

Кристаллография, 2020, T. 65, № 6, стр. 900-906
Ингибитор, нацеленный на границу между мономерами белка HU из Spiroplasma Melliferum, нарушает конформационную динамику и ДНК-связывающие свойства белка
Ю. К. Агапова 1, *, Д. А. Алтухов 1, Д. Э. Камашев 2, 3, В. И. Тимофеев 1, 4, Е. В. Смирнова 3, Т. В. Ракитина 1, 3, **
1 Курчатовский комплекс НБИКС-природоподобных технологий,
Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 Первый Московский государственный медицинский университет им. И.М. Сеченова Минздрава России
Москва, Россия
3 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
4 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
* E-mail: agapova.jk@gmail.com
** E-mail: taniarakitina@yahoo.com
Поступила в редакцию 03.06.2020
После доработки 03.06.2020
Принята к публикации 10.06.2020
Аннотация
Гистоноподобные HU-белки являются глобальными регуляторами топологии бактериального генома и участвуют в регуляции транскрипции. Они необходимы для выживания простейших микроорганизмов класса Mollicutes, ряда патогенных бактерий, включая Mycobacterium tuberculosis и Streptococcus pneumonia, и являются перспективной мишенью для разработки новых антибиотиков. Однако конкурентное ингибирование ДНК-связывающей способности HU-белков малоэффективно из-за высокой подвижности и большого размера ДНК-связывающего сайта. Поэтому использован альтернативный подход, основанный на поиске молекул, взаимодействующих с границей между двумя мономерами. Пространственная структура HU-белка из Spiroplasma melliferum (HUSpm) использована в качестве мишени для виртуального скрининга химической библиотеки, а также для моделирования комплекса HUSpm с ингибитором. Анализ молекулярно-динамической траектории, полученной для комплекса HUSpm–ингибитор, подтвердил, что связывание ингибитора на границе между мономерами приводит к нарушению конформационной динамики в области ДНК-связывающего домена. С помощью метода торможения в геле показано, что ингибитор действительно нарушает ДНК-связывающие свойства белка, что указывает на перспективность использования предложенного подхода.
ВВЕДЕНИЕ
Известно, что белковая кристаллография и рентгеноструктурный анализ активно используются для создания таргетных, направленных на определенную белковую мишень, лекарств. В данном процессе используют методы виртуального скрининга и молекулярного докинга, а также пространственную структуру белка-мишени, полученную с высоким разрешением [1, 2]. Кроме того, чтобы подтвердить взаимодействие найденной in silico молекулы с белком-мишенью, проводят кристаллизацию белка в комплексе с ингибитором. Если закристаллизовать комплекс не удается, то используют методы моделирования и молекулярной динамики (МД). На основании структурных данных, полученных для белка-мишени в свободной форме, сначала моделируют комплекс белка с ингибитором, а затем анализируют поведение комплекса с помощью МД-эксперимента.
Данная работа является наглядным примером использования описанного выше подхода для разработки химических ингибиторов гистоноподобных HU-белков, которые являются глобальными регуляторами топологии бактериального генома [3]. Ингибиторы HU-белков могут стать основой для получения новых антибиотиков [4]. Гистоноподобные HU-белки выполняют свою функциональную роль путем связывания и изгибания геномной ДНК бактерий [5]. Они участвуют в регуляции транскрипции и, как следствие, транскриптомного и протеомного профиля бактериальной клетки [6]. HU-белки необходимы для выживания простейших микроорганизмов класса Mollicutes [7], а также ряда других патогенных бактерий, включая Mycobacterium tuberculosis [8] и Streptococcus pneumonia [9].
Ранее найденные ингибиторы ДНК-связывающей способности HU-белков были направлены на ДНК-связывающий сайт [4]. Однако большой размер и подвижность ДНК-связывающего сайта, а также высокая аффинность HU-белков к ДНК существенно затрудняли поиск таких ингибиторов. Поэтому в данной работе использовали альтернативный подход, основанный на поиске молекул, взаимодействующих с границей между двумя мономерами HU-белка. Перспективность данного подхода и его преимущества по сравнению с разработкой ингибиторов, направленных на ДНК-связывающий сайт, была исследована ранее [10, 11].
Пространственная структура HU-белка из Spiroplasma melliferum (HUSpm) (PDB ID 5L8Z), полученная на синхротронном источнике НИЦ “Курчатовский институт” с разрешением 1.36 Å [12, 13], была использована в качестве мишени для виртуального скрининга химической библиотеки компании Vitas-M laboratory (https://vitasmlab.biz/), содержащей 1 400 000 соединений. Способность выявленных соединений подавлять ДНК-связывающую способность HUSpm была исследована in vitro методом торможения в геле [5]. Комплекс найденного ингибитора с HUSpm построен и изучен в МД-эксперименте, параметры которого были подобраны ранее при исследовании конформационной динамики микоплазменных HU-белков в свободной форме [14, 15].
Анализ МД-траектории показал, что связывание ингибитора в межграничной области димеров приводит к изменению конформационной динамики ДНК-связывающего домена, что в свою очередь влияет на эффективность ДНК-связывания. Снижение эффективности ДНК-связывания приводит к нарушению нормального функционирования HU-белка. Данный подход может быть использован при разработке низкомолекулярных ингибиторов целого ряда ДНК-связывающих мультимерных белков.
МАТЕРИАЛЫ И МЕТОДЫ
Виртуальный скрининг химической библиотеки. В качестве мишени для поиска химических ингибиторов, направленных на границу между двумя мономерами HU-белка, выбрана пространственная структура HUSpm (PDB ID 5L8Z), полученная методом рентгеноструктурного анализа с разрешением 1.36 Å [12, 13]. Библиотека химических соединений, содержащая 1 400 000 молекул, была получена от компании VitasM laboratories (http://www.vitasmlab.com/). Основные этапы скрининга: подготовка мономера HUSpm, скрининг библиотеки и структурная фильтрация результатов. Для моделирования по гомологии неразрешенных участков белка использовали программу Modeller [16], для оптимизации боковых радикалов – программу Mutate [17]. Молекулярный докинг проводили с помощью программы LeadFinder [18], структурную фильтрацию – с помощью программы dock_filter [19]. Визуализацию результатов моделирования проводили в программе VMD [20].
Валидация результатов виртуального скрининга методом торможения в геле. Метод основан на разности молекулярных масс комплекса белок–ДНК и свободной ДНК, которая обусловливает различную подвижность в не денатурирующем полиакриламидном геле при проведении электрофореза. При проведении анализа использовали два микоплазменных HU-белка HUSpm и HU-белок Mycoplasma gallisepticum (HUMgal), полученные как описано в [13, 21, 22], а также короткий двухцепочечный олигонуклеотидный дуплекс, полученный из олигонуклеотидов CrJ_A3 (5'-Cy3-CCGTCGTAGCAA GAAAGACTCAACTGCACT-3') и cr_D (5'-AGTGCAGTTGAGTCCTT GCTACGACGG-3'), Cy3 – флуоресцентная метка.
Электрофорез проводили в не денатурирующем полиакриламидном 10%-ном геле и 100 мМ Трис-боратном буфере. Детекцию результатов проводили на BIO RAD Faros FX Molecular Imager (532 nm EX, 605 nm BP), а анализ результатов с помощью программ Quantity One и GraphPad Prism 5.
Моделирование комплекса ингибитора с димером HUSpm и его исследование с помощью методов молекулярной динамики. Приготовление ДНК-белкового комплекса осуществляли с помощью интерактивной графической программы COOT [23]. Модель HUSpm была наложена на структуру HU-белка Borrelia burgdorferi в комплексе с ДНК (PDB ID 2NP2) [24], координаты ДНК были перенесены на модель HUSpm. После этого провели замену и удаление избытка олигонуклеотидов.
Параметризацию ингибитора проводили при помощи программного пакета ACPYPE [25].
Полученные комплексы изучены в МД-эксперименте. Для проведения МД-расчетов использовали программный пакет GROMACS [26]. В качестве силового поля выбрано поле AMBER99SB [27]. Комплексы были помещены в кубическую ячейку, заполненную водой с добавлением ионов в количестве, необходимом для того, чтобы суммарный заряд систем оставался нулевым. В качестве модели молекулы воды выбрана TIP3P.
На первом этапе МД-эксперимента провели минимизацию энергии комплекса для релаксации любых возможных искажений. Затем на подвижность атомов системы были наложены ограничения, при помощи симуляции системы в баростате и термостате установлены температура в 300 K и давление 1 атм. После снятия всех ограничений провели МД-эксперимент, параметры которого представлены в табл. 1.
Таблица 1.
Параметры молекулярно-динамического эксперимента
| Система в термостате | |
| Термостат | Berendsen |
| Т, K | 300 |
| Система в баростате | |
| Баростат | Parrinello-Rahman |
| Устанавливаемое давление, атм | 1 |
| Т, K | 300 |
| Продолжительность NVT- и NPT- динамики, пс | 100 |
| Шаг временной сетки, пс | 0.002 |
| Молекулярная динамика | |
| Шаг временной сетки dt, пс | 0.002 |
| Количество шагов | 50 000 000 |
| Длина траектории, нс | 100 |
| Интегратор | Leap-frog |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
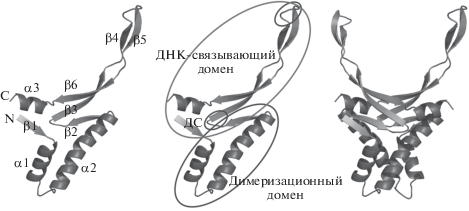
HU-белки представляют собой димеры. Каждый мономер в димере можно разделить на два домена: α-спиральный димеризационный домен и β-ленточный седлообразный ДНК-связываюший домен (рис. 1). В ДНК-связывающем домене можно выделить подвижные в отсутствие ДНК “руки”, состоящие из β-слоев 4 и 5 и примыкающих к ним петель, и стабильную ДНК-связывающую платформу, включающую в себя β-слои 3 и 6 и α-спираль 3, тогда как β-слой 2 и петля между β-слоями 2 и 3 участвуют в формировании границы между мономерами [5, 14, 15].
Рис. 1.
Модели мономера и димера HUSpm. На левой панели показаны элементы вторичной структуры, в центре – разделение мономера на домены. Окружностями обозначены функционально важные участки белка: димеризационный сигнал (ДС) и петля, интеркалирующая в малую бороздку ДНК.
Целью работы был поиск ингибиторов, взаимодействующих с границей между двумя мономерами. В связи с прочностью границы HUSpm [13] из результатов виртуального скрининга и докинга отобрали соединения, прочность связывания которых превосходила 10 ккал/моль. В результате анализа отобрали четыре молекулы, для которых структуры предсказанных комплексов не имели стерических ограничений. Структурные формулы потенциальных ингибиторов представлены на рис. 2, а энергия их связывания с белком в табл. 2.
Рис. 2.
Соединения из коллекции VitasM laboratories (http://www.vitasmlab.com/), отобранные при виртуальном скрининге.
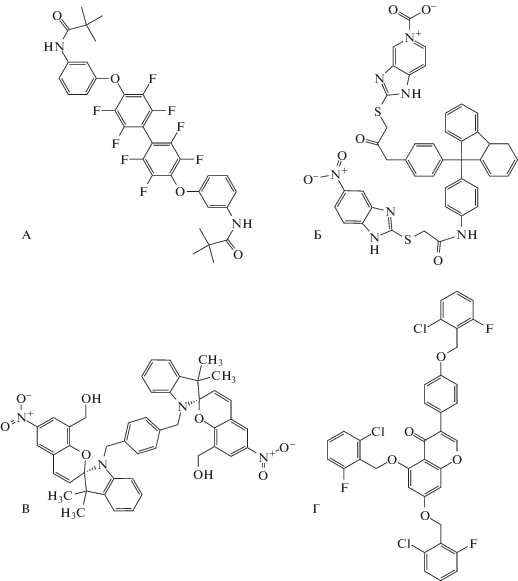
Таблица 2.
Соединения из коллекции VitasM laboratories (http://www.vitasmlab.com/), отобранные при виртуальном скрининге, и расчетные энергии связывания
| Панель на рис. 2 | Номер в коллекции | ΔGcalc, ккал/моль |
|---|---|---|
| A | STK045088 | –10.69 |
| Б | STK125332 | –10.72 |
| В | STK368440 | –10.29 |
| Г | STK888563 | –10.43 |
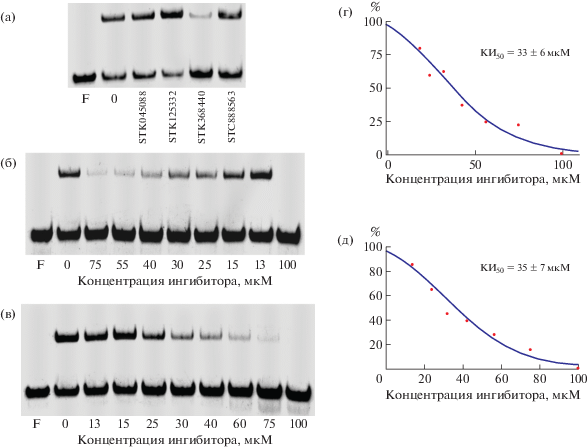
Все потенциальные ингибиторы протестировали на способность ингибировать ДНК-связывающую HUSpm c помощью метода торможения в геле (рис. 3). Как видно из рисунка, только соединение STK368440 обладало ингибирующей способностью в концентрации 100 мкM и было использовано для получения кривой дозовой зависимости и определения концентрации полумаксимального ингибирования (КИ50), снижающей эффективность связывания белка с ДНК на 50% (рис. 3б–3г). В данном эксперименте ингибирующую способность STK368440 исследовали в отношении двух микоплазменных белков HUSpm и HUMgal, полученные КИ50 составляли 33 ± 6 и 35 ± 7 мкМ для HUSpm и HUMgal соответственно.
Рис. 3.
Влияние соединений, отобранных при виртуальном скрининге, на ДНК-связывающую способность HU-белков, определенное методом торможения в геле: а – эффект 100 мкМ концентрации соединений на ДНК-связывающую способность HUSpm; б–д – концентрационные зависимости эффективности ингибирования ДНК-связывания соединением STK368440 (б, в), кривые дозовой зависимости и концентрации полумаксимального ингибирования (КИ50) (г, д), полученные для HUSpm (б, г) и HUMgal (в, д). F – свободная ДНК.
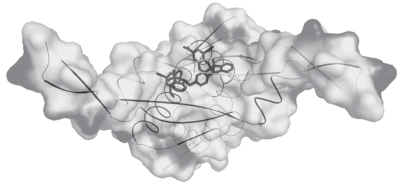
Несмотря на большой предварительный опыт кристаллизации HU-белков [12, 21, 28], в настоящей работе не удалось получить кристаллы комплексов HUSpm с STK368440, поэтому для исследования взаимодействия ингибитора с белком использовали методы моделирования и молекулярной динамики. Модель STK368440 на границе между двумя мономерами HUSpm представлена на рис. 4. МД-симуляция этой модели, с одной стороны, позволяла оценить устойчивость комплекса, а с другой стороны, узнать, как меняется конформационная динамика белка-мишени после связывания ингибитора.
Рис. 4.
Модель ингибитора STK368440, взаимодействующего с границей между двумя мономерами в области перехода α-спирального домена в ДНК-связывающий. Белок показан как заряженная поверхность с элементами вторичной структуры.
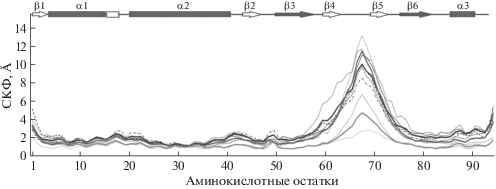
Был проведен анализ флуктуаций основной цепи HUSpm в свободном виде, в комплексе с модельной ДНК и в комплексе с ингибитором STK368440, наблюдаемых при 100 нс МД-симуляциях (рис. 4). Как видно из рисунка, в свободном HUSpm ДНК-связывающий домен, особенно его участок между β3- и β6-слоями, обладает значительной подвижностью. Именно такое поведение HU-белков в растворе наблюдалось при их исследованиях с помощью ЯМР-спектроскопии [14, 15]. В комплексе с ДНК подвижность ДНК-связывающего домена уменьшается на всем его протяжении (от β2-слоя до С-концевой α3-спирали). Это происходит из-за перехода молекулы в конформацию захвата ДНК, в которой подвижные β-ленточные “руки” обхватывают ДНК, а петля между β4- и β5-слоями интеркалирует в малую бороздку двойной спирали [24]. При взаимодействии с ингибитором наблюдается увеличение подвижности в области перехода α-спирального домена в ДНК-связывающий домен (предполагаемое место связывания ингибитора), а также в наиболее стабильных областях ДНК-связывающего домена (β3-, β6-слой и α3-спираль), которые обеспечивают адаптацию седловидной основы ДНК-связывающего домена при переходе белка из свободного состояния в конформацию захвата ДНК, что предположительно обеспечивает высокую прочность ДНК-белковых комплексов (константы диссоциации в наномолярной области) [5]. Наблюдаемое нарушение конформационной динамики сопровождается нарушением ДНК-связывающей способности (рис. 5), что подтверждает предположение о возможности контролировать ДНК-связывающие свойства HU-белков путем воздействия на границу между мономерами.
Рис. 5.
Среднеквадратичная флуктуация (СКФ в Å) Сα-атомов в течение 100 нс МД-симуляции, вычисленная для свободного HUSpm (темно-серые линии), HUSpm в комплексе с модельной ДНК (светло-серые линии) и в комплексе с ингибитором STK368440 (черные линии). Разрывными линиями показаны результаты независимых МД-экспериментов, сплошными – усредненные значения. Вторичная структура HUSpm, взятая из [15], показана сверху.
Вероятно, подобный подход может быть использован при разработке низкомолекулярных ингибиторов, специфичных к другим нуклеоид-ассоциированным белкам, которые вместе с белком HU поддерживают топологию бактериального генома.
Работы в части исследования комплексов HU-белков с ингибиторами выполнены при финансовой поддержке Российского фонда фундаментальных исследований (грант № 20-04-01001), в части поиска ингибиторов – Программы президиума РАН “Молекулярная и клеточная биология” (грант № 0101-2014-0086). Технология проведения МД-экспериментов разработана при поддержке НИЦ “Курчатовский институт” (приказ № 1363 от 25.06.2019), поиск условий кристаллизации комплексов проводили при частичной поддержке Министерства науки и высшего образования в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН.
Список литературы
Drews J. // Science. 2000. V. 287. P. 1960.
Anderson A. // Chem. Biol. 2003. V. 10. P. 787.
Stojkova P., Spidlova P., Stulik J. // Front. Cell. Infect. Microbiol. 2019. V. 9. P. 159.
Bhowmick T., Ghosh S., Dixit K. // Nature Commun. 2014. V. 5. P. 4124.
Kamashev D.E., Agapova Y.K., Rastorguev S. et al. // PLOS. 2017. V. 12. P. e188037.
Камашев Д.Э., Ракитина Т.В., Матюшкина Д.С. и др. // Биоорган. химия 2019. Т. 45. С. 524.
Glass J.I., Assad-Garcia N., Alperovich N. et al. // Proc. Natl. Acad. Sci. USA. 2006. V. 103. P. 425.
Griffin J.E., Jeffrey, Gawronsk D. et al. // PLoS Pathog. 2011. V. 7. P. e1002251.
Ferrándiz M.-J., Carreño D., Ayora S. et al. // Front. Microbiol. 2018. V. 9. P. 493.
Талызина А.А., Агапова Ю.К., Подшивалов Д.Д. и др. // Кристаллография. 2017. Т. 62 С. 903.
Agapova Y.K., Talyzina A.A., Altukhov D.A. et al. // Crystallography reports. 2019. V. 64. P. 602.
Boyko K.G., Rakitina T.V., Korzhenevskiy D.A. et al. // Acta Cryst. F. 2015. V. 71. P. 24.
Boyko K.M., Gorbacheva M.A., Rakitina T.V. et al. // Sci Rep. 2016. V. 3 P. 36366.
Altukhov D.A., Talyzina A.A., Agapova Y.K. et al. // J. Biomol. Struct. Dyn. 2016. V. 36. P. 45.
Timofeev V.I., Altukhov D.A., Talyzina A.A. // J. Biomol. Struct. Dyn. 2018. V. 36. P. 4392.
Sali A., Blundell T.L. // J. Mol. Biol. 1993. V. 234. P. 779.
Строганов О.В., Чилов Г.Г., Новиков Ф.Н. и др. // Программа Mutate. 2011. p. Свидетельство о государственной регистрации программы для ЭВМ 2011615051.
Stroganov O.V., Novikov F.N., Stroylov V.S. et al. // J. Chem. Inf. Model. 2008. V. 48. P. 2371.
Novikov F.N., Stroylov V.S., Stroganov O.V. et al. // J. Mol. Model. 2010. V. 16. P. 1223.
Humphrey W., Dalke A., Schulten K. // J. Mol. Graph. 1996. V. 14. P. 33.
Николаева А.Ю., Тимофеев В.И., Бойко К.М. и др. // Кристаллография. 2015. Т. 60. С. 922.
Алтухов Д.А., Агапова Ю.К., Власкина А.В. и др. // Вестн. МГУ. 2016. Т. 57. С. 226.
Emsley P., Lohkamp B., Scott W.G. et al. // Acta Cryst. D 2010. V. 66. P. 486.
Mouw K.W., Rice P.A. // Mol. Microbiol. 2007. V. 63. P. 1319.
Trott O., Olson A.J. // J. Comput. Chem. 2010. V. 31. P. 455.
Abraham R.G., Tanvir1 N.R., Santiago B.X. et al. // SoftwareX. 2015. P. 19.
Van Der Spoel D., Lindahl E., Hess B. et al. // J. Comput. Chem. 2005. V. 26. P. 1701.
Бойко К.М., Тимофеев В.И., Самыгина В.Р. и др. // Кристаллография. 2016. Т. 61. С. 691.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография