

Кристаллография, 2021, T. 66, № 1, стр. 13-31
“Гибкие” каркасные структуры и физические свойства соединений с переходными металлами, производных от элленбергита и β-тридимита
Л. В. Шванская 1, *, О. В. Якубович 1
1 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: lshvanskaya@mail.ru
Поступила в редакцию 13.07.2020
После доработки 29.07.2020
Принята к публикации 29.07.2020
Аннотация
Проанализированы особенности кристаллохимии и физических свойств фосфатов (арсенатов, ванадатов) щелочных и переходных металлов, производных от минералов элленбергерита (Mg,Ti,Zr,$\square $)2Mg6(A1,Mg)6(Si,P)2Si6O28(OH)10 и β-тридимита SiO2, в аспекте “гибкости” их принципиально различных каркасов. Обсуждается кристаллохимическая функция катионов переходных металлов в структурах элленбергерито- и тридимитоподобных аналогов. Приведены данные о методах их получения, термодинамических и электрических свойствах. Исследованы причины структурных различий тридимитоподобных фаз и связь полиморфизма с вариациями координационных полиэдров переходных металлов, стремящихся к наиболее устойчивой конфигурации.
ОГЛАВЛЕНИЕ
Введение
1. Кристаллохимия и физические свойства аналогов элленбергерита с октаэдрическими каркасами переходных металлов
2. β-тридимит и его производные: кристаллохимия и физические свойства соединений с тетраэдрическими каркасами на основе шестичленных колец
Заключение
ВВЕДЕНИЕ
Синтетические аналоги фосфатных минералов с переходными металлами широко используются в качестве катализаторов, сорбентов, биоматериалов, молекулярных сит, ионообменников, матриц электродов литиевых и натриевых батарей, а также нелинейно-оптических и магнитных материалов. На основе анализа химических формул более 200 минералов фосфора показано, что чаще всего в фосфатах встречаются катионы Fe2+ и Fe3+ (95 соединений), А13+ (60), Са2+ (56), Мn2+ и Mn3+ (45) [1]. Согласно “кластерной гипотезе” сохраняющиеся принципы формирования совокупностей многогранников (основных строительных блоков) в кристаллических структурах огромного количества фосфатных минералов относительно немногочисленны. Эти “кластеры”, чаще всего образующиеся при объединении октаэдров через общие вершины, ребра или грани в постройки различной размерности – от островных до каркасных, “сохраняются в пределах разумной геометрической репликации в довольно обширном диапазоне разнообразных структур” [2].
Кристаллические структуры фосфатов переходных металлов включают в себя нульмерные, одномерные, двумерные и трехмерные ассоциации магнитных ионов, обменные взаимодействия между которыми могут находиться под влиянием анизотропии и фрустраций. Материалы, характеризующиеся такими структурами, так называемые низкоразмерные магнетики, являются предметом интенсивного изучения, поскольку в объектах этого типа могут быть реализованы редкие основные состояния спинов атомов переходного металла, такие как спиновые жидкости, спиновые стекла, конденсация Бозе–Эйнштейна и другие. Проявления низкоразмерного магнетизма возможны в структурах, в которых кластеры магнитоактивных ионов окружены магнитоинертными группировками. Фосфаты пегматитов с переходными металлами отвечают этим требованиям. Однако многочисленные примеси, часто присутствующие в составе природных объектов, не позволяют использовать их в экспериментальных исследованиях и последующих технологических разработках. Эта проблема решается путем получения синтетических аналогов минералов в лаборатории в контролируемых условиях химического состава кристаллизационных сред, температуры (и давления).
В настоящей работе рассмотрены минералоподобные фосфаты переходных металлов двух семейств, в которых реализуются различные варианты магнитных взаимодействий. В первом разделе проанализированы кристаллические структуры минерала элленбергерита и его синтетических модификаций различного состава. Второй раздел посвящен обсуждению кристаллохимии фосфатов Co и Mn, кристаллические структуры которых производны от структуры β-тридимита. Магнитные свойства соединений интерпретированы в контексте их обусловленности особенностями строения и размерности магнитных подсистем.
1. КРИСТАЛЛОХИМИЯ И ФИЗИЧЕСКИЕ СВОЙСТВА АНАЛОГОВ ЭЛЛЕНБЕРГЕРИТА С ОКТАЭДРИЧЕСКИМИ КАРКАСАМИ ПЕРЕХОДНЫХ МЕТАЛЛОВ
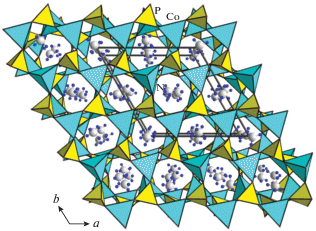
Элленбергерит, (Mg,Ti,Zr,$\square $)2Mg6(A1,Mg)6 (Si,P)2Si6O28(OH)10, акцессорный минерал метаморфических пород, образующийся в условиях высоких давлений (25–30 кбар) при температурах 700–800°С (параметры элементарной ячейки a = = b = 12.255 Å, c = 4.932 Å, пр. гр. P63) [3]. Основой его кристаллической структуры является каркас из связанных вершинами сдвоенных цепочек октаэдров алюминия и магния, делящих общие ребра и грани [3, 4] (рис. 1а, 1б). Каркас содержит каналы двух типов: узкие, ограниченные окнами треугольного сечения, и широкие, гексагонального типа. В узких каналах статистически располагаются тетраэдры PO4 и SiO4. Тетраэдры SiO4 находятся и в широких каналах. В центрах каналов гексагонального сечения вдоль оси с вытянуты цепочки из связанных гранями октаэдров, которые заселены атомами Mg, Ti и Zr и содержат 33% вакансий. В рамках данного структурного типа реализуются разновидности сложных составов: обогащенная фосфором фаза (Mg0.61Ti0.08$\square $0.31)2(Mg0.52Al0.43$\square $0.05)12[SiO3(O0.29OH0.71)]6 [(P0.71Si0.20$\square $0.09)O3OH]2(OH)6 [5] и фосфоэлленбергерит (Mg,Fe,$\square $)2Mg12(PO4, PO3OH,AsO4)6 (PO3OH,CO3)(OH)6 [6]. Для них наблюдается гетеровалентный изоморфизм Si4+ + Al3+ ↔ P5+ + Mg2+. Оба минерала кристаллизуются в высокосимметричной пр. гр. Р63mc по сравнению с элленбергеритом, в структуре которого атомы алюминия и магния упорядочены в цепочках сдвоенных октаэдров. В структуре фосфоэлленбергерита тетраэдры [HPO4] на осях третьего порядка частично замещены треугольными группами [СО3]; тетраэдрическая позиция внутри каналов шестиугольного сечения занята атомами фосфора и мышьяка в соотношении 1 : 1.
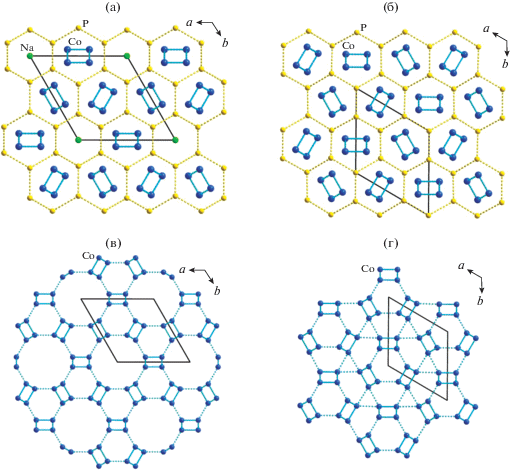
Рис. 1.
Кристаллические структуры: а – элленбергерита в проекции на плоскость ab; б – основные структурные фрагменты и обобщенная формула элленбергерит-дюмортьеритоподобных соединений; в – ориентация тетраэдров гексагональных и тригональных каналов в структурах элленбергеритов (слева) и Mn7 – 2/3z(OH)3(VO4)4 – 2z(V2O7)z (справа), закрашенные тетраэдры соответствуют заселенным позициям, пустые – вакансии в позиции кислорода О6; г – экатита в проекции на плоскость ab.
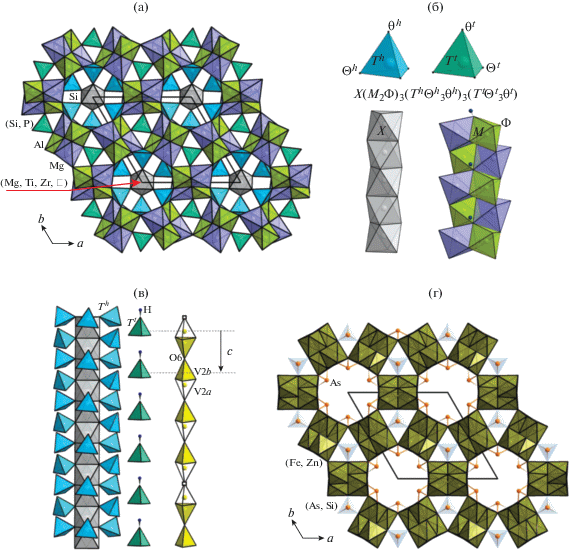
К семейству элленбергерита относят также арсенат экатит, (Fe3+,Fe2+,Zn)6(AsO3)3(AsO3,SiO3OH)(OH)3 [7], в структуре которого отсутствуют цепочки октаэдров в центре каналов шестиугольного сечения, а сдвоенные цепочки из полиэдров (Fe3+,Fe2+,Zn)O4(OH)2 соединяются с зонтичными группировками AsO3. Такие арсенитные группы наряду с кремниевыми тетраэдрами располагаются и в узких каналах структуры (рис. 1г). Было предложено называть минеральные и синтетические фазы групп элленбергерита и дюмортьерита (ромбического боросиликата, (Al,$\square $)Al6BSi3O16(O,OH)2, пр. гр. Pnma) “дюмортьеритоподобными материалами” [8]. Однако различия между кристаллическими структурами минералов групп элленбергерита и дюмортьерита не позволяют объединить их в одну супергруппу [7, 9, 10]. Эти различия связаны не только с составом катионных позиций в кислородных октаэдрах, треугольниках и тетраэдрах. Различаются также способы сочленения октаэдров в двойные цепочки, ориентация тетраэдров в широких каналах и ориентация сдвоенных цепочек, вытянутых вдоль [001]. Все перечисленное свидетельствует о малой вероятности перестройки ромбической структуры дюмортьерита в гексагональную структуру элленбергерита [9, 10].
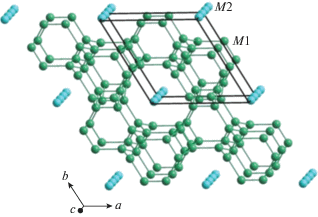
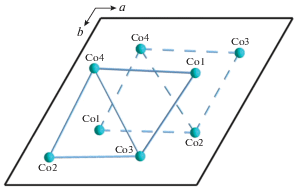
Наличие большого числа разновидностей внутри каждой минеральной группы (дюмортьерита и элленбергерита) означает высокую изоморфную емкость их каркасных структур, что определяет перспективность целенаправленного дизайна новых соединений на их основе. С точки зрения магнитных свойств элленбергеритоподобная топология катионного каркаса представляет собой весьма интересный случай сочетания двух взаимопроникающих подструктур различной размерности, которые потенциально могут быть образованы атомами переходных металлов одного или нескольких сортов (рис. 2).
К настоящему моменту синтезированы аналоги обсуждаемых минералов из разных химических классов. Это арсенаты, фосфаты, фосфиты, ванадаты, сульфато-ванадаты и халькогениды, большинство из которых рассмотрено в обзоре [8]. Все соединения получены в гидротермальных условиях и кристаллизуются в пр. гр. P63mc. Их химические формулы различны, однако могут быть связаны обобщенной формулой [11], которая релевантна для всех представителей семейств дюмортьерита и элленбергерита: X(M2Φ)3 (ThΘ$_{3}^{h}$θh)3(TtΘ$_{3}^{t}$θt) (катионы обозначены заглавными латинскими буквами, анионы – греческими) [8]. В этой формуле фактически фигурируют основные структурные элементы: M2Φ – двойные цепочки октаэдров; (ThΘ$_{3}^{h}$θh) и (TtΘ$_{3}^{t}$θt) – тетраэдрические/пирамидальные/треугольные группировки в каналах гексагонального (h) и треугольного (t) сечения, X – катионы в широких каналах (рис. 1б). Поскольку элленбергеритоподобные соединения различаются химическим составом и стехиометрией, изменение состава катионных позиций закономерно коррелирует с вариациями состава анионной части структуры. Это отсутствие или наличие апикальных атомов кислорода θh, θt и степень протонирования кислородных позиций. Полярный характер пр. гр. P63mc предполагает сонаправленную либо противоположно направленную ориентацию апикальных вершин групп (TtΘ$_{3}^{t}$θt) и (ThΘ$_{3}^{h}$θh). Первый случай реализуется в кристаллической структуре собственно элленбергерита [4] (рис. 1а, 1в).
В табл. 1 представлены элленбергеритоподобные соединения на основе фосфора, ванадия и мышьяка с магнитоактивными катионами переходных металлов, особенности кристаллохимии и физических свойств которых рассмотрены ниже. Первые элленбергеритоподобные фосфаты были получены [13] при исследовании возможности синтеза Ni- и Co-аналогов вторичных минералов гомологической серии вивианита, Fe3(H2О)n (PO4)2, формирующихся в низкотемпературных условиях зоны окисления месторождений металлов, в гранитных пегматитах, в глинах и глауконитах, а также в молодых аллювиальных отложениях, замещающих органический материал. В кристаллической структуре вивианита изолированные октаэдры и димеры из октаэдров железа связаны тетраэдрами фосфора и образуют слои, объединенные в трехмерную постройку водородными связями. Закономерно, что повышение температуры и давления в условиях эксперимента привело к образованию конденсированных фаз с трехмерным катионным каркасом, состоящим из связанных вершинами, ребрами и гранями октаэдров Со2+ или Ni2+. Позднее были получены арсенаты этих металлов и фосфатные фазы, содержащие Mg (табл. 1).
Таблица 1.
Кристаллографические характеристики и физические свойства P-, V-, As-элленбергеритоподобных фаз с атомами переходных металлов
| *Формула | Параметры элементарной
ячейки, Å α = β = 90°, γ = 120° |
Физические свойства | Заселен-ность Х | Литера-тура | ||
|---|---|---|---|---|---|---|
| a = b | с | V, Å3 | ||||
| Фосфаты и арсенаты **(Xу${{\square }_{{2/{v}--y}}}$)(M2OH)3[(TO4)$_{{1 + {v}y}}$(TO3OH)$_{{2--{v}y}}$](TO3OH), y – заселенность, ${v}$ – степень окисления; X = Na/Co/Ni/Mg, M = Co/Ni/(Mg, Ni), Т = As/P |
||||||
| #Na2 – xCo6(OH)3[HPO4][Hx/3PO4]3, x = 1.1 | 12.630(3) | 5.0170(10) | 693.1 | TN = 44 K | 0.9 | [12] |
| Co12 + xH4 – 2x(PO4)6(HPO4)2(OH)6, x = 1.22 | 12.580(2) | 5.0305(5) | 689.4 | TN = 15 K, аномалии ∼15, 50 K | 0.61 | [13, 14] |
| Ni12 + xH4 – 2x(PO4)6(HPO4)2(OH)6, x = 0.78 | – | – | – | не наблюдалось упорядочения до 2 K | 0.39 | [13] |
| Ni12 + xH6 – 2x(PO4)8(OH)6, x = 0.76 | 12.4697(3) | 4.9531(1) | 666.9 | нет данных | 0.38 | [14] |
| Mg8Ni4H6(PO4)8(OH)6 | 12.450(3) | 4.7491(4) | 637.5 | не наблюдалось упорядочения до 4.2 K | 0 | [15] |
| Ni12 + xH6 – 2x(AsO4)8(OH)6, x = 0.78 | 12.678(4) | 5.0259(4) | 699.6 | нет данных | 0.39 | [14] |
| Ni12 + xH6 – 2x(AsO4)8(OH)6, x = 1.16 | 12.7018(2) | 5.03223(9) | 703.11 | 0.58 | [16] | |
| Ni12 + xH6 – 2x(AsO4)8(OH)6, x = 1.33 | 12.6953(2) | 5.0311(1) | 702.23 | 0.66 | [16] | |
| #Сo1 – xCo6(OH)3(AsO4H2x/3)3(HAsO4), x = 1/3 | 12.8279(6) | 5.0919(2) | 725.64 | TN = 51 K | 0.66 | [17] |
| #Ni1 – xNi6(OH)3(AsO4H2x/3)3(HAsO4), x = 2/3 | 12.7021(2) | 4.9966(10) | 698.2 | TN = 42 K | 0.33 | [17] |
| Mg7.5Ni6H3(AsO4)8(OH)6 | 12.419(4) | 4.974(2) | 664.3 | не наблюдалось упорядочения до 4.2 K | 0.75 | [15] |
| Ванадаты $X_{{1 - 2/3z}}^{{3 + }}$$\square $2/3z (M2ОH)3(VО4)3[(VО4)1 – 2z(V2О7)z], z – степень полимеризации, Х = Mn3+, M = Mn2+ или Х2+(M2(OH)2/3(H2O)1/3))3(VO4)3(VO4), где Х = M = Co, Mg |
||||||
| Mn7 – 2/3z(OH)3(VO4)4 – 2z(V2O7)z, z = 0.2 | 13.229(1) | 5.255(1) | нет данных | 0.8 | [18, 19] | |
| #Co7V4O16(OH)2(H2O) | 12.917(2) | 5.0944 | 736.12 | TN = 71 K; метамагнитный переход |
не уточнялась | [20] |
| Фосфиты $\square $[(M1 – x$\square $x)2OH]3(HPO3)3(HPO3), x = 1/12, M = Mn, Fe, Co, Ni |
||||||
| Mn11(HPO3)8(OH)6 | 13.1957(6) | 5.1770(3) | 780.68 | вакантна | [21] | |
| #Fe11(HPO3)8(OH)6 | 12.9994(8) | 5.0932(3) | 745.36 | вакантна | [21] | |
| Co11(HPO3)8(OH)6 | 12.8244(4) | 4.9734(2) | 708.37 | нет данных | вакантна | [22] |
| Ni11(HPO3)8(OH)6 | 12.6329(4) | 4.9040(2) | 677.77 | вакантна | [22] | |
| Ni11(HPO3)8(OH)6 | 12.4589(1) | 4.9543(1) | 665.77 | TN = 67 K | вакантна | [23] |
Кристаллические структуры фосфатов и арсенатов различаются характером заполнения каналов с шестиугольными окнами, т.е. фактически особенностью заселения катионами позиции X в обобщенной формуле. Степень заселенности этой позиции имеет принципиальное значение, поскольку ее величина характеризует размерность катионной начинки каналов. Так, при факторе занятости (ф.з.) меньше 0.5 в широких каналах структуры, “центрированных” осью 63, располагаются изолированные октаэдры металла. В случае ф.з. = 1 формируются колонки октаэдров, делящих общие грани (рис. 1б, слева) и, наконец, при 0.5 < ф.з. < 1 каналы могут быть заполнены как изолированными октаэдрами, так и их димерами, тримерами и другими группировками кластерного типа. Вследствие сочленения октаэдров по граням расстояния между катионами в таких кластерах или колонках довольно короткие: Сo–Co ∼ 2.55 Å [17], Ni–Ni ∼ 2.51 Å [16]. Как видно из табл. 1, заселенность Х для кобальтовых аналогов изменяется от 0.61 до 0.66. Для никельсодержащих аналогов наблюдаются два интервала: 0.33–0.39 и 0.58–0.66. Последние значения были реализованы только в структурах арсенатов. По данным, уточненным методом Ритвельда, в кристаллической структуре Ni12 + xH6 – 2x (AsO4)8(OH)6 с максимальной концентрацией катионов Ni (х = = 0.66) позиции всех тяжелых атомов расщеплены, и такое разупорядочение приводит к увеличению расстояний между катионами Ni–Ni до 3.14 Å [16]. Попытки варьирования соотношения Ni : (As/P) в исходных растворах с целью получения фосфатов и арсенатов с разным количеством катионов никеля в позиции Х, в том числе гипотетического состава с вакантной позицией Х, не дали результатов [14, 16].
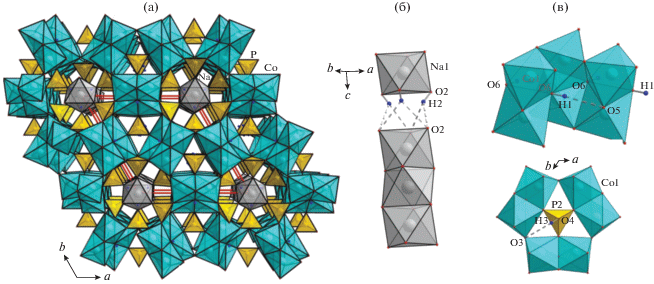
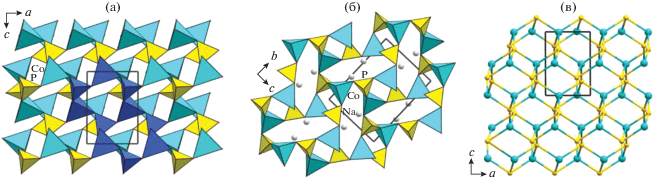
Ранее была исследована кристаллическая структура элленбергеритоподобного фосфата Na2 − xCo6(OH)3[HPO4][Hx/3PO4]3 (x ≈ 1.1) [12], синтезированного при температуре 280°С и давлении 70 бар. В широких каналах ее катионного каркаса, образованного полиэдрами Co2+ (рис. 3а), располагаются цепочки делящих общие ребра октаэдров Na, что определяет данное соединение в качестве первого представителя среди элленбергеритоподобных фаз с катионами щелочного металла в пустотах каркаса. Было показано, что заселенность позиции Na равна ∼0.9, а количество вакансий коррелирует с числом атомов водорода, протонирующих кислород фосфорных тетраэдров, располагающихся на стенках широких каналов. Такая корреляция отражается в формуле соединения и обеспечивает ее электронейтральность. На рис. 3б показана схема водородных связей, армирующих дефектную цепочку натрий–кислород: кислородная вершина натриевого октаэдра образует гидроксильную группу О2–Н2; О2 также является акцептором разупорядоченной бифуркированной водородной связи О2–Н2⋅⋅⋅О2'(О2''). Два других типа водородных связей –О6–Н1⋅⋅⋅О5 и О4–Н3⋅⋅⋅О3 (рис. 3в) – способствуют дополнительному укреплению структуры.
Рис. 3.
Кристаллическая структура Na2 − xCo6(OH)3[HPO4][Hx/3PO4]3 в аксонометрической проекции (а). Схема водородных связей, армирующих дефектную цепочку из NaО6-октаэдров (б), укрепляющих структуру (в).
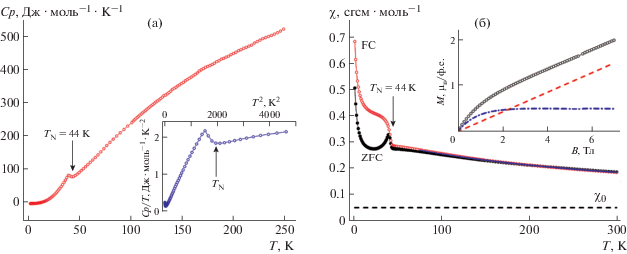
Температурные зависимости магнитной восприимчивости и теплоемкости свидетельствовали об установлении дальнего антиферромагнитного упорядочения при температуре Нееля TN = = 44 K (рис. 4). При данной температуре удельная теплоемкость демонстрировала плавный подъем, за которым следовал пик при T ∼ 39 K. Такой подъем является признаком усиления магнитных флуктуаций, предшествующих фазовому переходу второго рода. Разница в поведении магнитной восприимчивости, измеренной в режимах охлаждения при температурах T < TN в поле и в отсутствие поля (рис. 4а), указывала на наличие ферромагнитного компонента в магнитоупорядоченной фазе соединения Na2 − xCo6(OH)3[HPO4] [Hx/3PO4]3. Источником такого слабого ферромагнетизма может быть как анизотропия отдельных ионов двухвалентного кобальта, так и обменное взаимодействие типа Дзялошинского–Мория. Заметим, что недавно [24] была изучена магнитная структура Co6(OH)3(TeO3)4(OH) теллурата с аналогичным каркасом, и впервые показано, что слабый ферромагнетизм в магнитоупорядоченном состоянии (TN = 75.5 K) связан именно с наличием антисимметричных взаимодействий типа Дзялошинского–Мория в плоскости ab кристаллической структуры. Для этого соединения также установлено метамагнитное поведение с тремя различными антиферромагнитными состояниями со скошенными спинами. В случае Na2 − xCo6(OH)3[HPO4][Hx/3PO4]3 не наблюдалось каких-либо индуцированных полем переходов до 9 Тл. Полевая зависимость намагниченности была описана как сумма линейного и нелинейного вкладов (рис. 4б). Полагали, что линейный вклад вносит антиферромагнитная матрица элленбергеритоподобного фосфата кобальта, а нелинейный вклад может быть связан с парамагнитными примесными включениями и спонтанной намагниченностью.
Рис. 4.
Температурные зависимости: а – удельной теплоемкости соединения Na2 − xCo6(OH)3[HPO4][Hx/3PO4]3, на вставке – приведенная теплоемкость; б – магнитной восприимчивости, измеренные в режимах ZFC и FC, на вставке – полевая зависимость магнитной восприимчивости при Т = 2 K (кружки) с линейным (штриховая линия) и нелинейным вкладами (штрихпунктирная линия).
Схожие магнитные свойства демонстрировали структурно близкие с Na2 – xCo6(OH)3[HPO4] [Hx/3PO4]3 арсенаты M1 – xM$_{6}^{'}$(OH)3(AsO4H2x/3)3 (HAsO4), где M = M' = Co, Ni [17], с катионами переходных металлов в широких каналах. Оба соединения характеризуются парамагнитным поведением в области высоких температур; при ТN = 51 и 4 К соответственно Co- и Ni-арсенаты испытывают переход в антиферромагнитную фазу с небольшим ферромагнитным вкладом [17]. Поведение фосфатных аналогов указанных выше арсенатов, Co12 + xH4 – 2x(PO4)6(HPO4)2(OH)6 (x = 1.22) и Ni12 + xH4 – 2x(PO4)6(HPO4)2(OH)6 (x = 0.78), с близким содержанием переходных металлов в тех же каналах (табл. 1) несколько различалось. Кобальтовый фосфат демонстрировал антиферромагнитное упорядочение при 15 K, которому предшествовали две ферромагнитные аномалии при 50 K и в районе температуры упорядочения, видимые на кривой температурной зависимости χ(Т). В случае фосфата Ni дальнее магнитное упорядочение вплоть до 2 K не было обнаружено [13].
Фосфат и арсенат смешанных составов, Mg7.5Ni6H3(AsO4)8(OH)6 и Mg8Ni4H6(PO4)8(OH)6, были синтезированы в интервале температур 200–600°С при давлении 600 атм. [15]. Наличие примесей не позволило провести уточнение структуры фосфатной фазы. В кристаллической структуре арсената был установлен статистический характер заселения октаэдров двойных цепочек катионами Mg2+ и Ni2+ в соотношении 0.49 : 0.51, а широких каналов – катионами Mg2+ на 76%. При измерении магнитной восприимчивости обоих соединений не наблюдалось дальнего магнитного порядка в интервале 4.2–300 K. Отрицательная температура Кюри–Вейса, полученная в результате обработки температурной зависимости магнитной восприимчивости выше 80 K, свидетельствовала о наличии антиферромагнитных взаимодействий [15].
С учетом того, что атомы металлов в октаэдрическом окружении, формирующие двойные цепочки и заселяющие широкие каналы структуры, могут быть разного сорта, общая формула из [8] для всех фосфатов и арсенатов может быть модифицирована следующим образом: (Ху${{\square }_{{2/{v}--у}}}$) (M2OH)3[(TO4)$_{{1 + {v}у}}$(TO3OH)$_{{2--{v}у}}$](TO3OH), где у – заселенность позиции Х и ${v}$ – степень окисления атома в этой позиции. Кристаллические структуры ванадатов с элленбергеритоподобными каркасами не могут быть описаны указанной выше формулой, поскольку для них характерны полимеризованные конструкции из тетраэдров VO4.
Среди четырех структурно изученных на сегодняшний день элленбергеритоподобных ванадатов два являются Mg-содержащими (Mg7V4O16 (OH)2(H2O) [25], Mg13.4(OH)6(HVO4)2(H0.2VO4)6 [26]) и два содержат магнитоактивные катионы: Mn1 – 2/3zMn6(OH)3 (VO4)4 – 2z(V2O7)z (z = 0.20) [18, 19] и Co7V4O16(OH)2(H2O) [20]. Ванадат диванадат марганца в состоянии с двумя степенями окисления, Mn7 – 2/3z(OH)3(VO4)4 – 2z(V2O7)z (z = 0.20), был получен в гидротермальных условиях при 165°С в присутствии органического компонента – гидроксида тетраэтиламмония [18, 19]. Элленбергеритоподобный каркас его кристаллической структуры образован Mn2+-центрированными октаэдрами. На стенках широких каналов находятся VO4-тетраэдры, а в центрах каналов располагаются дефектные цепочки октаэдров Mn3+O6 (заселенность позиции 80%), связанных вершинами. Характер заполнения узких каналов уникален по сравнению с описанными выше фазами. Тетраэдрическая позиция катиона в этих каналах расщеплена на две подпозиции (V2a, V2b). Фактически реализуется статистическое распределение тетраэдров VO4 (80%) и пированадатных групп V2O7 (20%), количество которых коррелирует с количеством кислородных вакансий в позиции O6 (рис. 1в, справа). Благодаря такому распределению в узких каналах присутствуют ванадиевые тетраэдры двух ориентаций: апикальные вершины изолированных тетраэдров сонаправлены с вершинами тетраэдров широких каналов, вершины тетраэдров пированадатных групп противоположны им (рис. 1в, слева). Коэффициент z в химической формуле этого соединения характеризует степень полимеризации ванадиевых тетраэдров [8]. Как отмечено в [18, 19], величина z может зависеть от условий синтеза: крайние значения 0 и 1/2 могут быть реализованы в соединениях с изолированными тетраэдрами в узких каналах Mn7(OH)3(VO4)4 и с пированадатными группами Mn7 – 1/3(OH)3(VO4)3(V2O7)1/2 соответственно.
При температуре 200°С в гидротермальных условиях было синтезировано соединение Co7V4O16(OH)2(H2O) [20], изоструктурное Mg7V4O16(OH)2(H2O) [25]. В обеих кристаллических постройках октаэдры в широких каналах не содержат вакансий в отличие от структуры Mn7 – 1/3(OH)3(VO4)3(V2O7)1/2. Тетраэдры в узких каналах имеют различную ориентацию относительно оси с и не связаны между собой. Поскольку атомы водорода не были локализованы при расшифровке структуры, ОН/H2O-группы были “приписаны” атому кислорода, на котором сходятся четыре атома Co/Mg в двойных цепочках, в соответствии с данными расчета баланса валентных усилий и электронейтральности формулы. Нельзя исключить, что в структурах этих ванадатов возможно протонирование тетраэдров VO4, как в структурах элленбергеритоподобных фосфатов, арсенатов [8] и ванадата Mg13.4(OH)6(HVO4)2 (H0.2VO4)6 [26].
Измерения магнитной восприимчивости и теплоемкости Co7V4O16(OH)2(H2O) [20] указывали на установление скошенного антиферромагнитного порядка при температуре TN ∼ 71 K, наибольшей по сравнению с температурами фазовых переходов в структурно близких фосфате и арсенате. Изотерма намагниченности при 2 K демонстрировала линейный рост до 1 Тл, затем намагниченность медленно увеличивалась и после 5 Тл становилась более пологой, достигая 0.41 μВ в поле с индукцией 8 Тл, что было далеко до значения намагниченности насыщения. Такое поведение в слабых полях характерно для антиферромагнетика со скошенными спинами, а нелинейное увеличение восприимчивости в сильных полях могло свидетельствовать о метамагнитном поведении в описываемой системе.
Еще одна группа синтетических соединений с элленбергеритоподобными каркасами и общей формулой M11(HPO3)8(OH)6, M = Fe, Mn, Zn, Co, Ni [21–23], относится к химическому классу фосфитов – солей фосфористой кислоты. Анионная группа [HPO3]2– представляет собой псевдотетраэдр, центрированный катионом P3+, с тремя атомами O и атомом H в вершинах. В структурах фосфитов тетраэдрические группы широких и узких каналов являются фосфит-ионами, т.е. Θh(Θt = = О2–) и θh(θt = Н–). Фосфиты получены преимущественно из растворов фосфористой кислоты или гипофосфитов щелочных металлов при температурах от 170 до 212°С. В отличие от элленбергеритоподобных синтетических фосфатов, арсенатов и ванадатов в кристаллических структурах фосфитов позиция в центре каналов шестиугольного сечения вакантна, как в структуре экатита. Кроме того, дефектной является позиция переходного металла, причем количество вакансий в этой позиции ∼1/12. В зависимости от сорта двухвалентного металла фосфитные псевдотетраэдрические группы в узких каналах по-разному ориентированы относительно НРО3-групп широких каналов. Так, в структурах Fe2+- и Mn2+-содержащих аналогов [21] все фосфитные группы имеют одинаковую ориентацию, в случае Ni2+-фазы [22] НРО3-группировки больших и малых каналов направлены в противоположные стороны. В Co-аналоге фосфитные группы в тригональных каналах присутствуют в обеих ориентациях ввиду разупорядочения позиций Tt и θt [8, 22]. Хотя целенаправленному синтезу этих соединений посвящено много работ, ориентированных на получение наноматериалов различной морфологии (стержни, гантели, сферы, крестообразные звезды и другие) [27–29], практически отсутствует информация об их термодинамических свойствах, таких как магнитная восприимчивость и теплоемкость. В [23] сообщалось о сложном магнитном поведении Ni11(HPO3)8(OH)6, отличном от поведения обычного трехмерного антиферромагнетика. Соединение демонстрировало переход в антиферромагнитное упорядоченное состояние при 67 K с дополнительным ферромагнитным вкладом. Полевые зависимости намагниченности, измеренные при 5 K, имели S-образную форму, что подтверждало наличие ферромагнитных взаимодействий. В поле с индукцией 5 Tл намагниченность приближалась к 2.5μВ, что значительно ниже ожидаемого значения намагниченности насыщения для 11 ионов Ni2+ на формулу Ni11(HPO3)8(OH)6. Отметим, что параметр гексагональной элементарной ячейки а = 12.4589 Å этой фазы, синтезированной в виде порошка в присутствии фосфорной кислоты, меньше соответствующих значений для ранее полученных фосфитов Ni (а ∼ 12.63 Å) [22] и близок к параметру ячейки фосфата никеля, Ni12 + xH6 – 2x (PO4)8(OH)6, x = 0.76 (a = 12.4697(3) Å) [14]. В [27] измерения магнитной восприимчивости порошкового образца Ni11(HPO3)8(OH)6 (а = = 12.70, b = 4.94 Å) в температурном интервале от 5 до 300 K указывали на парамагнитное поведение материала.
Двойные цепочки связанных ребрами и гранями октаэдров переходного металла, являющиеся основными структурными фрагментами элленбергеритоподобного каркаса, характеризуют кристаллическую структуру полиморфной разновидности фосфата кобальта γ-Co2(PO4)(OH) [30]. Это соединение кристаллизуется в тригональной сингонии (пр. гр. P31m). Параметры его элементарной ячейки (а = 11.2514(4), b = 4.9967(4), c = = 547.81(5) Å, V = 547.81(5) Å3, Z = 6) близки к параметрам структур элленбергеритового типа. Сравнение кристаллических построек γ-Co2(PO4)(OH) и Na2 − xCo6(OH)3[HPO4][Hx/3PO4]3 показывает, что в обоих структурных типах атомы фосфора образуют гексагональную сетку (рис. 5). Топологически идентичные ленточные фрагменты, состоящие из октаэдров кобальта, располагаются в центре “фосфорных” гексагональных колец. В структуре элленбергеритоподобного фосфата двойные цепочки кобальта чередуются с дефектными цепочками натриевых полиэдров в соотношении 3 : 1 (рис. 5а), в то время как в структуре γ-Co2(PO4)(OH) каждое гексагональное кольцо содержит двойные цепочки СоО6-октаэдров (рис. 5б). Связанные вершинами двойные кобальтовые цепочки образуют в структуре элленбергерита в проекции на ось с одну из 11 архимедовых мозаик, описываемую символикой (4, 6, 12) согласно [31]. Цифры в скобках отражают типы многоугольников, встречающихся в одной вершине. Интересно, что топология сетки из атомов Со фазы γ-Co2(PO4)(OH) уникальна (рис. 5). Она имеет вершины двух видов: в одной сходятся трех-, шести-, четырех- и шестиугольники; в другой – четырехугольник и два шестиугольника, так что символ может быть записан как (3, 6, 4, 6) (4, 62) [30]. Такой мотив расположения магнитоактивных катионов, как показали исследования физических свойств γ-Co2(PO4)(OH), не приводит к установлению дальнего магнитного порядка. Было показано, что на кривых температурных зависимостей теплоемкости, измеренных в нулевом поле и поле с индукцией 5 Тл, отсутствуют пики λ-типа вплоть до 2 K, а совпадение кривых свидетельствует об отсутствии каких-либо индуцированных полем магнитных переходов. Измерения магнитной восприимчивости при переменном и постоянном токе указывали на поведение спинового стекла при температурах ниже 10 K [30].
Рис. 5.
Топология решеток на основе расположения атомов P и Co в структурах элленбергеритоподобного фосфата кобальта Na2 − xCo6(OH)3[HPO4][Hx/3PO4]3 (а, в) и γ-Co2(PO4)(OH) (б, г).
Оценивая современное состояние исследований кристаллохимических особенностей и свойств элленбергеритоподобных соединений, можно сказать, что в этих областях еще достаточно пробелов. Практически отсутствует структурная информация о характере водородных связей, о причинах различной ориентации тетраэдрических/пирамидальных групп в узких каналах, о способах контроля степени заполнения широких каналов структуры катионами переходных и/или щелочных металлов, о возможности их упорядочения. Как отмечено в [23, 24], отличная от “классических” антиферромагнетиков картина магнитного поведения и полярный характер структуры элленбергеритоподобных соединений требуют изучения их диэлектрических характеристик с целью установления возможных взаимосвязей между магнитной и электрической подсистемами.
2. β-ТРИДИМИТ И ЕГО ПРОИЗВОДНЫЕ: КРИСТАЛЛОХИМИЯ И ФИЗИЧЕСКИЕ СВОЙСТВА СОЕДИНЕНИЙ С ТЕТРАЭДРИЧЕСКИМИ КАРКАСАМИ НА ОСНОВЕ ШЕСТИЧЛЕННЫХ КОЛЕЦ
Кристаллические структуры минералов, основой которых являются анионные каркасы смешанного типа, образованные тетраэдрами разного сорта, часто обнаруживают разнообразные физические свойства, такие как ферро- и ферримагнитные, пьезо- и пироэлектрические, ионно-обменные, суперионные, нелинейно-оптические, каталитические, цеолитные и многие другие. Для них характерны хиральные кристаллические постройки, двойники, полиморфные модификации, модулированные структуры, обусловленные типами катионов в составе минерала и условиями формирования. Фазовые переходы связаны с повышением температуры и характеризуются перестройкой либо искажением кристаллической структуры за счет атомных смещений [32].
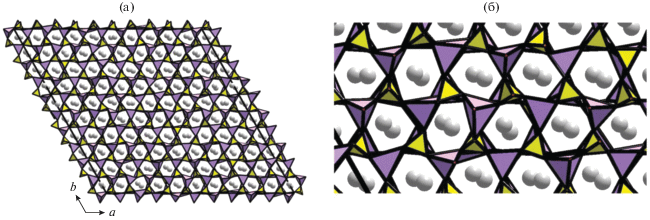
На примере анализа силикатов было установлено, что замещение атомов кремния в тетраэдрах на атомы меньшей валентности в структурах, производных от кварца, тридимита или кристобалита, необходимо компенсировать вхождением дополнительных атомов, радиус которых должен соответствовать размеру пустот конкретной кристаллической постройки. Такие модифицированные структуры (например, минералов кальсилита KAlSiO4 и нефелина Na3KAl4Si4O16) были названы “производными внедрения”, а дополнительные атомы в пустотах и полостях тетраэдрических каркасов – “внедренными атомами” [33]. В кристаллической структуре β-тридимита и его производных тетраэдры в шестичленных кольцах развернуты (относительно плоскости колец) через один вершинами вверх (up – U) и вниз (down – D) по схеме UDUDUD (рис. 6а).
Рис. 6.
Шестичленные кольца в кристаллической структуре, конформация: UDUDUDU, β-тридимит SiO2 (а); тринефелин NaAlSiO4 (б); UUUDDD, цеолит Li-A(BW) LiAlSiO4.2H2O (в); восьми- и четырехчленные кольца, образованные тетраэдрами (наряду с шестичленными), – отличительная особенность ABW-структур (г).
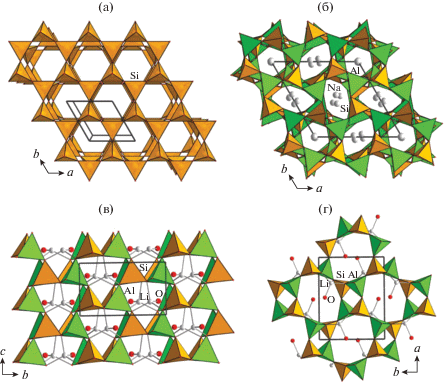
Альтернативная конформация шестичленных колец из тетраэдров была обнаружена в цеолитоподобных анионных 3D-постройках смешанного типа ABW [34–36]. В таких структурах разворот тетраэдров в шестичленных кольцах в слое описывается последовательностью UUUDDD (рис. 6б). Изменение расположения апикальных вершин тетраэдров относительно плоскости сеток из шестичленных колец по сравнению с вершинами в структурах производных внедрения на основе β-тридимита приводит к формированию каркаса с каналами, ограниченными четырех-, шести- и восьмичленными окнами, в которых размещаются ионы A+ (рис. 6в). Любопытно, что ABW-структуры могут различаться химическим составом позиций (сортом атомов) в тетраэдрической координации и внекаркасных, а также симметрией. Взаимоотношения восьми пространственных групп, характерных для структур этого семейства, в аспекте группа–подгруппа при понижении от максимально возможной топологической симметрии Imam были проанализированы в [37].
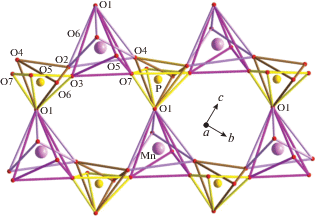
Кристаллические структуры семейства фосфатов с общей формулой A[MPO4], где M = Be, Zn, Co, Mn, Cu, Mg и A = Li, Na, K, Rb, Cs, Tl, Ag, NH$_{4}^{ + }$, также основаны на трехмерных постройках P- и M-центрированных тетраэдров, в которых выделяются слои шестичленных колец. Тетраэдры MO4 и РО4 чередуются в шестичленных кольцах, составляющих основу анионного тетраэдрического каркаса смешанного типа. Было показано [38, 39], что в рамках этого семейства реализуются структурные типы, различающиеся ориентацией тетраэдров в шестичленных кольцах относительно плоских сеток из этих колец. В качестве примеров соединений с анионными каркасами смешанного типа, образованными шестичленными кольцами тетраэдров, рассмотрим фосфаты с магнитоактивными ионами Co2+ и Mn2+ (табл. 2).
Таблица 2.
Состав, кристаллографические характеристики, структурный тип и физические свойства силикатов и производных фосфатов морфотропной серии AM[PO4] (A+ = Cs, Rb, K, (NH4), Na в каналах тетраэдрического каркаса; M2+ = Co или Mn в кислородных тетраэдрах)
| Формула/ минерал | a, b, c, Å β(γ), град |
Пр. гр. V, Å3, Z |
Конформация тетраэдров в шестичленных кольцах | Генезис/физические свойства/структурные особенности | Литература |
|---|---|---|---|---|---|
| Примеры производных внедрения на основе высокотемпературной гексагональной модификации SiO2–β-тридимита (пр. гр. P63/mmc) и конформация тетраэдров в шестичленных кольцах UDUDUD | |||||
| KAlSiO4/кальсилит | 5.157(1), 8.706(3) |
P31c 200.52 2 |
UDUDUD | Метасоматический минерал из гнейсов гранулитовой фации | [40] |
| NaAlSiO4/тринефелин | 9.995(2), 24.797(4) |
P61 (P65) 2145.3 24 |
UDUDUD | Метаморфический минерал на основе жадеита; по мере прохождения реакции заменяет нефелин | [41, 42] |
| Архетип – цеолит Li-A(BW), (Li · 2H2O)AlSiO4 и конформация тетраэдров в шестичленных кольцах UUUDDD | |||||
| Ромбические фосфаты кобальта со структурным типом RbAlSiO4 (ABW) | |||||
| CsCoPO4 530 K |
9.1572(2), 5.4973(1), 9.4360(2) |
Pnma 475.01 2 |
UUUDDD | Фазовый переход в ацентричную ромбическую модификацию при T = 512 К | [43] |
| CsCoPO4 495 K |
9.2027(2), 5.4763(1), 9.4007(2) |
Pn21a 473.76 4 |
UUUDDD | Фазовый переход в моноклинную модификацию при T = 481 К | [43] |
| Моноклинные фосфаты Co структурного типа RbZnPO4 (ABW) | |||||
| CsCoPO4 | 18.4281(2), 5.47254(5), 9.2897(2) 90.365(1) |
P121/a1 936.83 8 |
UUUDDD | Существует в температурном интервале 481–311 K, фазовый переход первого рода при T = 311 K в стабильную ацентричную модификацию | [44] |
| CsCoPO4 250 K |
9.2063(2), 5.4734(1), 9.2738(2) 90.394(1) |
P1211 467.30 4 |
UUUDDD | Диэлектрическая аномалия около T = 245°C, сегнетоэлектрик (спонтанная поляризация) | [43, 45] |
| RbCoPO4 | 8.8377(8), 5.4150(5), 8.9723(8), 90.212(2) |
P1211 429.38 4 |
UUUDDD | Синтез из водного раствора (при изменении pH в интервале 5.7–8.9 и при T = 150 и 180°C) | [39] |
| NH4CoPO4 | 8.7968(7), 5.4621(4), 9.0105(7) 89.983(2) |
P1211 432.95 4 |
UUUDDD | Парамагнитное поведение в интервале температур 12–300 K, θ отрицательная, s = 3/2, ферромагнитное упорядочение при ТС = 3 K, g = 2.43 | [39] |
| Гексагональные фосфаты Co и конформация тетраэдров в шестичленных кольцах UDUDUD и UUUDDD | |||||
| Структурный тип NH4ZnPO4 | |||||
| NH4CoPO4 | 10.7189(6), 8.7096(6) |
P63 866.62 8 |
UDUDUD и UUUDDD в соотношении 1 : 3 | Парамагнитное поведение в интервале температур 12–300 K, θ отрицательная, s = 3/2, ферромагнитное упорядочение при ТС = 3 K, g = 2.32 | [39] |
| Структурный аналог минерала мегакальсилита KAlSiO4 | |||||
| KCoPO4 | 18.230(1), 8.5208(7) |
P63 2452.4 24 |
UDUDUD и UUUDDD в соотношении 1 : 3 | Парамагнитное поведение в интервале температур 12–300 K, θ отрицательная, s = 3/2, ферромагнитное упорядочение при ТС = 3 K, g = 2.44 | [39] |
| Структурный тип NaCoPO4 | |||||
| NaCoPO4 | 10.166(1), 23.881(5) |
P65 2137.4 24 |
UDUDUD и UUUDDD в соотношении 1 : 3 | Парамагнитное поведение в интервале температур 12–300 K, θ – отрицательная, s = 3/2, антиферромаг-нитное упорядочение при ТN = 3 K, g = 2.17, фрустрированный магнитоэлектрический антиферромагнетик | [46, 39, 47, 48 ] |
| Структурный тип NaZnPO4 и конформация тетраэдров в шестичленных кольцах UDUDDD и UDDDDD | |||||
| NaCoPO4 | 5.221(1), 9.983(1), 7.388(1), 90.210(4) |
P121/n1 385.07 4 |
UDUDDD и UDDDDD в соотношении 1 : 1; NaZnPO4 | Получен сольвотермальным методом при Т = 413–433 K | [49, 39 ] |
| Высокотемпературная и низкотемпературная модификация CsMnPO4 (ABW) | |||||
| α-CsMnPO4 | 9.128(3), 9.575(2), 5.595(1) |
Pna21 489.01 4 |
UUUDDD | Высокотемпературная модификация, три из четырех симметрийно независимых атома O расщеплены | [50] |
| β-CsMnPO4 | 11.0699(4), 11.0819(6), 9.1106(3) 119.480(5) |
P11a 972.94 8 |
UUUDDD | Низкотемпературная модификация, соразмерно модулированная структура, векторы модуляции: q1 = 0.4a* и q2 = 0.4b*, антиферромагнитное упорядочение при TN = 4.5 K | [51] |
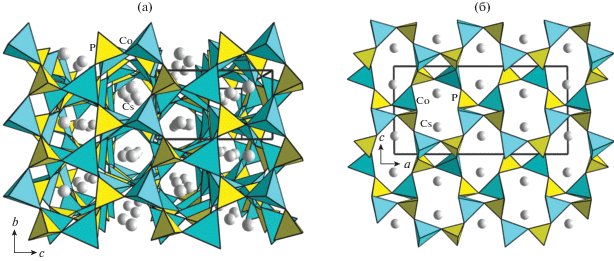
Наибольшее разнообразие структурных типов в этом ряду описано для фосфата Cs и Co, характеризующегося четырьмя модификациями. Полиморфные превращения соединения CsCoPO4 были зафиксированы при изменении температуры: трем эндотермическим пикам на кривой нагревания отвечали три экзотермических максимума при охлаждении при T = 311, 481 и 512 K, свидетельствующие о необратимых фазовых переходах [45, 43 ]. Синхротронный экспериментальный материал, полученный на порошке при четырех температурах (530, 495, 350 и 250 K), позволил уточнить кристаллические структуры модификаций CsCoPO4 методом Ритвельда, используя в качестве стартовых координаты базисных атомов изотипных Cs,Zn-фосфатов. Как отмечено выше, основа кристаллических структур всех четырех модификаций CsCoPO4 – 3D-постройка из делящих кислородные вершины чередующихся тетраэдров CoO4 и PO4. В структурах выделяются сетки-слои, образованные шестичленными кольцами тетраэдров, в которых разворот апикальных вершин относительно плоскости слоев подчиняется закону UUUDDD. Объединение таких слоев вдоль третьей кристаллографической оси приводит к образованию широких каналов с окнами, образованными восьмичленными кольцами, в которых размещаются атомы Cs.
Однако при сохранении одной и той же конформации тетраэдров в шестичленных кольцах кристаллические структуры различаются симметрией: они реализуются в рамках ромбических (структурный тип RbAlSiO4) (рис. 7) и моноклинных (структурный тип RbZnPO4) модификаций. Согласно [44] крупные катионы Cs+ выполняют функцию темплата, вокруг которого “собираются” анионные тетраэдрические [CoPO4]–ABW-каркасы, не содержащие молекул H2O (рис. 8), в отличие от “водных” ABW-фосфатов LiZnPO4 ⋅ H2O [52, 53] с внекаркасными мелкими атомами лития, дополнительно координированными молекулами H2O в каналах. Тем не менее сильное искажение Co-центрированных тетраэдров в цеолитоподобных фосфатах в сочетании с высокой гибкостью ABW-каркаса допускает участие катионов меньшего размера (Rb, K, NH4) в формировании таких структур [44].
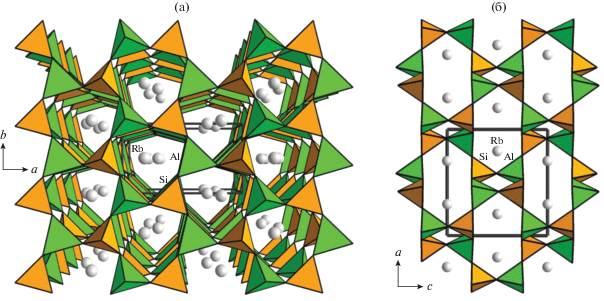
Рис. 8.
Кристаллическая структура моноклинной центросимметричной (пр. гр. P21/a) модификации CsCoPO4 в аксонометрии вдоль оси а (а) и в проекции на плоскость ас (б).
Методами адиабатической и дифференциальной сканирующей калориметрии была измерена теплоемкость кристаллов CsCoPO4 для расчета стандартных термодинамических функций в диапазоне температур от 0 до 650 K и стандартной энтропии образования при 298.15 K, которая оказалась равной –386.40 ± 1.38 Дж·K–1·моль–1 [54]. На кривой емкости образца CsCoPO4 как функции температуры в диапазоне 20–600°C была зафиксирована диэлектрическая аномалия при T = = 245°C, связанная с фазовым переходом соединения в ацентричную модификацию (табл. 2), для которой были установлены спонтанная поляризация и пироэлектрические свойства [45].
Рубидиевый аналог RbCoPO4, изоструктурный этой модификации Cs,Co-фосфата, был получен в “мягких” гидротермальных условиях при T = 180°C [39]. Закономерное изменение параметров элементарной ячейки Rb-фазы по сравнению с параметрами Cs-изотипа (табл. 2) обусловлено существенным уменьшением ионного радиуса щелочного катиона Rb+ (1.60 Å) относительно ионного радиуса Cs+ (1.74 Å). Аналогично для изоструктурной разновидности NH4CoPO4 характерны значительно меньшие параметры моноклинной элементарной ячейки по сравнению с CsCoPO4; соответственно, эти параметры сопоставимы с параметрами соединений RbCoPO4 и NH4CoPO4.
Заметим, что содержащий аммоний фосфат кобальта обладает диморфизмом. Второй формульной разновидности присущи гексагональная симметрия и более сложный тип конформации шестичленных колец из тетраэдров, аналогичный конформации в структуре природного силикатного аналога – минерала мегакальсилита KAlSiO4 [55]. Гексагональные кристаллические структуры мегакальсилита и NH4CoPO4 “собраны” из шестичленных колец двух типов, а именно UDUDUD и UUUDDD в соотношении 1 : 3 (рис. 9). Любопытно, что аналогичная конформация шестичленных колец характерна для всех гексагональных фосфатов кобальта этого семейства, хотя параметры их элементарных ячеек могут меняться в зависимости от типа внекаркасного катиона щелочного металла в каналах тетраэдрического каркаса (табл. 2). Так, мегакальсилиту KAlSiO4 изоструктурен фосфат KCoPO4, а также KGeAlO4 [56, 57] и фосфаты Zn, содержащие в составе щелочные катионы K+ [58, 59].
Рис. 9.
Аксонометрическая проекция гексагональной модификации кристаллической структуры NH4CoPO4. Параллельные плоскости ab слои образованы шестичленными кольцами двух типов: UDUDUD и UUUDDD.
Аналогично в гексагональной кристаллической структуре β-NaCoPO4 четвертая часть шестерных колец UDUDUD из чередующихся Co- и P-центрированных тетраэдров и три четверти колец UUUDDD-типа заселены атомами Na; причем кольца обеих конформаций характеризуются дитригональным искажением [46]. Атомы Na сдвинуты из центра колец UUUDDD на 0.45–0.65 Å, что свидетельствует об их недостаточном размере для симметричной координации в сравнении с атомами K. Эта формальная замена (K на Na) приводит также к сильному искажению тетраэдров CoO4, наклону тетраэдров каркаса обоих типов и формированию гофрированных сеток из шестичленных колец, перпендикулярных оси гексагональности. Результирующая хиральная кристаллическая структура оказывается менее симметричной и к тому же полярной (пр. гр. P61 либо P65), нежели аналоги с ионами K+ или NH4+, а параметр c = 23.88 Å β-NaCoPO4 приблизительно отвечает утроенному соответствующему параметру элементарной ячейки β-тридимита. Измерения намагниченности и удельной теплоемкости β-NaCoPO4 показали дальнее антиферромагнитное упорядочение с небольшим ферромагнитным компонентом при TN ≈ 3.2 K. Корреляции спинов ближнего порядка обнаружены при температуре около 15 K, что указывает на магнитную фрустрацию из-за псевдотреугольной геометрии спиновой системы (рис. 10) [47]. Существенно, что изменение электрической поляризации индуцируется дальним и ближним магнитным упорядочением, а также приложением внешнего магнитного поля. Показано, что β-NaCoPO4 является редким примером полярного и хирального магнитоэлектрического антиферромагнетика. Чувствительное к коротким спиновым корреляциям внутреннее магнитоэлектричество может быть использовано для исследования таких систем [48].
Рис. 10.
Два уровня треугольной сетки из магнитных ионов Co2+, размножающейся осью 65, в проекции на плоскость ab кристаллической структуры β-NaCoPO4.
Еще одна полиморфная разновидность соединения NaCoPO4, в кристаллической структуре которой атомы Co имеют тетраэдрическую координацию, описана в [49]. Эта фаза изотипна цинковому формульному аналогу NaZnPO4 [60]. Как и в случае β-NaCoPO4, участие мелкого атома Na в формировании моноклинной кристаллической постройки приводит к искажению тетраэдрического каркаса, вплоть до изменения в данной структуре разворота тетраэдров в шестичленных кольцах в сетках, перпендикулярных оси моноклинности (рис. 11а). В результате в структурах этих двух изотипных фосфатов реализуется еще одна конформация шестичленных колец, а именно структуры “собраны” из колец двух типов: UDUDDD и UDDDDD в соотношении 1 : 1. Окна эллипсоидальной формы из восьмичленных колец, ограничивающие каналы, вытянуты параллельно направлениям диагоналей грани (100) (рис. 11б). В каналах тетраэдрического каркаса размещаются атомы Na. Интересно, что для рассматриваемой моноклинной модификации NaCoPO4 характерны вытянутые в двух направлениях пересекающиеся лестничные конструкции катионной подструктуры (рис. 11в), описанные впервые в связи с минералом цельзианом BaAl2Si2O8 [61].
Рис. 11.
Кристаллическая структура моноклинной модификации NaCoPO4 в проекции на плоскости ас (а) и bc (б). Выделены шестичленные кольца двух типов, формирующие 3D-постройку. Лестничная конструкция катионной подструктуры каркаса (в): крупными шарами показаны атомы Co, мелкими – P.
Из приведенных выше данных видно, что для соединений рассматриваемого семейства характерно наличие полиморфных модификаций, об-условленных структурными трансформациями при изменении температуры. Еще одним примером аналогичного преобразования является ортофосфат Cs и Mn. Ромбическая фаза α-CsMnPO4 была получена в гидротермальных условиях при T = 623 K и P = 100 МПа. Ее ацентричная кристаллическая структура образована шестичленными кольцами чередующихся тетраэдров MnO4 и PO4 (конформация UUUDDD) [50], однако она отличается от структуры рассмотренного ранее фосфата CsCoPO4 с близкими параметрами элементарной ячейки и той же пространственной группой [43] (табл. 2). Особенностью структуры α-CsMnPO4 является расщепление позиций трех из четырех независимых атомов кислорода, которое сопровождается разворотом Mn- и P-центрированных тетраэдров каркаса приблизительно на 30° вокруг одной кислородной вершины (рис. 12). Расщепление атомных позиций и связанные с ним колебания тетраэдров каркаса в структуре α-CsMnPO4 в отличие от структуры также высокотемпературного Co-аналога обусловлено значительным увеличением размера катиона переходного металла Mn2+ (0.83 Å) по сравнению с радиусом Co2+ (0.74 Å). Колебания тетраэдров вызваны стремлением атомов Mn оказаться в более привычной для него пятивершинной или октаэдрической координации.
Рис. 12.
Разворот шестичленных колец вокруг вершины O1, образованных тетраэдрами MnO4 и PO4 в кристаллической структуре α-CsMnPO4. Ребра альтернативных тетраэдров показаны цветом разной интенсивности.
Очевидно, что структура в целом стремится к энергетически более выгодному состоянию, которое реализуется в рамках моноклинной сингонии низкотемпературной модификации β-CsMnPO4, кристаллы которой были получены в гидротермальном эксперименте при T = 553 K и P = 7 МПа. Геометрическое взаимоотношение элементарных ячеек α- и β-фаз представлено на рис. 13. Подчеркнем, что плоскость скользящего отражения a (b) перпендикулярна оси элементарной ячейки ∼9.1 Å в обоих случаях. 3D-сверхструктура β-CsMnPO4 была уточнена как соразмерно модулированная с параметрами элементарной ячейки a' = 5a, b' = 5b, c' = c (пр. гр. P11a) с учетом псевдомероэдрического двойникования исследованного кристалла осью второго порядка [110 ] , которая отвечает оси 21, параллельной оси с ромбической элементарной ячейки α-фазы [51]. Важно отметить, что пр. гр. $C_{{2{v}}}^{9}$ = Pna21 (α-CsMnPO4) является надгруппой симметрии по отношению к группе $C_{s}^{2}$ = P1a1, характерной для β-CsMnPO4.
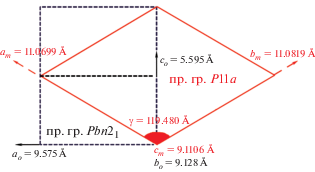
Основное различие кристаллических структур α- и β-модификаций соединения CsMnPO4 состоит в отсутствии расщепления позиций атомов кислорода во втором случае. Все позиции атомов O в структуре β-фазы заселены целиком, т.е. не содержат вакансионных дефектов, как в случае α-CsMnPO4. Упорядочение атомов кислорода приводит к увеличению параметров a и b элементарной ячейки β-CsMnPO4 в 5 раз, т.е. псевдопериоды в этих направлениях в основном нарушаются за счет разворота Mn- и P-центрированных тетраэдров (рис. 14). Трехмерная постройка из делящих вершины чередующихся тетраэдров MnO4 и PO4 и атомов Cs в каналах каркаса присуща обеим модификациям. Сетки-слои из гексагональных колец конформации UUUDDD характеризуют обе структуры. Все сказанное выше позволяет предположить фазовый переход второго рода между α- и β-модификациями CsMnPO4, связанный с колебаниями тетраэдров каркаса. В обоих случаях стремление атомов Mn оказаться в комфортной координации обусловливает формирование вакансионных дефектов в позициях атомов кислорода (высокотемпературная фаза α-CsMnPO4) либо возникновение модуляций (низкотемпературная фаза β-CsMnPO4). Симптоматично, что подобных эффектов не наблюдается в структурах высокотемпературного ромбического и низкотемпературного моноклинного ацентричных кобальтовых аналогов CsCoPO4.
Рис. 14.
Кристаллическая структура β-CsMnPO4 в проекции на плоскость ac (а) и фрагмент структуры в той же проекции, демонстрирующий разворот тетраэдров анионного каркаса (б). Тетраэдры MnO4 крупные, тетраэдры PO4 мелкие, атомы Cs показаны шарами.
По данным [62] кристаллические CsMnPO4 и CsCoPO4 могут служить контейнерами для захоронения радиактивного цезия при комнатной температуре. Термодинамические свойства высокотемпературной ромбической модификации CsMnPO4 были исследованы в температурном интервале 5–650 K. Согласно данным [63] соединение претерпевает два фазовых перехода при температуре 6.9 и 127 K. На кривой магнитной восприимчивости α-CsMnPO4 при изменении температуры от 2 до 300 K были выявлены два пика, которым отвечают два максимума на кривой теплоемкости. Соединение демонстрирует парамагнитное поведение ниже T = 150 K. Магнитная восприимчивость выше T = 150 K следует закону Кюри–Вейса с Θ = – (16.0 ± 0.5) K и С = (4.528 ± ± 0.009) моль–1·см3. Эффективный магнитный момент, рассчитанный по константе Кюри, равен 5.96 мкБ; это значение отвечает высокоспиновому состоянию иона Mn2+. По данным экспериментальных исследований α-модификации были рассчитаны стандартные термодинамические функции и энтропия образования при T = 298.15 K [63].
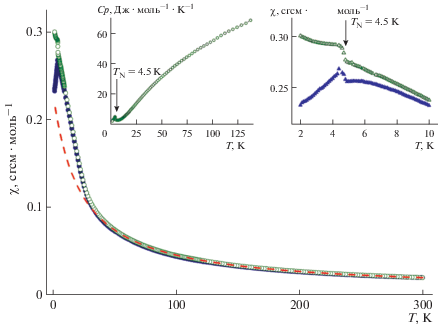
На рис. 15 показаны графики температурных зависимостей магнитной восприимчивости χ = M/B при B = 0.1 Тл, измеренные для β-CsMnPO4 в режимах FC (охлаждение в поле) и ZFC (охлаждение в нулевом поле). В области высоких температур зависимости χ(Т) следуют модифицированному закону Кюри–Вейса: χ = χ0 + C/(T – Θ). Аппроксимация кривой FC в интервале температур 200–300 K с g-фактором 1.99, оцененным по результатам измерений электронного парамагнитного резонанса при комнатной температуре, дает следующие значения: температурно-независимый вклад χ0 = 5.84 × 10–3 сгсм·моль–1, постоянная Кюри C = 4.331 сгсм·K·моль–1 и температура Кюри–Вейса Θ = –2 K. Отрицательное значение температуры Кюри–Вейса свидетельствует о преобладании антиферромагнитных обменных взаимодействий. Отклонение кривой χ(T) от экстраполяции закона Кюри–Вейса при более низких температурах (рис. 15, штриховая линия) означает усиление ферромагнитных обменных взаимодействий при приближении к TN = 4.5 K. Как видно на правой вставке рис. 15, кривые ZFC и FC расходятся в окрестности температуры магнитного упорядочения, что может указывать на присутствие слабого ферромагнитного компонента в намагниченности β-CsMnPO4, что подобно поведению KNiPO4 [64]. Формирование дальнего магнитного порядка подтверждают также результаты измерения теплоемкости: при TN = 4.5 K кривая Cp(T) демонстрирует острую аномалию λ-типа (рис. 15, левая вставка).
Рис. 15.
Температурные зависимости магнитной восприимчивости β-CsMnPO4, полученные в режимах FC (незакрашенные кружки) и ZFC (закрашенные кружки) при B = 0.1 Тл. Штриховая линия аппроксимирует экспериментальные данные по закону Кюри–Вейса. Левая вставка представляет температурную зависимость удельной теплоемкости β-CsMnPO4. На правой вставке показана низкотемпературная область кривых χ(T) при большем увеличении.
Интересно, что для рубидиевого формульного аналога RbMnPO4 были зафиксированы мультиферроэлектрические свойства при низких температурах [65]. На основе нейтронного дифракционного эксперимента и термодинамических измерений показано, что эта фаза представляет собой пример слабоферромагнитного полярного соединения с температурой Нееля TN = 4.7 K. При T < TN RbMnPO4 демонстрирует свойства скошенного антиферромагнетика с ферромагнитным компонентом в плоскости ас. Вследствие эффектов магнитной фрустрации образуется промежуточная магнитная фаза в температурном диапазоне 4.7–5.1 K, что позволяет характеризовать соединение как мультиферроик.
Морфотропный ряд фосфатов марганца с общей формулой AMnPO4 включает в себя помимо рассмотренных выше Cs и Rb соединений K- и Ag-содержащие фазы. Однако в отличие от формульных аналогов с атомами других переходных металлов большинство марганцевых представителей в этом семействе не обладает структурами, в основе которых лежит каркас, собранный из тетраэдров, связанных в шестичленные кольца. Так, в кристаллических структурах RbMnPO4 [66] и KMnPO4 [67] атомы Mn в двух независимых структурных позициях находятся в тетраэдрической и пятивершинной координации. Кристаллохимическое различие формульных Rb- и K-аналогов проявляется в различной симметрии их элементарных ячеек (триклинной в первом случае и моноклинной во втором) и обусловлено разным строением ленточных фрагментов, образованных полиэдрами MnO5 и PO4. В кристаллической структуре AgMnPO4 [68] атомы Mn находятся в центрах тригональных бипирамид и октаэдров, которые, объединяясь ребрами и вершинами, образуют широкие ленты, вытянутые параллельно одной из осей триклинной элементарной ячейки. Эти примеры подтверждают отмеченную тенденцию ионов Mn2+ в кислородных соединениях к увеличению координационного числа до 5 и 6, стремление к которым проявляется в статистическом развороте тетраэдров анионного каркаса структуры высокотемпературного α-CsMnPO4 либо в возникновении модуляций в случае низкотемпературного β-CsMnPO4.
ЗАКЛЮЧЕНИЕ
Представленные в рамках обзора два семейства фосфатов и их арсенатные и ванадатные аналоги с катионами щелочных и переходных металлов принципиально различаются между собой с точки зрения особенностей их кристаллохимии. Основа кристаллических структур элленбергеритового типа – катионные каркасы, образованные атомами переходного металла, как правило, с низшей степенью окисления, в октаэдрическом окружении из атомов кислорода. Октаэдры делят общие грани, ребра и вершины и формируют трехмерные постройки, которые могут содержать вакансионные дефекты. Основным магнитным состоянием всех исследованных на сегодняшний день фосфатов, арсенатов и ванадатов кобальта со структурой элленбергерита является антиферромагнитное состояние со скошенными спинами. Очевидно, такое упорядочение связано с магнитными взаимодействиями между катионами двойных цепочек, образующими каркас. Поведение никелевых аналогов заслуживает дальнейшего изучения, поскольку исследованные представители разных химических классов и с разным содержанием Ni2+ в магнитной подсистеме демонстрируют как дальнее магнитное упорядочение, так и его отсутствие при низких температурах.
Группа соединений, рассмотренная во второй части, объединила синтетические фосфаты с кристаллическими структурами, производными от структуры гексагонального высокотемпературного минерала β-тридимита SiO2. Их дизайн основан на шестичленных кольцевых конструкциях из чередующихся тетраэдров PO4 и MO4 (M = Co, Mn), изменение конформации которых обусловливает формирование структурных особенностей конкретного соединения и соответствующие физические свойства. Для производных β-тридимита характерны анионные каркасы смешанного типа, образованные кислотными тетраэдрическими оксокомплексами (силикатными, германатными, фосфатными, сульфатными) и оксотетраэдрами, центрированными атомами переходного металла, либо другими атомами с амфотерными свойствами (Al, Be, Ge). Таким образом, в кристаллических структурах фосфатов этого типа атомы переходных металлов первого ряда выполняют функцию анионообразователя в отличие от их катионной функции в структурах элленбергеритового типа. Важно подчеркнуть, что топология катионных подструктур из атомов переходного металла тридимитоподобных фосфатов близка у различных представителей этого семейства и обусловливает сходство их магнитного поведения. Гибкость построек, образованных делящими кислородные вершины тетраэдрами, позволяет катионам щелочных металлов различного размера размещаться в каналах каркаса, не разрушая его. Для рассмотренных фосфатов характерны температурные фазовые переходы с образованием структурных модификаций как различной симметрии, так и различающихся лишь наличием центра инверсии. Они часто образуют двойники, модулированные и дефектные кристаллические структуры.
Показано, что рассматриваемым семействам свойственна широкая изоморфная емкость их кристаллических структур как в катионной, так и в анионной частях. Это свидетельствует о способности их каркасных построек к значительным геометрическим деформациям, т.е. об их “гибкости” или приспосабливаемости к изменениям химического состава в результате изоморфных замещений. Перспективным видится изучение ионно-проводящих свойств обсуждаемых в работе соединений. Для производных β-тридимита эти свойства весьма вероятны в силу особенностей кристаллической структуры с каналами из шести- и восьмичленных колец, в которых могут располагаться атомы щелочных металлов различного размера от Li до Cs. Установленная возможность внедрения атомов щелочного металла в трубчатые каналы структуры элленбергеритового типа [12] также напоминает об их потенциале проявления ионно-проводящих свойств, о которых упоминалось еще в [13, 16].
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (проект № 18-03-00908а).
Список литературы
Fisher D.G. Environmental Phosphorus Handbook / Ed. Griffith E. et al. New York: Wiley, 1973.
Moore P.B. // Phosphate Minerals / Ed. Nriagu J.O., Moore P.B. Berlin; Heidelberg: Springer, 1984. P. 155.
Chopin C., Klaska R., Medenbach O., Dron D. // Contributions to Mineralogy and Petrology. 1986. V. 92. P. 316.
Comodi P., Zanazzi P.F. // Eur. J. Mineral. 1993. V. 5. P. 819.
Zubkova N.V., Pushcharovsky D.Yu., Pasero M. et al. // Crystallography Reports 2007. V. 52. P. 199.
Raade G., Rømming C., Medenbach O. // Mineral. Petrol. 1998. V. 62. P. 89.
Keller P. // Eur. J. Mineral. 2001. V. 13. P. 769.
Evans R.J., Groat L.A. // Can. Mineral. 2012. V. 50. P. 1197.
Ferraris G., Ivaldi G., Chopin C. // Eur. J. Mineral. 1995. V. 7. P. 167.
Pieczka A., Evans R.J., Grew E.S. et al. // Mineral. Magazine. 2013. V. 77. № 6. P. 2825.
Hawthorne F.C. // Acta Mineral. Petrogr. Abstr. Ser. 2010. V. 6. P. 734.
Yakubovich O.V., Kiriukhina G.V., Dimitrova O.V. et al. // Dalton Trans. 2015. V. 44. № 26. P. 11827.
Pizarro J.L., Arriortua M.I., Lezama L., Rojo T. // Solid State Ionics. 1993. V. 63–65. P. 71.
Marcos M.D., Amorós P., Beltrán A., Beltrán D. // Solid State Ionics. 1993. V. 63–65. P. 87.
Rojo J.M., Mesa J.L., Pizarro J.L. et al. // High Press. Res. 2002. V. 22. P. 569.
Marcos M.D., Amorós P., Beltrán D. et al. // J. Mater. Chem. 1995. V. 5. P. 917.
Hughes R.W., Gerrard L.A., Price D.J., Weller M.T. // Inorg. Chem. 2003. V. 42. P. 4160.
Zhang F., Zavalij P.Y., Whittingham M.S. // J. Mater. Chem. 1999. V. 9. P. 3137.
Pecharsky V.K., Zavalij P.Y. Fundamentals of Powder Diffraction and Structural Characterization of Materials, Second Edition. Ch. 23. Springer, 2009. https://doi.org/10.1007/978-0-387-09579-0
Hu T., Lin J.-B., Kong F., Mao J.-G. // Inorg. Chem. Commun. 2008. V. 11. P. 1012.
Aitfield M.P., Morris R.E., Cheetham A.K. // Acta Cryst. C. 1994. V. 50. P. 981.
Marcos M.D., Amorós P., Beltrán-Porter A. et al. // Chem. Mater. 1993. V. 5. P. 121.
Poienar M., Maignan A., Sfirloaga P. et al. // Solid State Sci. 2015. V. 39. P. 92.
Poupon M., Barrier N., Pautrat A. et al.// J. Solid State Chem. 2019. V. 270. P. 147.
Zhang S.-Y., Guo W.-B., Yang M. et al. // J. Solid State Chem. 2015. V. 225. P. 78.
Đorđević T., Karanović Lj., Tillmanns E. // Cryst. Res. Technol. 2008. V. 43. P. 1202.
Gu Z., Zhai T., Gao B. et al. // Crystal Growth Design. 2007. V. 7. № 4. P. 825.
Jin L., Hong J.B., Ni Y. // Mater. Chem. Phys. 2010. V. 123. P. 337.
Ni Y., Liao K., Hong J., Wei X. // Cryst. Eng. Commun. 2009. V.11. P. 570.
Cui M., Wang N., Chen S. et al. // J. Alloys Compd. 2019. V. 785. P. 1009.
Grünbaum B., Shephard G.C. Tilings and Patterns. New York, 1987.
Wallez G., Luca F., Souron J.-P., Quarton M. // Mater. Res. Bull. 1999. V. 34. P. 1251.
Buerger M.J. // J. Chem. Phys. 1947. V. 15. P. 1.
Kerr I.S. // Z. Krist. 1974. B. 139. S. 186.
Anderson E.K., Ploug-Sorensen G.Z. // Z. Krist. 1986. B. 176. S. 67.
Baerlocker Ch., McCusker L.B., Olson D.H. Atlas of Zeolite Framework Types. Elsevier, 2007.
Kahlenberg V., Fisher R.X., Baur W.H. // Z. Krist.2001. B. 216. S. 489.
Сандомирский П.А., Белов Н.В. Кристаллохимия смешанных анионных радикалов. М.: Наука, 1984. 205 с.
Feng P., Bu X., Tolbert S.H., Stucky G.D. // J. Am. Chem. Soc. 1997. V. 119. P. 2497.
Cellai D., Bonazzi P., Carpenter M.A. // Am. Mineral. 1997. V. 82. P. 276.
Ferraris C., Parodi G.C., Pont S. Et al. // Eur. J. Mineral. 2014. V. 26. P. 257.
Kahlenberg V., Böhm H. // Am. Mineral. 1998.V. 83. P. 631.
Kawaji H., Ishihara Y., Nidaira A. et al.// J. Therm. Analysis Calorimetry. 2008. V. 92. P. 451.
Henry P.F., Hughes E.M., Weller M.T. // Dalton Trans. 2000. V. 4. P. 555.
Blum D., Peuzin J.C., Henry J.Y. // Ferroelectrics. 1984. V. 61. P. 265.
Hammond R., Barbier J. // Acta Cryst. B. 1966. V. 52. P. 440.
Raju N.P., Greedan J.E. // Can. J. Phys. 1995. V. 73. P. 658.
Kimura K., Kimura Ts. // J. Phys. Soc. Jpn. 2015. V. 84. 003705.
Chippindale A.M., Cowley A.R., Chen J. et al. // Acta Cryst. C. 1999. V. 55. P. 845.
Yakubovich O.V., Simonov N.N., Belov N.V. // Sov. Phys. Cryst. 1990. V. 35. P. 22.
Bolotina N., Yakubovich O., Shvanskaya L. et al. // Acta Cryst. B. 2019. V. 75. P. 822.
Harrison W.T.A., Gier Th.E., Nicol J.M., Stucky G.D. // J. Solis State Chem.1995. V. 114. P. 249.
Jansen T.R. // Dalton Trans. 1998. V. 13. P. 2261.
Korchemkin I.V., Pet’kov V.I., Markin A.V. et al. // J. Chem. Thermodynamics. 2016. V. 96. P. 34.
Khomyakov A.P., Nechelyustov G.N., Sokolova E. et al. // Can. Mineral. 2002. V. 40. P. 961.
Sandomirskii P.A., Meshalkin S.S., Rozhdestvenskaya I.V. et al. // Soviet Physics Crystallography. 1986. V. 31. P. 522.
Lampert G., Boehme R. // Z. Krist. 1986. B. 176. S. 29.
Andratschke M., Range K.-J., Haase H., Klement U. // Z. Naturforsch. B. 1992. B. 47. S. 1249.
Yakubovich O.V., Mel’nikov O.K. // Soviet Physics Crystallography. 1989. V. 34. P. 34.
Ng H.Y., Harrison W.T.A. // Micropor. Mesopor. Mater. V. 23. P. 197.
Taylor W.H. // Z. Krist.1933. B. 85. S. 425.
Borovikova E.Yu., Kurazhkovskaya V., Ksenofontov D. et al. // Eur. J. Mineral. 2012. V. 24. P. 777.
Korchemkin I.V., Pet’kov V.I., Markin A.V. et al. // J. Chem. Thermodinamics. 2014. V. 78. P. 114.
Luján M., Rivera J.-P., Kizhaev S. et al. // Ferroelectrics. 1994. V. 161. P. 77.
Nénert G., Bettis J.Jr., Kremer R. et al. // Inorg. Chem. 2013. V. 52. P. 9627.
Yahia H.B., Gaudin E., Darriet J. // J. Alloys Compd. 2007. V. 442. P. 74.
Luján M.F., Kubel F. // Z. Naturforsch. 1995. B. 50. S. 1210.
Yahia H.B., Gaudin E., Darriet J. // Z. Naturforsch. 2009. B. 64. S. 875.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография