассеяния в нулевой угол I(0) и на основе объема Порода VP (MMPorod) [25]. Последнее значение было определено с учетом того, что эмпирическое соотношение между VP и молекулярной массой белка приблизительно равно 1.65 [26].
Функция парных расстояний p(r) и максимальный размер рассеивающих частиц Dmax рассчитаны с помощью программы GNOM [27]:
Формы низкого разрешения белков dh-PPase получены с помощью метода ab initio и программы DAMMIN [28], которая использует алгоритм имитации отжига для построения моделей, соответствующих экспериментальным данным Iexp(s), что минимизирует расхождение
(3)
${{{\chi }}^{2}} = \frac{1}{{N - 1}}\mathop \sum \limits_j {{\left[ {\frac{{{{I}_{{{\text{exp}}}}}({{s}_{j}}) - c{{I}_{{{\text{calc}}}}}({{s}_{j}})}}{{{\sigma }({{s}_{j}})}}} \right]}^{2}}{\text{,}}$Анализ неоднозначности полученных ab initio-моделей выполнен с помощью программы AMBIMETER [29]. Прогнозирование форм белков и их структурную классификацию проводили с помощью DATCLASS [26].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для исключения влияния на интерпретацию данных МУРР возможного межчастичного взаимодействия в растворе кривые малоуглового рассеяния dh-PPазы измеряли при нескольких концентрациях белка в диапазоне значений от 1 до 3 мг/мл. В указанном интервале концентрационной зависимости не наблюдалось. Поэтому с целью уменьшения экспериментальных ошибок для структурного анализа и вычисления макромолекулярных характеристик белка по данным МУРР использовали кривые, измеренные при концентрации 3 мг/мл (50 мкМ), т.е. кривые с минимальными экспериментальными шумами. Для изучения структурных перестроек при взаимодействии с лигандами в раствор исходной dh-PPазы вводили AMФ или Ap4A с конечным содержанием лигандов 1000 и 100 мкМ соответственно. Полученные кривые малоуглового рассеяния представлены на рис. 1.
Рис. 1.
Экспериментальные кривые рассеяния белка dh-PPазы (а): без лиганда (1), с AМФ (2), с Aр4А (3). Графики Гинье (б, в, г) и Кратки (д, е, ж) для dh-PPазы без лиганда (б, д), с AМФ (в, е), с Aр4А (г, ж).
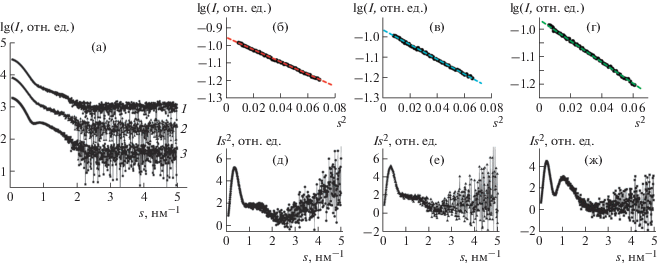
Как видно из рис. 1, профили экспериментальных кривых МУРР исходной dh-PPазы и кривых рассеяния этим ферментом при взаимодействии с лигандами AMФ и Ap4A такие же, как в случае монодисперсных соединений – на них не наблюдается увеличения интенсивности рассеяния в самых малых углах. О монодисперсности образцов свидетельствуют также графики в координатах Гинье (рис. 1б–1г): для этих образцов приближение Гинье выполняется в достаточно большом интервале углов при sRg = 0.47–1.29 для исходной dh-PPазы, 0.46–1.29 для dh-PPазы в комплексе с AМФ и 0.42–1.29 для dh-PPазы в комплексе с Aр4А.
Графики Кратки (рис. 1д–1ж) для всех изучаемых dh-PPаз имеют характерный колоколообразный вид, что указывает на то, что все образцы в основном структурированы и имеют ограниченное число разупорядоченных, гибких областей (линкеров) [30]. Наибольшее количество разупорядоченных регионов в белке в соответствии с графиком Кратки наблюдается в исходной dh-PPазе – заметно большее увеличение Is2 при увеличении вектора рассеяния s. Вероятно, присоединение лигандов в какой-то степени дополнительно структурирует фермент. Обращает на себя внимание график в координатах Кратки для комплекса dh-PPазы с Aр4А. Резко выраженный вторичный максимум на этом графике может свидетельствовать о формировании компактной, но полой структуры. Это предположение будет рассмотрено ниже при обсуждении моделирования ab initio формы dh-PPазы в растворе. Кроме того, именно для комплекса dh-PPазы с Aр4А наблюдается наиболее значительное изменение профиля кривой МУРР, что говорит о существенных конформационных изменениях фермента при связывании Ар4А (рис. 1а, кривая 3).
В табл. 1 представлены макромолекулярные характеристики (инварианты рассеяния) исследуемых CBS-РРаз. Молекулярные массы, определенные по экспериментальной кривой рассеяния, исходя из оценки объема Порода [25] и обобщенного подхода Байеса [24], равны в среднем 242 кДа, что соответствует молекулярной массе тетрамера. Эта величина практически совпадает с теоретическим значением молекулярной массы этого белка, рассчитанным по аминокислотной последовательности мономера (UniProt: B8FP42), умноженной на четыре, т.е. равной 241 кДа.
Таблица 1.
Макромолекулярные характеристики dh-PPазы с лигандами Ар4А и АМФ и без них
| Образец | Rg, нм | VP, нм3 | Dmax, нм | MMPorod, кДа | MMBayesian, кДа |
|---|---|---|---|---|---|
| dh-PPаза | 4.96 ± 0.45 | 410 ± 40 | 18 | 248 ± 20 | 242 |
| dh-PPаза с AМФ | 5.06 ± 0.23 | 388 ± 40 | 18.5 | 235 ± 20 | 243 |
| dh-PPаза с Ap4A | 5.22 ± 0.03 | 403 ± 40 | 17 | 244 ± 20 | 242 |
Радиусы инерции Rg как свободной dh-PPазы, так и в комплексах с АМФ и Ар4А оказались близкими по величине в пределах ошибки определения. В то же время наблюдается тенденция к увеличению Rg при присоединении лигандов, причем Rg образца dh-PPазы в комплексе с Aр4А, т.е. в случае присоединения тетрафосфата, несколько больше, чем при взаимодействии с монофосфатом (АМФ). Возможно, это объясняется стерическим фактором, поскольку размер Ap4A больше размера АМФ, что приводит к связыванию только одной молекулы тетрафосфата на модуль Бейтмана, в то время как туда могут поместиться две молекулы АМФ [15].
Породовский объем VP в пределах ошибки измерения одинаков для всех образцов. Этот параметр характеризует суммарное рассеяние образцом, и вклад небольших молекул лигандов практически не сказывается на его значении. Основной вывод этой части исследования заключается в констатации факта образования устойчивых тетрамеров dh-PPаз, в том числе в случае взаимодействия этого фермента с разными лигандами.
Для определения ab initio форм низкого разрешения dh-PPазы с помощью программы DAMMIN [28] были рассчитаны функции парных расстояний p(r) (рис. 2б–2г). Чтобы избежать ошибочной интерпретации, функции парных расстояний p(r) были рассчитаны по укороченным кривым малоуглового рассеяния в интервале векторов 0.027 < s < < 1.6 нм–1. Выбор такого интервала обусловлен тем, что использование полных кривых рассеяния может привести к появлению в восстановленных формах ложных мелких деталей. Метод восстановления ab initio, основанный на имитации отжига, позволяет получить форму рассеивающего объекта с разрешением порядка 2 нм. Для этого используют начальную часть кривой малоуглового рассеяния, длина которой определяется ее информационной составляющей: число шенноновских каналов Ns = smaxDmax/2π должно быть порядка десяти [31]. В данном случае для восстановления структуры низкого разрешения dh-PPазы использовали девять шенноновских каналов, для комплекса с АМФ – десять, а с Ар4А – девять. Поскольку исследуемая dh-PPаза представляет собой тетрамер, образованный двумя гомодимерами, ее структуру определяли с использованием симметрии Р2.
Рис. 2.
Экспериментальные кривые рассеяния (точки) и модельные кривые (сплошные линии), рассчитанные с помощью программы DAMMIN для форм низкого разрешения CBS-пирофосфатаз (а): dh-PPаза без лиганда (1), с AМФ (2), с Aр4А (3). Кривые смещены по вертикали на одну логарифмическую единицу для лучшей визуализации. Функции парных расстояний (б, в, г) и восстановленные формы белка (д, е, ж) dh-PPазы без лиганда (б, д), с АМФ (в, е) и Ap4A (г, ж).
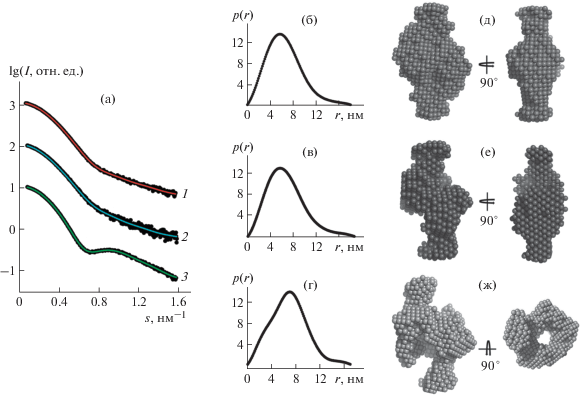
Восстановленные таким образом формы представлены на рис. 2д–2ж. О хорошем качестве и надежности восстановления свидетельствуют значения χ2 = 1.21 для исходной dh-PPазы, 1.08 и 0.94 для комплексов с АМФ и Ар4А соответственно. Dh-PPаза при связывании Ар4А формирует полую структуру, представленную восстановленной формой этого белка (рис. 2ж). Возможность формирования полой структуры dh-PPазы в комплексе с Ар4А была предсказана при анализе графика Кратки, а также с помощью программы DATCLASS [25], тогда как для dh-PPазы без лигандов или с АМФ программой DATCLASS были предсказаны просто компактные структуры, показанные на рис. 2д, 2е.
Поскольку решение обратной задачи восстановления трехмерных форм по одномерной кривой рассеяния неоднозначно, важно подчеркнуть, что анализ неоднозначности полученных ab initio-моделей, проведенный с помощью программы AMBIMETER [29], показал, что индекс неоднозначности найденных моделей меньше 1.5. Этот результат указывает на практически уникальные модели белка.
Таким образом, при использовании функции парных расстояний, симметрии Р2 и метода ab initio восстановления формы, реализованного в программе DAMMIN [28], были получены формы низкого разрешения белка исходной dh-PPазы и в комплексе с различными лигандами. Наиболее заметные конформационные изменения в четвертичной структуре белка происходят при связывании Ар4А. Можно предположить, что такие изменения вызваны природой Ap4A, поскольку этот лиганд связывается с CBS-доменами своими двумя аденозиновыми фрагментами, формируя более открытую конформацию регуляторной вставки по сравнению с АМФ [15]. Лиганд Ap4A, по-видимому, расталкивает другие фрагменты белка, переводя его в активное состояние. Это заключение отчасти подтверждается тем, что Ap4A активирует CBS-PPазу, тогда как AMФ ингибирует ее [12, 13]. То есть полученную полую структуру фермента в комплексе с Ар4А можно рассматривать в качестве активной конформации dh-PPаз, а в комплексе с АМФ – в качестве малоактивной. В целом взаимодействия субъединиц должны быть надлежащим образом сбалансированы, чтобы обеспечить передачу конформационных изменений между ними [17].
Участие регуляторных CBS-доменов в образовании не только димера, но и тетрамера соотносится с информацией о том, что канонические РРазы семейства II (содержащие только каталитические домены) являются гомодимерами [9].
ЗАКЛЮЧЕНИЕ
Полученная методом МУРР информация об олигомерной структуре dh-PPазы (тетрамер, образованный двумя гомодимерами) подтверждает многоуровневый механизм регуляции CBS-РРазы. Результаты проведенных структурных исследований могут служить базисом для понимания механизма передачи информации между регуляторными и каталитическими центрами.
Авторы выражают благодарность профессору Р. Лахти (Университет Турку, Турку, Финляндия) за предоставленную плазмиду с геном dh-PPазе.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (проект № 18-34-00918) и Министерства науки и высшего образования РФ в рамках Государственного задания ФНИЦ “Кристаллография и фотоника” РАН.
Список литературы
Heinonen J. Biological Role of Inorganic Pyrophosphate. M.: Kluwer Academic Publishers, 2001. 250 p.
Shintani T., Uchiumi T., Yonezawa T. et al. // FEBS Lett. 1998. V. 439. P. 263.
Young T., Kuhn N., Wadeson A. et al. // Microbiology. 1998. V. 144. P. 2563.
Anashkin V.A., Aksenova V.A., Salminen A. et al. // BBRC. 2015. V. 517. P. 266.
Baykov A.A., Anashkin V.A., Salminen A. et al. // FEBS Lett. 2017. V. 591. P. 3225.
Merckel M., Fabrichniy I., Salminen A. et al. // Structure. 2001. V. 94. P. 289.
Ahn S., Milner A., Fütterer K. et al. // J. Mol. Biol. 2001. V. 313. P. 797.
Rantanen M., Lehtiö L., Rajagopal L. et al. // Acta Cryst. D. 2007. V. 63. P. 738.
Aravind L., Koonin E. // Trends Cell Biol. 1998. V. 23. P. 17.
Ignoul S., Eggermont J. // Am. J. Physiol. Cell Physiol. 2005. V. 289. P. 1369.
Ereno-Orbea J., Oyenarte I., Martınez-Cruz L.A. // Arch. Biochem. Biophys. 2013. V. 540. P. 70.
Jamsen J., Tuominen H., Salminen A. et al. // Biochem. J. 2007. V. 408. P. 327.
Anashkin V.A., Salminen A., Tuominen H.K. et al. // J. Biol. Chem. 2015. V. 290. P. 27594.
Salminen A., Anashkin V.A., Lahti M. et al. // J. Biol. Chem. 2014. V. 289. P. 22865.
Tuominen H., Salminen A., Oksanen E. et al. // J. Mol. Biol. 2010. V. 398. P. 400.
Anashkin V.A., Salminen A., Osipova E. et al. // ACS Omega. 2019. V.4. P. 15549.
Anashkin V.A., Salminen A., Vorobjeva N.N. et al. // Biochem. J. 2016. V. 473. P. 2097.
Svergun D.I., Koch M.H.J., Timmins P.A. et al. Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules. Oxford: Oxford University Press, 2013. 358 p.
Korasick D.A., Tanner J.J. // Protein Sci. 2018. V. 27. P. 814.
Parfenyev A., Salminen A., Halonen P. et al. // J. Biol. Chem. 2001. V. 276. P. 24511.
Gasteiger E., Hoogland C., Gattiker A. et al. The Proteomics Protocols Handbook. N.J.: Humana Press, 2005. 607 p.
Jeffries C.M., Graewert M.A., Svergun D.I. et al. // J. Synchr. Rad. 2015. V. 22. P. 273.
Konarev P.V., Volkov V.V., Sokolova A.V. et al. // J. Appl. Cryst. 2003. V. 36. P. 1277.
Hajizadeh N.R., Franke D., Jeffries C.M. et al. // Sci. Rep. 2018. V. 8. P. 7204.
Porod G., Glatter O., Kratky O. et al. Small angle X-ray Scattering. London: Academic Press, 1982. 51 p.
Franke D., Petoukhov M.V., Konarev P.V. et al. // J. Appl. Cryst. 2017. V. 50. P. 1212.
Svergun D.I. // J. Appl. Cryst. 1992. V. 25. P. 495.
Svergun D.I. // Biophys. J. 1999. V. 76. P. 2879.
Petoukhov M.V., Svergun D.I. // Acta Cryst. D. 2015. V. 71. P. 1051.
Jacques D.A., Guss J.M., Svergun D.I. et al. // Acta Cryst. D. 2012. V. 68. P. 620.
Konarev P.V., Svergun D.I. // IUCr J. 2015. V. 2. P. 352.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография