

Кристаллография, 2021, T. 66, № 5, стр. 708-722
Структурные исследования мембранного гликопротеина-предшественника бета-амилоида и белков, участвующих в процессе его протеолиза
А. С. Урбан 1, 2, 3, *, Я. В. Бершацкий 1, 2, К. В. Павлов 3, Э. В. Бочаров 1, 2
1 Институт биоорганической химии им. М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
2 Московский физико-технический институт
Долгопрудный, Россия
3 Федеральный научно-клинический центр физико-химической медицины
Москва, Россия
* E-mail: anatoly.urban@gmail.com
Поступила в редакцию 07.10.2020
После доработки 14.10.2020
Принята к публикации 23.11.2020
Аннотация
Болезнь Альцгеймера – самый распространенный вид нейродегенеративного заболевания в мире. Ее клинические проявления объясняются избирательной дегенерацией нейронов в участках коры головного мозга, ответственных за когнитивное восприятие и память. Вне нервных клеток в местах контакта нейронов накапливается амилоидный пептид, который собирается в упорядоченные тяжи, фибриллы, образующие так называемые амилоидные бляшки. Установлено, что амилоидный пептид (бета-амилоид или Aβ1–38–Aβ1–43), агрегирующий и формирующий амилоидные бляшки в мозге, является продуктом последовательного расщепления мембранного гликопротеина-предшественника β-амилоида β- и γ-секретазами в плазматической мембране нейронов. В обзоре освещены результаты структурных исследований данного процесса и основных белков, участвующих в нем.
ОГЛАВЛЕНИЕ
Введение
1. Многодоменная структура белка-предшественника бета-амилоида
1. Многодоменная структура белка-предшественника бета-амилоида
1.1. GFLD/HBD
1.2. CuBD
1.3. E1
1.4. KPI
1.5. E2
1.6. Полноразмерный эктодомен
1.7. AICD
1.8. ТМ домен
1.9. Aβ
1.10. Олигомеры
2. Α-секретаза
3. Β-секретаза
4. Γ-секретаза
Заключение
ВВЕДЕНИЕ
Стремительное увеличение доли пожилых людей, нуждающихся в уходе и медицинской поддержке, является серьезной социальной и экономической проблемой в развитых странах. Процесс старения включает в себя различные нейродегенеративные заболевания, нарушающие сложную сеть метаболических и сигнальных путей. Болезнь Альцгеймера – самый распространенный в мире вид нейродегенеративного заболевания, которым ежегодно заболевает несколько миллионов человек [1]. С увеличением продолжительности жизни вероятность развития этого вида деменции возрастает многократно. В патогенезе болезни Альцгеймера задействованы различные пептиды и белки, в том числе амилоидогенные, и прежде всего бета-амилоидный пептид (Aβ) и его белок-предшественник, а также мембранные секретазы, расщепляющие его до различных Aβ-изоформ. Настоящий обзор посвящен структурным исследованиям данного процесса современными методами структурной биологии, такими как рентгеноструктурный анализ (РСА), малоугловое рассеяние рентгеновских лучей (МУРР), ядерный магнитный резонанс (ЯМР) и криоэлектронная микроскопия.
1. МНОГОДОМЕННАЯ СТРУКТУРА БЕЛКА-ПРЕДШЕСТВЕННИКА БЕТА-АМИЛОИДА
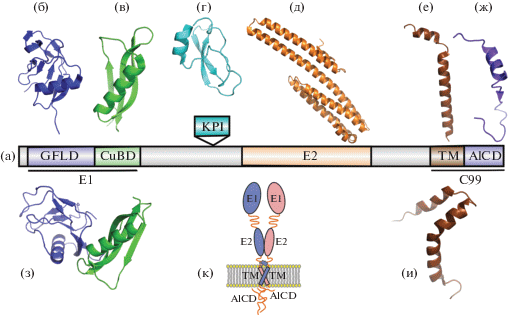
Гликопротеин-предшественник β-амилоида (APP – amyloid precursor protein) экспрессируется почти во всех животных клетках, исследованных на сегодня, причем с выраженным эволюционным консерватизмом. Хотя изучению белка АРР посвящены тысячи работ, его клеточные функции до сих пор полностью неясны. Был сделан ряд предположений, что белок АРР является рецептором холестерина в рафтах мембран нейронов [2], регулирует метаболизм железа в нейронах [3], а также является частью системы врожденного иммунитета в нервной системе человека [4]. Это типичный многодоменный мембранный белок первого типа, пронизывающий мембрану один раз, т.е. он имеет внеклеточную, внутриклеточную и трансмембранную части (рис. 1к). Как и многие мембранные рецепторы типа I, белок АРР димеризуется латерально в плазматической мембране. Полноразмерный белок АРР включает в себя следующие структурные домены: цистин-богатый домен, имеющий сходство с фактором роста (GFLD – growth factor-like domain), гепарин-связывающий домен (HBD), медьсвязывающий домен (CuBD), вместе образующие домен E1, кислый домен Ac, домен, гомологичный ингибиторам сериновых протеаз типа Кунитц (KPI), домен E2, трансмембранный домен, содержащий последовательность Aβ, и внутриклеточный домен AICD (APP intracellular domain) (рис. 1а). Вследствие большого размера (∼750 аминокислотных остатков), димеризации в мембране и подвижности доменов друг относительно друга структура высокого разрешения полноразмерного белка АРР не получена до сих пор, поэтому пространственные структуры доменов были определены отдельно различными структурными методами.
Рис. 1.
Схема строения APP человека (а) и структуры его отдельных доменов (б–з): б – GFLD – домен, подобный фактору роста (PDB:4PQD); в – CuBD – медьсвязывающий домен (PDB:2FJZ); г – KPI – домен, гомологичный ингибиторам протеаз типа Кунитц (PDB:1AAP); д – домен E2 (PDB:3NYL); е – трансмембранный домен (PDB:2LLM); ж – AICD – внутриклеточный домен (PDB:2ROZ); з – полноразмерный домен E1, различными цветами показаны входящие в него поддомены GFLD и CuBD (PDB:4PWQ); и – фрагмент C99, состоящий из трансмембранного и внутриклеточного доменов, показана структура трансмембранного домена, так как внутриклеточный домен находится в неупорядоченном состоянии (PDB:2LP1); к – схематическое изображение общей доменной организации APP.
1.1. GFLD/HBD
Для цистин-богатого домена, имеющего сходство с фактором роста (28–123), содержащего сайт связывания гепарина (HBD) [5], методом РСА определена структура с разрешением 1.8 Å (PDB:1MWP, рис. 1б) [6]. Структура домена состоит из девяти β-тяжей и одной α-спирали, которые складываются в компактный глобулярный домен. Большинство тяжей очень короткие, шесть из них содержат только три остатка. В основе домена лежит пятицепочечный антипараллельный β-лист. Центральный лист покрыт с одной стороны двумя двухцепочечными листами и с другой – спиралью и длинной петлей. Домен содержит три дисульфидных мостика, которые связывают его части вместе [6]. Поверхность домена в целом имеет положительный заряд, показано ее взаимодействие с фосфоинозитол бифосфатами и трифосфатами [7]. Структура этого домена в свободном состоянии также была определена с разрешением 1.33 Å (PDB:4PQD, не опубликовано). Домен способен образовывать комплекс с двухвалентным ионом меди, структура и свойства которого описаны в [8] (PDB:4JFN, 1.75 Å).
1.2. CuBD
Структура изолированного медьсвязывающего домена CuBD была определена методом ЯМР в растворе ([9], PDB:1OWT) и позднее методом РСА с разрешением 1.61 Å ([10], PDB:2FJZ, рис. 1в). Структура состоит из α-спирали (остатки 147–159) и β-листа из трех тяжей (остатки 133–139, 162–167 и 181–188). Дисульфидная связь между Cys-133 и Cys-187 соединяет тяжи 1 и 3, а между Cys-158 и Cys-186 – α-спираль с тяжем 3. Дисульфидная связь Cys-144–Cys-174 соединяет две петли на другом конце молекулы. Ввиду малого количества гидрофобных остатков в окрестностях этой дисульфидной связи можно предположить, что она очень важна для стабилизации структуры в области, которая не имеет вторичной структуры. В дополнение к трем дисульфидным связям в домене есть небольшое гидрофобное ядро (Leu136, Trp150, Val153, Ala154, Leu165, Met170, Val182 и Val185), стабилизирующее структуру. Поверхность CuBD сильно заряжена с несколькими областями отрицательного (Glu156, Glu160 и Glu183, Asp167 и Asp131) и положительного (Lys132, Lys134, Lys161, His147, His151 и Lys155) электростатического потенциала.
В [9] на основе данных электронного парамагнитного резонанса предложен механизм восстановления иона меди Cu2+ до Cu+, опосредованный взаимодействием ионов меди с CuBD. Так как сайт связывания меди имеет достаточно жесткую тетраэдрическую геометрию, предпочтительную для связывания иона Cu+, в то время как для Cu2+ энергетически выгодной является гексаэдрическая геометрия, связывание меди сопровождается восстановлением Cu2+ до Cu+. В отсутствие других восстановителей это приводит к окислению Met170, входящего в сайт связывания. Однако вряд ли имеет место такая реакция in vivo, так как приводит к ослаблению взаимодействия сайта с ионом, а восстановителем могут выступать аскорбат или тиолы.
1.3. E1
Ряд работ посвящен исследованию полноразмерного домена E1. Была получена кристаллическая структура гепарин-индуцированного димера домена E1 [11] (PDB:3KTM). Низкие B-факторы остатков на границе доменов, сравнимые с B-факторами внутренних остатков, заметно сниженные по сравнению с независимо определенными структурами отдельных поддоменов, говорят о том, что домен E1 образует единую структурную единицу и не состоит из двух независимых доменов, соединенных гибким линкером. Сам линкер, являясь высоко консервативным фрагментом молекулы, не принимает никакой стандартной вторичной структуры, но соединяет оба субдомена, как зиппер, и является частью гидрофобной междоменной границы раздела. Граница раздела стабилизирована одним солевым мостиком и несколькими сетями водородных связей. Также интересно, что Cu2+-связывающий сайт находится вблизи границы раздела между GFLD и CuBD. В [12] была получена структура E1-домена в комплексе с ионом меди (PDB:4JFN), где он скоординирован с атомами Nε2 двух консервативных остатков гистидина (His108 и His110). Она подтвердила, что сайт связывания иона находится на границе раздела доменов. Интересно, что связывание меди вызывает восстановление дисульфидного мостика (Cys98–Cys105), хотя очистку и кристаллизацию не проводили в восстанавливающих условиях, а также частичную дестабилизацию домена E1, как показано с помощью анализа B-факторов и спектроскопии кругового дихроизма. Также было подтверждено наличие двух пар остатков гистидина, которые координируют медь в GFLD (His108/His110) и CuBD (H147/H151), и что связывание ими ионов меди вызывает димеризацию E1-домена как in vitro, так и in vivo.
Структура домена E1 субатомного разрешения определена в [13] (PDB:4PWQ, 1.4 Å, рис. 1з). На основе ее анализа, а также данных динамического рассеяния света и точечного мутагенеза было установлено, что упаковка субдоменов GFLD и CuBD внутри E1 носит pH-зависимый характер. При нейтральных и слабощелочных значениях pH образуется более открытая конформация комплекса, а при кислых – более компактная. Специфические водородные связи между двумя карбоксилатами Asp177 и Glu87, а также между Asn89 и His147 являются основными детерминантами этого рН-управляемого конформационного переключения в APP–E1.
1.4. KPI
Первым кристаллизованным фрагментом APP был домен, гомологичный ингибиторам протеаз типа Кунитц (KPI), присутствующий только в не нейронных изоформах (sAPPα751 и sAPPα770) [14]. Поскольку этот домен кодируется одним экзоном и вставляется в середину последовательности APP, а также с учетом того, что он довольно гомологичен другим ингибиторам протеазы типа Кунитц, вероятно, что он представляет собой независимую единицу фолгинга и что структура изолированного домена хорошо представляет структуру домена в контексте полноразмерного APP. Домен ингибирует сериновые протеазы, как и гомологичные ему ингибиторы, структура его комплекса с бычьим трипсином была решена в [15] (PDB:1AAP, рис. 1г).
1.5. E2
Еще одна область внеклеточной части APP с определенной пространственной структурой и высокой консервативностью – E2-домен – присутствует, в том числе, и в нейрональной изоформе APP695. Первая структура этого сегмента APP была определена методом ЯМР в растворе (PDB:1TKN). Она включает в себя остатки 460–569, что соответствует поддомену E2, устойчивому к протеолизу трипсином [16]. Структура состоит из четырех α-спиралей, соединенных короткими петлями. Позднее были опубликованы структуры полноразмерного домена E2, определенные методом РСА (PDB:3NYL [17], PDB:3NYJ [17], рис. 1д). Полноразмерный домен E2 состоит из шести α-спиралей. N-концевая часть включает спирали 1 и 2, половину спирали 3. Ее наиболее заметной особенностью является двуспиральный элемент (colied-coil), образованный спиралями 2 и 3. Отсутствие какого-либо видимого поворота спиралей друг относительно друга может являться следствием нарушения в гептадном повторе, характерном для канонических двуспиральных белков. С-концевая часть содержит вторую половину спирали 3 и спирали 4, 5 и 6. Эта структура состоит в основном из трехцепочечных антипараллельных двуспиральных элементов: спирали 3 и 5 параллельны, тогда как 4 находится в противоположной ориентации. С-концевой пучок спиралей обладает левосторонней закрученностью в отличие от N-концевого. Эти два структурных элемента, объединенные непрерывной спиралью 3, находятся на ее противоположных сторонах и напрямую не взаимодействуют друг с другом. Центральная спираль 3 включает 56 аминокислот и имеет длину 90 Å. Она слегка изогнута в средней части, хотя не содержит остатков пролина. Изгиб может быть следствием взаимодействия со спиралями 1/2 и 4/5 либо образования димера E2. Обе описанные кристаллические структуры E2 содержат домен в виде антипараллельного димера и почти полностью повторяют друг друга. Кроме того, в [17] показано, что домен E2 образует димер в растворе, и димеризация усиливается в присутствии гепарина. В дополнение к этому высокое разрешение структуры позволило идентифицировать в ней два сайта связывания фосфат-анионов, которые в физиологических условиях участвуют в связывании гепарина.
Как и E1, домен E2 обладает способностью связывать ионы Zn2+ и Cu2+. Структуры комплексов решены в [18] (PDB:3UMI, 3UMK). Связывание ионов происходит в двух сайтах с разной спецификой. Аффинность к ионам меди оказывается гораздо больше, чем к ионам цинка. Данные поверхностного плазмонного резонанса, спектроскопии кругового дихроизма и ограниченного протеолиза показали, что связывание металлов в растворе приводит к значительным конформационным изменениям. Анализ структуры комплексов подтверждает это. Связывание ионов требует определенной координационной геометрии и, следовательно, определенной конформации домена. Так, при связывании меди происходит поворот N-концевого субдомена на 12°. Координация меди требует ориентации четырех координирующих гистидинов (His313, His382, His432 и His436) в отличие от цинка, в случае которого молекула воды заменяет в качестве лиганда His313. Таким образом, домен E2 может находиться по крайней мере в трех разных конформационных состояниях, различающихся гибкостью: наиболее гибкая свободная форма; более жесткая форма, связанная с цинком; и еще более ограниченный во внутримолекулярных движениях комплекс с медью [18].
1.6. Полноразмерный эктодомен
Полноразмерный эктодомен APP является самостоятельной биологически активной молекулой, биологические функции которой определяются фрагментами sAPPα и sAPPβ, продуцируемыми α- и β-секретазами соответственно. Структура высокого разрешения этого фрагмента до сих пор не определена. Однако в [19] методом МУРР и молекулярного моделирования была исследована структура фрагментов sAPPα695 sAPPα770. Из кривых рассеяния были определены функции попарного распределения расстояний P(r) для этих молекул, анализ которых позволил установить максимальные размеры белков Dmax и их радиусы вращения Rg. Максимумы длины, полученные из функций P(r), оказались равными 135 и 150, а радиусы вращения 42.6 ± 0.3 и 46.5 ± 2 Å для sAPPα695 и sAPPα770 соответственно. Практически такие же результаты дал анализ графиков Гинье, также подтвердивший монодисперсность исследованных образцов. Из соотношения между Dmax и Rg, а также общей формы распределения P(r) можно сделать вывод о вытянутой форме исследованных частиц. На основе тех же данных МУРР было проведено моделирование ab initio полноразмерного sAPPα с использованием в качестве модели цепочки псевдоостатков и с применением метода симулированного отжига для минимизации расхождения между расчетной и экспериментально измеренной кривой P(r) [20]. Для проверки моделей по ним были рассчитаны отношения гидродинамических объемов и молекулярных масс sAPPα и sAPPβ, которые показали хорошую сходимость к экспериментально измеренным значениям.
Известные кристаллические структуры доменов GFLD, CuBD, KPI и E2 были вписаны в структуру низкого разрешения, полученную по данным МУРР методом моделирования твердого тела [21], что позволило впервые рассмотреть модель структуры полноразмерного эктодомена APP. Также в [15] методом МУРР показано, а с помощью аналитической гель-фильтрации и ультрацентрифугирования подтверждено, что при взаимодействии с высокомолекулярным гепарином sAPPα695 образует комплекс с соотношением белок:гепарин 2:1, т.е. происходит гепарин-индуцированная димеризация эктодомена APP в растворе.
1.7. AICD
Внутриклеточный домен APP, содержащий около 50 аминокислотных остатков в свободном состоянии, не имеет стабильной упорядоченной структуры, что было показано методом ЯМР в растворе [22] и что типично для внутриклеточных доменов многих трансмембранных белков. Однако нестабильные элементы вторичной структуры, о наличии которых свидетельствуют измеренные химические сдвиги HN и Hα, могут являться зачатками структуры, которую принимает домен при взаимодействии с внутриклеточными лигандами. Структурные изменения в AICD при взаимодействии с белком Fe65 были зарегистрированы методом ЯМР, а структура комплекса из 31-аминокисотного фрагмента AICD и домена PTB2 Fe65 решена методом РСА [23] (PDB:3DXD). При связывании с Fe65 C-концевая часть AICD образует структуру, состоящую из двух α-спиралей, соединенных коротким β-листом. На основе анализа структуры мутантов T668E и T668A предполагаем механизм разрушения комплекса при фосфорилировании T668, согласно которому присоединение фосфата приводит к нарушению кэп-бокса N-концевой спирали AICD и ее дестабилизации. Аналогичная структура была получена с 32-аминокислотным C-концевым пептидом AICD и белком мыши Fe65L1 методом ЯМР [24] (PDB:2ROZ, рис. 1ж).
1.8. Трансмембранный домен
Полноразмерный трансмембранный домен APP включает в себя остатки Gly700-Leu723, часто к нему добавляют примембранный регион Gln686-Lys699, так как они образуют общую структурную единицу, взаимодействующую с мембраной. Пептид слишком мал, гидрофобен и подвижен, поэтому его сложно использовать как для получения кристаллов, так и исследовать методом криоэлектронной микроскопии. Основным методом исследования этого домена стала спектроскопия ЯМР. Пептид нерастворим в воде, поэтому необходим подбор мембраномоделирующей среды для поддержания его в нативной конформации. Так как размеры липосом и нанодисков не позволяют эффективно использовать ЯМР-спектроскопию для их изучения, структуры трансмембранного домена APP определены с использованием детергентных мицелл как мембраномоделирующей среды.
Первая структура полноразмерного трансмембранного домена опубликована в [25] (PDB:2LLM, рис. 1е) (использованы мицеллы додецилфосфохолина в качестве мембраномоделирующей среды), она включала в себя остатки Gln686–Lys726, т.е. весь трансмембранный и примембранный участки. Структура состоит из двух α-спиралей: короткой примембранной (Lys687–Asp694), взаимодействующей с поверхностью мембраны, и длинной трансмембранной спирали (Gly700–Leu723), связанных через небольшую подвижную петлю. Вблизи остатков Gly708–Gly709 трансмембранной спирали – небольшой 20°-ный изгиб. Топология структуры была подтверждена с помощью анализа динамики и метода спиновых меток. Также косвенно с помощью анализа изменений химических сдвигов при добавлении в среду холестерил гемисукцината – растворимого аналога холестерина – было подтверждено наличие в петле, соединяющей две спирали, сайта связывания холестерина [2]. В [26] опубликована структура димера Gln686–Lys726 (PDB:2LOH), в которой трансмембранные α-спирали образуют параллельный левозакрученный димер – расширенный гептадный мотив I702X3M706X2G709X3A713X2I716X3I720X2I723. Примембранная спираль, хотя и не участвует в димеризации непосредственно, также испытывает влияние перехода домена в димерную форму, что подтверждается анализом изменений химического сдвига. На другой стороне спирали находится GG4, который часто является мотивом димеризации трансмембранных доменов односпиральных трансмембранных белков [27], однако в данном димере он не задействован, и, возможно, участвует во взаимодействии трансмембранного домена с холестерином. Димер имеет площадь контакта ∼630 Å2, равномерно распределенную вдоль трансмембранной спирали, и энергию димеризации ∼1.4 ккал/моль.
Еще одна структура [28], решенная методом ЯМР, включает трансмембранный домен вместе с внутриклеточным доменом AICD (PDB:2LP1, рис. 1и). В качестве мемебраномоделирующей среды были использованы мицеллы LMPG. Структура состоит из неупорядоченного N-концевого сегмента, переходящего в примембранную спираль, взаимодействующую с поверхностью мембраны. Она, в свою очередь, соединена с трансмембранной спиралью с характерным изгибом в центре, как и в [25]. Далее идут C-петля и C-концевая спираль из девяти аминокислотных остатков, погруженная в мембрану. N-концевая часть структуры в целом повторяет (PDB:2LLM), а C-концевая представляет собой почти неупорядоченное состояние AICD. Также, но уже при использовании в качестве среды бицелл DHPC–DMPC с содержанием холестерина 0–20 мол. % в [28] показано связывание холестерина в районе N-концевой части трансмембранной спирали и N-концевой межспиральной петли с константой релевантной физиологической концентрации холестерина в мембране клеток. Связывание вовлекает незадействованный в димеризации GG4-мотив, что проливает свет на его физиологическую роль. Однако в [29] при исследовании изолированного трансмембранного фрагмента, лишенного примембранных спирали и петли, была получена альтернативная правозакрученная структура димера с ассоциацией через мотив GX3A (PBD:2LZ3) с углом между спиралями 22°. Также была показана роль наследственной патогенной мутации V715M, дестабилизирующей спиральную структуру. Для другой наследственной мутации L723P установлен эффект дестабилизации C-концевой части трансмембранной спирали с увеличением доступа воды к пептидному остову, что может приводить к заглублению N-концевой стороны спирали и ускорять ее протеолитическое расщепление γ-секретазой [30].
1.9. Aβ
β-амилоидные пептиды – наиболее часто исследуемые фрагменты APP. Это связано с тем, что именно β-амилоидный пептид – основной компонент амилоидных бляшек, обнаруживаемых в тканях мозга, пораженных болезнью Альцгеймера [31]. Более точно, основными компонентами амилоидных отложений являются пептиды Aβ1–40 и Aβ1–42, или, в другой нумерации, APP672-711 и APP672-713. Первая опубликованная структура Aβ1–40 была определена методом ЯМР в растворе (PDB:1AML, рис. 2а) [32]. В ней выделяются две α-спиральные области: от Gln15 до Asp23 и от Ile31 до Met35. Однако в качестве среды была использована смесь 40% трифторэтанола и 60% воды, которая предотвращает агрегацию пептида, но далека от его нативного мембранного окружения. В частности, она может способствовать образованию искусственно индуцированной α-спиральной структуры [33], следствием может быть достаточно большое среднеквадратичное отклонение 4.8 Å по 20 лучшим рассчитанным моделям. Структура того же пептида была определена с использованием додецилсульфата натрия (SDS) как мембраномоделирующей среды (PDB:1BA4, рис. 2б) [34]. В этой структуре наблюдается только одна протяженная α-спираль Gln15–Val36 с изгибом в области Gly25–Asn27. Хотя две части спирали проявляют заметную подвижность друг относительно друга, среднеквадратичное отклонение по остаткам 15–36 составляло ∼1.5 Å, а N-концевая часть структуры находится в неупорядоченном состоянии. В [35] удалось провести структурные исследования пептида Aβ1–40 в водном окружении, использовав специальный протокол дезагрегации и работая с очень малыми – 77 мкM – концентрациями пептида. Такие условия наиболее приближены к условиям in vivo. В полученной методом ЯМР структуре (PDB:2LFM, рис. 2г) центральная часть пептида H13–D23 образует спираль 310, что подтверждается наличием i,i+2 контактов, выявленных методом спектроскопии ядерного эффекта Оверхаузера (ЯЭО). Эта спираль имеет гидрофобные участки с обеих сторон, с которыми взаимодействуют гидрофобные остатки в почти неструктурированных N- и С-концах пептида. Хотя на N- и C-концах отсутствует стабильная вторичная структура, они не являются полностью не структурированными и содержат много коротких элементов α- и β-структур, образовавшихся в результате локальных взаимодействий между боковыми цепями концов и центральной спиральной структурой. Такая структура может быть переходным звеном между α-спиральной структурой, характерной для трансмембранного домена APP, и β-структурой – основой β-амилоидных олигомеров и фибрилл [36]. Для пептида Aβ17–34 была показана α-спиральная конформация в областях Phe20–Ser26 и Lys28–Gly33 с перегибом около остатков Ser26–Asn27–Lys28 (PDB:2MJ1). Для стабилизации пептида в растворимом состоянии на N- и C-концы были добавлены по два остатка глютаминовой кислоты [37], что позволило провести измерения с концентрацией пептида в образце ~3 мг/мл в физиологически релевантных условиях без использования детергентов или органических растворителей.
Рис. 2.
Различные структуры β-амилоидных пептидов (а–г), образованных ими фибрилл (д–ж) и олигомеров (з, и): а – Aβ1–40 в смеси TFE:H2O 40:60 (PDB:1AML); б – Aβ1–40 в мицеллах SDS (PDB:1BA4); в – Aβ1–42 в смеси HFIP:H2O 30:70 (PDB:1Z0Q); г – Aβ1–40 в воде при концентрации пептида 77 μM (PDB:2LFM); д – структура фибриллы Aβ1–42, определенная методом ЯМР в растворе (PDB:2BEG); е – структура фибриллы Aβ1–40 с трехосевой симметрией, построенная на основе данных ЯМР и просвечивающей электронной микроскопии (PDB:2LMQ); ж – структура фибриллы, построенная основе данных ЯМР в твердом теле с использованием в качестве затравки биоматериала больных болезнью Альцгеймера (PDB:2M4J); з – структура додекамера Aβ17–36, полученная методом рентгеновской кристаллографии (PDB:5HOY); и – структура тетрамера Aβ1–42 в мицеллах DPC, полученная методом ЯМР в растворе, цветом выделены мономеры с различной структурой.
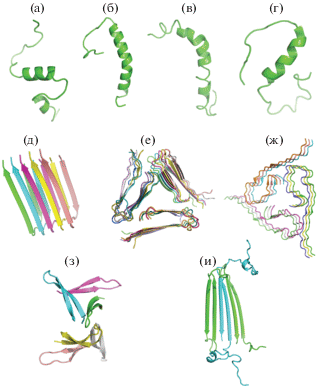
Перегиб в середине пептида, который разбивает его на две спирали, предположительно, позволяют пептиду претерпевать структурные изменения от α-спирали к β-листу, когда две половинки сближаются. 42-членный пептид Aβ1–42 является более сложным объектом для изучения. Имея два дополнительных гидрофобных остатка на C-конце, он еще более склонен к быстрой агрегации в растворе, чем Aβ1–40. Тем не менее, растворив пептид в гексафторэтаноле (HFIP) и добавляя необходимое количество воды, можно получить Aβ1–42 в растворенном состоянии в почти водной среде и исследовать его свойства. При варьировании доли воды в смеси от 20 до 99% с помощью спектроскопии кругового дихроизма и ЯМР удалось наблюдать постепенный переход от α-спиральной конформации к β-структурной [38]. Большая часть молекул переходит к β-структурной конформации при концентрации воды около 85%, но поскольку за этим следует достаточно быстрая агрегация пептида, для получения структуры методом ЯМР была выбрана концентрация воды 70%, что соответствует молярной доле воды 92.9%. Структура (PDB:1Z0Q, рис. 2в) состоит из двух α-спиралей, соединенных петлей: N-концевая спираль достаточно устойчива, со среднеквадратичным отклонением 1.67 Å; C-концевая спираль теряет стабильность, укорачивается, распространяясь лишь до остатка Gly33, и для некоторых рассчитанных конформеров начинает проявлять свойства, характерные для β-укладки.
Все структуры, содержащие α-спиральные элементы, имеют сходство с интактным трансмембранным доменом APP, также обладающим α-спиральной конформацией, и, по всей видимости, представляют разные стадии переходного процесса от α-спирального трансмембранного домена к β-структурным димерам, олигомерам и фибриллам, который реализуется после расщепления полноразмерного APP секретазами. Первая структура фибрилл была получена методом ЯМР в растворе в [39]. Для ее получения образцы фибрилл растворяли в DMSO/TFA, что позволило получить агрегаты, достаточно мелкие для изучения методом ЯМР. Так как комплексы, полученные диссоциацией фибрилл по такому протоколу, все еще слишком большие для получения ограничений методом ЯЭО, для установления водородных связей и солевых мостиков, участвующих в стабилизации внутри- и межмолекулярных взаимодействий, был применен обмен H/D, сайт-направленный мутагенез и данные твердотельного ЯМР. Остатки 1–17 в данной структуре (PDB:2BEG, рис. 2д) неупорядочены, а остатки 18–42 образуют мотив β-тяж–поворот–β-тяж. β-тяжи соседних мономеров образуют два параллельных β-слоя. Межмолекулярная связь стабилизирована солевым мостиком между Asp23 и Lys28 соседних мономеров. Межмолекулярные контакты боковых цепей образуются между нечетными остатками тяжа 1 n-й молекулы и четными остатками тяжа 2n–1-й молекулы. Это приводит к неспаренной валентности для взаимодействия на концах фибрилл и объясняет их рост. Фибриллы другой структуры, состоящие из пептидов Aβ1–40, были исследованы методом твердотельного ЯМР [40]. Фибриллы содержат параллельные β-тяжи из остатков 11–22 и 30–39, соединенные неупорядоченной вставкой 23–29. На основе данных ЯМР и просвечивающей электронной микроскопии была построена модель фибриллы, имеющая трехосевую симметрию относительно оси ее роста (PDB:2LMQ, рис. 2е). Похожая структура (PDB:2M4J, рис. 2ж) была получена также с помощью данных ЯМР в твердом теле, но в качестве стартовых точек роста фибрилл использовали биологический материал, выделенный из мозга пациентов с клинически подтвержденной болезнью Альцгеймера [41]. Это является важным свидетельством в пользу биологической релевантности результатов, так как молекулярная структура и свойства фибрилл, полученных in vitro, повторяют структуру и свойства тех образований, которые являлись начальными точками роста [42].
1.10. Олигомеры
На данный момент преобладает мнение, что наиболее токсичными для клеток формами β-амилоидных пептидов являются олигомеры [43–45]. Изучение структуры и свойств олигомерных форм осложняется трудностью получения их монодисперсных образцов, достаточно стабильных для проведения структурных исследований. В ряде работ по данным молекулярного моделирования, атомно-силовой микроскопии, электронной микроскопии, электрофизиологии и других методов были предложены модели образования олигомеров и мембранных пор и каналов на их основе. Так, была предложена модель формирования гексамера Aβ1–42, принимающего форму β-бочонка. Собираясь в 36-членный агрегат, способный пронизывать мембрану, он увеличивает ее проводимость для ионов, в частности Ca2+ [46]. Электрофизиологические измерения показывают, что пептиды Aβ1–42 способны образовывать как минимум три типа каналов с различной проводимостью, в то время как Aβ1–40 каналов не образует [47].
Олигомерное состояние пептидов Aβ является, по всей видимости, наиболее токсичным и, по крайней мере, в условиях in vitro транзиентным с характерным временем жизни несколько часов или дней в зависимости от условий эксперимента [48, 49]. Для стабилизации олигомерных агрегатов и исследования их структуры применяют различные экспериментальные методики. В случае пептида Aβ17–36, дополнительно стабилизировав дисульфидным мостиком изгиб, необходимый для взаимодействия его N- и C-концевых частей, удалось определить структуру додекамера методом РСА (PDB:5HOY, рис. 2з). Структура додекамера Aβ17–36 состоит из четырех тримерных блоков. В центральной полости, образованной четырьмя тримерными единицами, две молекулы Jeffamine M-600, использованного при кристаллизации, стабилизируют додекамерную структуру за счет гидрофобных взаимодействий с остатками в Aβ17–36 [50]. Для уточнения структуры было проведено моделирование методом молекулярной динамики с использованием крупнозернистого (coarse grained) силового поля и модели диссипативной динамики частиц (DPD – dissipative particle dynamics) пептида Aβ9–42 с экспериментально определенной структурой в качестве начального состояния. В этом моделировании C-концевые области спонтанно вписываются в центральную полость без каких-либо дополнительных ограничений на С-концевых участках. Таким образом, они участвуют в формировании гидрофобного ядра, стабилизирующего додекамерную структуру [51]. Додекамеры, в свою очередь, способны собираться в пору. Ее внешний диаметр составляет около 11 нм, а диаметр отверстия в центре поры ~2 нм. Толщина поры составляет ~5 нм, что сопоставимо с толщиной двухслойной липидной мембраны. Пять додекамеров собираются, образуя кольцевую структуру поры [50]. Олигомеры, использованные для кристаллизации, связываются с антителом A11, которое взаимодействует с олигомерными формами Aβ, в том числе с додекамерами массой 56 кДа, обнаруженными in vivo и обладающими токсичностью [52], но не взаимодействует с фибриллярными формами [53]. Это позволяет говорить о годности примененной модели и релевантности результатов.
Совсем другая структура олигомерной субъединицы, сформированной Aβ1–42, была получена методом ЯМР в растворе с использованием детергента DPC в качестве мембраномоделирующей среды [54]. В таких условиях пептиды формировали тетрамеры, состоящие из структурно различных субъединиц. Один тип субъединиц Aβ1–42 формирует β-шпильку из двух β-цепей, β1 и β2 – от G9 до Ala21 и от Gly29 до Val40. В субъединице второго типа остатки от Leu17 до Phe20 образуют короткую α-спираль, тогда как остатки Gly29–Ile41 образуют β-цепь β3. Таким образом, ядро тетрамера состоит из шести β-тяжей, соединенных всего двумя β-поворотами, а также двух коротких и двух длинных, содержащих α-спиральный сегмент, гибких N-концов. Среднеквадратичное отклонение структуры (PDB:6RHY, рис. 2и), рассчитанное по атомам основной цепи, составляет 0.77 Å. Чтобы отнести сигналы и уточнить характер взаимодействия между субъединицами в различных конформациях, были проведены эксперименты с использованием смеси Aβ1–42 и Aβ17–42, лишенного сегмента, образующего α-спираль в субъединице второго типа и не способного сложиться в β-шпильку первого типа. Тетрамеры, структура которых была определена, способны самопроизвольно образовывать октамеры со структурой β-сэндвича, как показано методами эксклюзионной хроматографии/спектрометрии ионной подвижности и с помощью моделирования методом молекулярной динамики [54]. Также с помощью моделирования и электрофизиологических измерений была показана способность таких олигомеров формировать поры в липидном бислое.
2. Α-СЕКРЕТАЗА
Α-секретазы – протеазы, расщепляющие APP в α-сайте, что приводит к образованию неамилоидогенного пептида p3 и внеклеточной части sAPPα. Такую активность проявляют белки семейства ADAM (A Disintegring And Metalloproteinase). В геноме человека закодировано 22 белка, относящихся к семейству ADAM. Все они расщепляют трансмембранные белки вблизи поверхности клетки в ходе процесса, называемого шеддингом. Субстраты ADAM включают факторы роста, цитокины, хемокины, рецепторы и молекулы адгезии. Таким образом, белки ADAM играют важную роль в межклеточной адгезии и передаче сигналов [55]. Хотя активность в отношении APP способны проявлять ADAM9 и ADAM17, основным путем, приводящим к p3 и sAPPα, является протеолиз ADAM10 [56, 57]. Помимо APP биологически важными субстратами ADAM10 являются Notch [58], эфриновый рецептор [59], проэпидермальный фактор роста [60], HER2 [61], TNF-α [62] и другие белки. Металлопротеиназы ADAM представляют собой трансмембранные белки I типа с модульной доменной организацией. ADAM10, как и его ближайший гомолог – ADAM17 (также известен как TACE – TNF-α converting enzyme), имеет эктодомен, состоящий из N-концевой сигнальной последовательности. Далее следует так называемый продомен, за ним металлопротеиназный, дезинтегриновый и цистеин-богатый домены. После них идет трансмембранный домен, пронизывающий мембрану клетки, и небольшой цитоплазматический домен.
Как и многие другие члены семейства ADAM, ADAM10 экспрессируется как профермент, включающий инактивирующий продомен 10–213, а удаление при протеолизе конвертазой PC7 и фурином продомена в ходе созревания фермента приводит к его активации [63]. Продомен является селективным ингибитором ADAM10 с IC50 около 50 нм и не взаимодействует с другими белками того же семейства [64]. Изолированный продомен образует димер в растворе и по данным спектроскопии кругового дихроизма имеет стабильный фолд, состоящий из α- и β-структурных элементов [64]. По всей видимости, продомен также играет роль шаперона, необходимого для созревания ADAM10, так как экспрессия укороченного варианта белка, лишенного продомена, приводит к протеолитически неактивному продукту, а коэкспрессия изолированного продомена вместе с ним восстанавливает протеолитическую функцию [63]. Был описан ряд мутаций, затрагивающих продомен ADAM10, ассоциированных с поздним развитием болезни Альцгеймера [65]. На данный момент нет структурных данных о механизмах ингибирования протеолитической активности и специфичности этого взаимодействия.
Впервые структура дезинтегринового и цистеин-богатого доменов (PDB:2AO7) была определена для ADAM10 из Bos taurus методом РСА с разрешением 2.9 Å [59]. Пары доменов образуют непрерывную удлиненную, слегка изогнутую структуру длиной около 75 Å. Дезинтегриновый домен содержит два β-тяжа, а цистеин-богатый домен – семь коротких β-тяжей и три короткие α-спирали. Помимо небольших гидрофобных ядер в цистеин-богатом домене и на границе между двумя доменами общая структура в основном стабилизируется набором дисульфидных связей. Дезинтегриновый домен ADAM10 (остатки 455–550) обладает структурной гомологией с дезинтегринами триместатином [66] и ингибитором свертывания крови [67]. Дисульфидные связи в дезинтегриновом домене ADAM10 топологически идентичны связям в триместатине. Богатый цистеином домен (остатки 551–646) вместе с дезинтегриновым доменом образует небольшое гидрофобное ядро, формируя непрерывную структуру, которая обладает небольшой междоменной гибкостью, а дисульфидный мостик (Cys530–Cys562) между двумя доменами дополнительно стабилизирует их взаимодействие. Цистеин-богатый домен имеет характерный для ADAM-белков α/β-фолд, стабилизированный пятью дисульфидными связями. Один из трех его β-листов состоит из трех центральных β-тяжей, которые контактируют с тремя α-спиралями, а два других β-листа состоят из двух N-концевых и двух C-концевых β-тяжей соответственно. N-концевой β-лист контактирует с α-спиралью цистеин-богатого домена с одной стороны и с дезинтегриновым доменом с другой. На поверхности цистеин-богатого домена находится достаточно большой отрицательно заряженный карман, через который происходит взаимодействие с субстратом [59].
Для изучения взаимодействия механизма специфичности субстрата и роли каждого из доменов в [68] методом РСА была определена структура полноразмерного экстодомена ADAM10 человека (PDB:6BE6, рис. 3а). Внеклеточный домен ADAM10 принимает компактную конформацию, напоминающую острие стрелы, где каталитический – металлопротеиназный – домен окружен дезинтегриновым и цистеин-богатым доменами. Структура, вероятно, представляет аутоингибированную конформацию, поскольку расположение богатого цистеином домена в устье пептид-связывающей бороздки частично перекрывает активный сайт. Как и в структуре из [59] (PDB:2AO7), дезинтегриновый и цистеин-богатый домены содержат мало вторичной структуры и принимают чашевидную форму, а их структура стабилизируется сетью дисульфидных связей. Каталитический домен ADAM10 похож на активные центры других ферментов из семейств ADAM, матриксных металлопротеаз и металлопротеиназ из яда змей. Одна сторона кармана, в котором расположен активный сайт, образована пятицепочечным β-листом, вторая – центральной спиралью. Эта спираль включает мотив HEX2H, часть каталитического центра HEX2HX2GX2H [69], содержащий два гистидина, которые координируют связанный ион цинка (His383 и His387) и остаток глутамата (Glu384), участвующий в кислотном катализе. Третий гистидин цинксвязывающего мотива (His393) расположен на петле, следующей за этой спиралью.
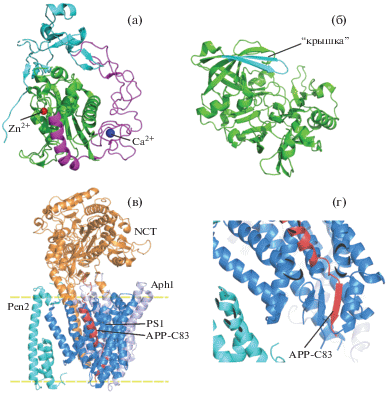
Рис. 3.
Структура эктодомена ADAM10 (PDB:6BE6), определенная методом РСА (а), сферами обозначены координированные ионы цинка и кальция. Структура эктодомена свободного BACE1 (PDB:1SGZ) (б): отмечена “крышка”. Структура γ-секретазного комплекса (в), содержащего никастрин (NCT), Aph1, Pen2, пресенилин (PS1), APP-C83, линиями схематически изображена поверхность мембраны. Увеличенный фрагмент структуры γ-секретазного комплекса, на котором показано образование β-слоя сегментами APP-C83 и PS1 (г).
Интересной особенностью данной структуры является то, что каждый активный центр фермента в кристалле занят С-концом соседней белковой субъединицы, мимикрируя процесс узнавания субстрата. Один из центров координации цинка занят C-концевым атомом кислорода из остатка Gly655. То есть C-концевой гистидиновый таг, используемый для очистки белка, был отщеплен во время выделения или кристаллизации, что привело к случайному формированию комплекса фермент–продукт. Из анализа структуры следует, что цистеин-богатый домен препятствует доступу белковых субстратов к карманам S1' и S2', что приводит к автоингибированию. Наложение структуры эктодомена ADAM10 на структуру комплекса дезинтегринового и цистеин-богатого домена с модулирующим активность ADAM10 антителом 8C7 [70] (PDB:5L0Q) показывает, что каталитический домен ADAM10 и тяжелая цепь 8C7 будут перекрываться, что приведет к значительному стерическому конфликту. То есть связывание антитела 8C7 приведет к увеличению доступности активного сайта и снятию автоингибирования. По биохимическим данным связывание 8C7 действительно увеличивает активность ADAM10 [68].
3. Β-СЕКРЕТАЗА
Β-секретаза – первый фермент, осуществляющий процессинг APP по амилоидогеннному пути. Именно с расщепления APP в β-сайте начинается серия протеолитических реакций, приводящих к образованию пептидов Aβ40 и Aβ42. Ферментативная активность β-секретазы определяется двумя близкими гомологами BACE1 и BACE2. Активность BACE1 превалирует в нервной ткани и повышается с возрастом, и в случае развития болезни Альцгеймера [71] в последнее время упрочняется мнение, что BACE2 также может вносить вклад в развитие патологии [72]. BACE1 экспрессируется преимущественно в нейронах, и ее активность важна для нормальной миелинизации аксонов [73], что опосредовано, по всей видимости, активностью BACE1 в отношении субстрата Neuregulin 1 (Nrg1) [74]. Среди других субстратов BACE1 важные биологические функции выполняют лиганд рецептора Notch Jag1 [75], натриевый потенциал-зависимый канал β2 Navβ2 [76], калиевые потенциал-зависимые каналы KCNE1 и KCNE2 [77], ассоциированный с судорогами 6-подобный белок Sez6L [78]. Методом протеомики было идентифицировано 68 потенциальных субстратов BACE1 [79], позднее с помощью более детального анализа секретома к ним было добавлено еще 23 [78].
BACE1 представляет собой интегральный трансмембранный гликопротеин I типа с отщепляемой сигнальной последовательностью из 21 остатка, продомен из 23 остатков с большим эктодоменом из 421 остатка, одним трансмембранным доменом из 22 аминокислот и коротким цитоплазматическим сегментом из 24 остатков [80]. Продомен проявляет лишь слабые свойства ингибитора активности BACE1 в отличие от похожих доменов других аспартатных протеаз, однако помогает фолдингу фермента [81]. Известно, что взаимодействие BACE1 с липидным бислоем критически важно для протеолиза APP и BACE1 с делетированным трансмембранным доменом и не активно в отношении APP in vivo [82].
Структура эктодомена BACE1 в комплексе с ингибитором пептидной природы изучена в [83]. Структура была получена методом молекулярного замещения с использованием в качестве начальной модели человеческого пепсина, который на 22% гомологичен BACE1. Протеазный домен ВАСЕ1 (PDB:1FKN) имеет двулопастную структуру, типичную для аспартатных протеаз, и демонстрирует высокую степень консервативности с пепсином и ВАСЕ2 [84] (PDB:2EWY). Субстрат, связывающий карман и содержащий активный сайт, расположен между N- и C-концевыми долями и частично прикрыт “крышкой”, представляющей собой антипараллельную шпильку, которая контролирует доступ субстрата к активному сайту и протеолитическую специфичность. В целом структура почти полностью повторяет структуру пепсина, за исключением восьми коротких вставок, образующих петли или спирали на поверхности глобулы и несколько увеличивающих общую поверхность молекулы. Из 21 остатка продомена только последние шесть видны на картах электронной плотности. Остальные, вероятно, подвижны, что согласуется с тем, что неструктурированный просегмент вытесняется из щели активного сайта ингибитором. В структуре свободного эктодомена (PDB:1SGZ, рис. 2б) “крышка” оказывается зафиксированной в еще более открытом состоянии водородной связью между Tyr41 и Gly74, в то время как в структуре со связанным ингибитором Tyr41 взаимодействует с его боковыми цепями P1 и P2'. При этом остается пространство 6.5 Å, достаточное для ограниченного прохода субстрата к активному центру [85].
Структура проливает свет на патологический механизм “шведской” наследственной мутации APP KM670/671NL. Подсайты BACE1 S1 и S3, состоящие в основном из гидрофобных остатков, имеют конформации, сильно отличающиеся от пепсина из-за отсутствия пепсиновой спирали 111–114. Боковые цепи ингибитора P3 – валин – и P1 – лейцин – плотно упакованы друг относительно друга и имеют сильные гидрофобные контакты с ферментом, особенно P1, который взаимодействует с Tyr71 и Phe108. В APP дикого типа остатки P2 и P1, смежные с сайтом расщепления секретазой, – это лизин и метионин соответственно. “Шведский” мутант APP содержит аспарагин и лизин в этих положениях, что приводит к более сильному межмолекулярному взаимодействию и 60-кратному увеличению Kcat/Km APP [86].
Трансмембранный домен BACE1 является крайне интересным объектом, так как содержит уникальный мотив M462X3C466X3M470X3C474X3C478, способный к тримеризации и взаимодействию с ионами меди [87]. Поскольку активность BACE1 является ключевым звеном в патогенезе болезни Альцгеймера, определение его структуры дало начало большому количеству исследований, цель которых – найти клинически эффективные и безопасные ингибиторы этого фермента. На данный момент методом РСА получено более 400 пространственных структур комплексов BACE1 с различными ингибиторами. Ряд из них находится на поздних стадиях клинических исследований.
4. Γ-СЕКРЕТАЗА
Γ-секретаза завершает протеолитический процессинг APP, предварительно расщепленного α- или β-секретазой. Ее активность приводит к образованию фрагмента p3 и β-амилоидных пептидов соответственно, а также внутриклеточного фрагмента AICD. Γ-секретаза обладает несколькими характерными и крайне интересными особенностями. Во-первых, в отличие от α- и β-секретаз, активные домены которых растворимы, протеолиз γ-секретазой происходит в толще мембраны [88]. Во-вторых, γ-секретаза, по всей видимости, не имеет ярко выраженной субстратной специфичности и выполняет роль “трансмембранной протеасомы”, расщепляя трансмембранные домены различных белков [89, 90]. Всего насчитывается более 90 трансмембранных белков I типа, трансмембранные домены которых расщепляет γ-секретаза, после того как их эктодомены были отщеплены шеддазами, такими как α- и β-секретазы [90]. В-третьих, γ-секретаза способна взаимодействовать со своим субстратом несколько раз, на каждом последующем этапе отщепляя от него по три–четыре аминокислотных остатка и таким образом создавая набор продуктов различной длины [91, 92]. Именно так образуются пептиды Aβ1–40 и Aβ1–42, изменения в абсолютном и относительном количестве которых сопровождают патогенез болезни Альцгеймера. Кроме того, именно в последовательности PS1–компонент γ-секретазного комплекса находится большинство мутаций, ассоциированных с ранним развитием болезни Альцгеймера [93].
Таким образом, γ-секретаза является ключевым ферментом, участвующим в образовании β-амилоидных пептидов, и изучение ее структуры необходимо для понимания молекулярных механизмов развития этого заболевания.
Γ-секретаза, а точнее γ-секретазный комплекс, является мультисубъединичным ферментом и состоит из четырех трансмембранных белков: PS (presenilin, представлен белками PS1 или PS2), NCT (nicastrin), Aph1 (anterior pharynx defective 1), Pen2 (presenilin enhancer 2) [94]. Комплекс включает в себя 20 трансмембранных спиралей и имеет молекулярную массу ∼170 кДа, в результате гликозилирования никастрина к нему добавляются еще 30–70 кДа [88]. Пресенилин осуществляет каталитическую функцию, никастрин – функцию распознающего субстрат центра, остальные субъединицы действуют как необходимые кофакторы [95]. В ходе созревания комплекса происходит эндопротеолиз пресенилина с образованием N- и C-концевых фрагментов, являющихся частями активной γ-секретазы [96]. Так как компоненты комплекса претерпевают сложный процесс созревания и по отдельности не проявляют биологической активности (в частности, не удается получить каталитически активный изолированный пресенилин), важным шагом на пути к пониманию структурных основ организации γ-секретазы стала разработка системы экспрессии функциональной γ-секретазы в клетках CHO (Chinese hamster ovary – клетки яичника китайского хомячка) [97], позволяющей получать достаточные для структурных исследований количества γ-секретазы.
Следующим важным шагом на пути решения этой задачи стало развитие метода криоэлектронной микроскопии, который позволил получить структуру функционального γ-секретазного комплекса с субатомным разрешением. Структура была опубликована почти одновременно двумя независимыми группами: (PDB:4UIS) [98] с разрешением 4.4 Å и (PDB:5A63) [99]. В обеих работах для построения структуры пресенилина использовали структуру белка PSH – гомолога пресенилина из архей, совпадающего по последовательности с пресенилином на 19%, – полученную методом РСА. Структура γ-секретазы имеет подковообразную форму. Внеклеточная область γ-секретазы в основном состоит из внеклеточного домена никастрина. Единственная трансмембранная (ТМ) спираль никастрина взаимодействует с TM1, TM5, TM7 Aph-1. Pen-2 содержит три трансмембранные спирали, две из которых не проходят через мембрану насквозь, а соединяются в ее толще. TM1 и TM3 Pen-2 взаимодействуют с TM4 из пресенилина. Основная часть подковы состоит из 16 трансмембранных спиралей пресенилина и Aph-1. Девять трансмембранных спиралей пресенилина значительно различаются по длине: так, TM9 содержит 30 остатков, а TM7 – только 18. Расстояние между атомами Сα двух каталитических остатков пресенилина Asp257 и Asp385 – около 10.6 Å, что значительно больше, чем в других аспартатных протеазах, но они находятся на разном расстоянии от поверхности мембраны. Каталитический центр расположен рядом с мотивом PAL на TM9, который, как считается, играет роль в распознавании субстрата [100]. Вероятно, связывание вызывает выравнивание этих двух остатков аспартата и последующий катализ. Расположение каталитических остатков свидетельствует о том, что белки-субстраты могут получать доступ к активному центру латерально с выпуклой стороны подковы. Мутации в гене пресенилина, ассоциированные с болезнью Альцгеймера, затрагивают 135 аминокислот, из которых на 53 приходится две или более мутаций (http://www.alzforum.org/mutations). Среди этих 53 остатков 35 находятся в областях с разрешенной электронной плотностью и структурой. Таких остатков восемь в TM5, семь в TM6, но ни одного в TM1. На эти 35 аминокислот приходится в общей сложности 101 патогенная мутация. Эти остатки группируются в два кластера. Первый находится на внутренней поверхности трансмембранных спиралей ТМ2–ТМ5. Среди 20 затронутых остатков в этих трансмембранных спиралях боковые цепи 18 обращены к внутренней стороне TM 2–5. То есть затрагивается только одна сторона каждой спирали. Второй кластер расположен на внутренней стороне TM6–ТМ9 вблизи каталитических остатков Asp257 и Asp385.
На данный момент кульминацией структурных исследований γ-секретазы стала работа [101], в которой методом криоэлектронной микроскопии с разрешением 2.6 Å определена структура комплекса γ-секретазы с ее субстратом – трансмембранным доменом APP (PDB:6IYC, рис. 3в). Для стабилизации комплекса фермент–субстрат в обе последовательности были введены остатки цистеина: мутация Q112C для PS1 и V695C для APP-C83, а чтобы избежать расщепления субстрата, каталитический остаток Asp385 в PS1 был мутирован на Ala. Так как каталитическая активность необходима для автопротеолиза пресенилина, он был экспрессирован в виде N- и C-концевых фрагментов. По сравнению с γ-секретазой, не содержащей субстрата, гибкая трансмембранная спираль 2 (TM2) PS1 упорядочивается при связывании с APP–C83 и участвует в его распознавании, а C-концевой участок TM6 PS1 распадается на петлю и короткую спираль. Tрансмембранная спраль APP тесно взаимодействует с пятью окружающими трансмембранными спиралями (TM2, TM3, TM5, TM6 и TM7) пресенилина, в том числе с теми, на которые приходится большая часть патогенных мутаций. C-концевой сегмент APP образует β-тяж, который вместе с двумя индуцированными β-тяжами пресенилина формирует β-лист (рис. 3г). В результате пептидная связь, непосредственно предшествующая β-листу, оказывается подставлена каталитическому сайту γ-секретазы для расщепления.
ЗАКЛЮЧЕНИЕ
Развитие методов изучения пространственной структуры биомолекул позволило полностью описать на молекулярном уровне процесс образования β-амилоидных пептидов, в том числе описать каскад событий, происходящих при последовательном протеолизе белка АРР. Полученные структурные данные позволяют выявить ряд факторов, приводящих к развитию болезни Альцгеймера, и составляют практическую основу для разработки эффективных целевых фармакологических соединений для ее эффективной терапии и ранней диагностики.
Работа выполнена при поддержке Российского научного фонда: раздел 1 (Многодоменная структура белка-предшественника бета-амилоида) – грант № 20-64-46027, разделы 2–4 (α-,β-, γ-секретазы) – грант № 19-15-00427.
Список литературы
O’Brien R.J., Wong P.C. // Annu. Rev. Neurosci. 2011. V. 34. P. 185. https://doi.org/10.1146/annurev-neuro-061010-113613
Beel A.J., Mobley C.K., Kim H.J. et al. // Biochemistry. 2008. V. 47 (36). P. 9428. https://doi.org/0.1021/bi800993c
Duce J.A., Tsatsanis A., Cater M.A. et al. // Cell. 2010. V. 142 (6). P. 857. https://doi.org/10.1016/j.cell.2010.08.014
Soscia S.J., Kirby J.E., Washicosky K.J. et al. // PLoS ONE / Ed. Bush A.I. 2010. V. 5 (3). P. e9505. https://doi.org/10.1371/journal.pone.0009505
Small D.H., Nurcombe V., Reed G. et al. // J. Neurosci. J. Soc. Neurosci. 1994. V. 14 (4). P. 2117.
Rossjohn J., Cappai R., Feil S.C. et al. // Nat. Struct. Biol. 1999. V. 6 (4). P. 327. https://doi.org/10.1038/7562
Dawkins E., Gasperini R., Hu Y. et al. // J. Neurosci. Res. 2014. V. 92 (11). P. 1478. https://doi.org/10.1002/jnr.23422
Baumkötter F., Schmidt N., Vargas C. et al. // J. Neurosci. 2014. V. 34 (33). P. 11159. https://doi.org/10.1523/JNEUROSCI.0180-14.2014
Barnham K.J., McKinstry W.J., Multhaup G. et al. // J. Biol. Chem. 2003. V. 278 (19). P. 17401. https://doi.org/10.1074/jbc.M300629200
Kong G.K.-W., Adams J.J., Harris H.H. et al. // J. Mol. Biol. 2007. V. 367 (1). P. 148. https://doi.org/10.1016/j.jmb.2006.12.041
Dahms S.O., Hoefgen S., Roeser D. et al. // Proc. Natl. Acad. Sci. 2010. V. 107 (12). P. 5381. https://doi.org/10.1073/pnas.0911326107
Baumkötter F., Schmidt N., Vargas C. et al. // J. Neurosci. 2014. V. 34 (33). P. 11159. https://doi.org/10.1523/JNEUROSCI.0180-14.2014
Hoefgen S., Dahms S.O., Oertwig K., Than M.E. // J. Mol. Biol. 2015. V. 427 (2). P. 433. https://doi.org/10.1016/j.jmb.2014.12.005
Hynes T.R., Randal M., Kennedy L.A. et al. // Biochemistry. 1990. V. 29 (43). P. 10018. https://doi.org/10.1021/bi00495a002
Scheidig A.J., Hynes T.R., Pelletier L.A. et al. // Protein Sci. 1997. V. 6 (9). P. 1806. https://doi.org/10.1002/pro.5560060902
Dulubova I., Ho A., Huryeva I. et al. // Biochemistry. 2004. V. 43 (30). P. 9583. https://doi.org/10.1021/bi049041o
Lee S., Xue Y., Hu J. et al. // Biochemistry. 2011. V. 50 (24). P. 5453. https://doi.org/10.1021/bi101846x
Dahms S.O., Könnig I., Roeser D. et al. // J. Mol. Biol. 2012. V. 416 (3). P. 438. https://doi.org/10.1016/j.jmb.2011.12.057
Gralle M., Oliveira C.L.P., Guerreiro L.H. et al. // J. Mol. Biol. 2006. V. 357 (2). P. 493. https://doi.org/10.1016/j.jmb.2005.12.053
Svergun D.I., Petoukhov M.V., Koch M.H. // Biophys. J. 2001. V. 80 (6). P. 2946.
Konarev P.V., Petoukhov M.V., Svergun D.I. // J. Appl. Cryst. 2001. V. 34 (4). P. 527. https://doi.org/10.1107/S0021889801006100
Ramelot T.A., Gentile L.N., Nicholson L.K. // Biochemistry. 2000. V. 39 (10). P. 2714. https://doi.org/10.1021/bi992580m
Radzimanowski J., Simon B., Sattler M. et al. // EMBO Rep. 2008. V. 9 (11). P. 1134. https://doi.org/10.1038/embor.2008.188
Li H., Koshiba S., Hayashi F. et al. // J. Biol. Chem. 2008. V. 283 (40). P. 27165. https://doi.org/10.1074/jbc.M803892200
Nadezhdin K.D., Bocharova O.V., Bocharov E.V., Arseniev A.S. // Acta Naturae. 2011. V. 3 (1). P. 69.
Nadezhdin K.D., Bocharova O.V., Bocharov E.V., Arseniev A.S. // FEBS Lett. 2012. V. 586 (12). P. 1687. https://doi.org/10.1016/j.febslet.2012.04.062
Mackenzie K.R. // Chem. Rev. 2006. V. 106 (5). P. 1931. https://doi.org/10.1021/cr0404388
Barrett P.J., Song Y., Van Horn W.D. et al. // Science. 2012. V. 336 (6085). P. 1168. https://doi.org/10.1126/science.1219988
Chen W., Gamache E., Rosenman D.J. et al. // Nat. Commun. 2014. V. 5 (1). P. 3037. https://doi.org/10.1038/ncomms4037
Bocharov E.V., Nadezhdin K.D., Urban A.S. et al. // ACS Chem. Biol. 2019. V. 14 (1). P 1573. https://doi.org/10.1021/acschembio.9b00309
Hardy J., Allsop D. // Trends Pharmacol. Sci. 1991. V. 12 (10). P. 383.
Sticht H., Bayer P., Willbold D. et al. // Eur. J. Biochem. 1995. V. 233 (1). P. 293. https://doi.org/10.1111/j.1432-1033.1995.293_1.x
Fraga A.S., Esteves A.C., Micaelo N. et al. // Int. J. Biol. Macromol. 2012. V. 50 (2). P. 323. https://doi.org/10.1016/j.ijbiomac.2011.12.018
Coles M., Bicknell W., Watson A.A. et al. // Biochemistry. 1998. V. 37 (31). P. 11064. https://doi.org/10.1021/bi972979f
Vivekanandan S., Brender J.R., Lee S.Y., Ramamoorthy A. // Biochem. Biophys. Res. Commun. 2011. V. 411 (2). P. 312. https://doi.org/10.1016/j.bbrc.2011.06.133
Fezoui Y., Teplow D.B. // J. Biol. Chem. 2002. V. 277 (40). P. 36948. https://doi.org/10.1074/jbc.M204168200
Fonar G., Samson A.O. // Biosci. Rep. 2014. V. 34 (6). P. e00155. https://doi.org/10.1042/BSR20140094
Tomaselli S., Esposito V., Vangone P. et al. // Chem. Bio. Chem. 2006. V. 7 (2). P. 257. https://doi.org/10.1002/cbic.200500223
Lührs T., Ritter C., Adrian M. et al. // Proc. Natl. Acad. Sci. 2005. V. 102 (48). P. 17342. https://doi.org/10.1073/pnas.0506723102
Paravastu A.K., Leapman R.D., Yau W.-M., Tycko R. // Proc. Natl. Acad. Sci. 2008. V. 105 (47). P. 18349. https://doi.org/10.1073/pnas.0806270105
Lu J.-X., Qiang W., Yau W.-M. et al. // Cell. 2013. V. 154 (6). P. 1257. https://doi.org/10.1016/j.cell.2013.08.035
Kodali R., Williams A.D., Chemuru S., Wetzel R. // J. Mol. Biol. 2010. V. 401 (3). P. 503. https://doi.org/10.1016/j.jmb.2010.06.023
Zhao L.N., Long H., Mu Y., Chew L.Y. // Int. J. Mol. Sci. 2012. V. 13 (6). P. 7303. https://doi.org/10.3390/ijms13067303
Sengupta U., Nilson A.N., Kayed R. // E Bio. Medicine. 2016. V. 6. P. 42. https://doi.org/10.1016/j.ebiom.2016.03.035
Rauk A. // Dalton Trans. Camb. Engl. 2008. № 10. P. 1273. https://doi.org/10.1039/b718601k
Shafrir Y., Durell S., Arispe N., Guy H.R. // Proteins. 2010. V. 78 (16). P. 3473. https://doi.org/10.1002/prot.22853
Bode D.C., Baker M.D., Viles J.H. // J. Biol. Chem. 2017. V. 292 (4). P. 1404. https://doi.org/10.1074/jbc.M116.762526
Yu Y., Yin T., Peng Q. et al. // ACS Sens. 2019. V. 4 (2). P. 471. https://doi.org/10.1021/acssensors.8b01493
Shea D., Hsu C.-C., Bi T.M. et al. // Proc. Natl. Acad. Sci. 2019. V. 116 (18). P. 8895. https://doi.org/10.1073/pnas.1820585116
Kreutzer A.G., Hamza I.L., Spencer R.K., Nowick J.S. // J. Am. Chem. Soc. 2016. V. 138 (13). P. 4634. https://doi.org/10.1021/jacs.6b01332
Kawai R., Chiba S., Okuwaki K. et al. // ACS Chem. Neurosci. 2020. V. 11 (3). P. 385. https://doi.org/10.1021/acschemneuro.9b00602
Lesne S.E. // Swiss Med. Wkly. 2014. V. 144. № 4546. P. w14021. https://doi.org/10.4414/smw.2014.14021
Ono K., Yamada M. // J. Neurochem. 2011. V. 117 (1). P. 19. https://doi.org/10.1111/j.1471-4159.2011.07187.x
Ciudad S., Puig E., Botzanowski T. et al. // Nat. Commun. 2020. V. 11 (1). P. 3014. https://doi.org/10.1038/s41467-020-16566-1
Weber S., Saftig P. // Development. 2012. V. 139 (20). P. 3693. https://doi.org/10.1242/dev.076398
Asai M., Hattori C., Szabó B. et al. // Biochem. Biophys. Res. Commun. 2003. V. 301 (1). P. 231. https://doi.org/10.1016/s0006-291x(02)02999-6
Kuhn P.-H., Wang H., Dislich B. et al. // EMBO J. 2010. V. 29 (17). P. 3020. https://doi.org/10.1038/emboj.2010.167
Kovall R.A., Gebelein B., Sprinzak D., Kopan R. // Dev. Cell. 2017. V. 41 (3). P. 228. https://doi.org/10.1016/j.devcel.2017.04.001
Janes P.W., Saha N., Barton W.A. et al. // Cell. 2005. V. 123 (2). P. 291. https://doi.org/10.1016/j.cell.2005.08.014
Arduise C., Abache T., Li L. et al. // J. Immunol. 2008. V. 181 (10). P. 7002. https://doi.org/10.4049/jimmunol.181.10.7002
Feldinger K., Generali D., Kramer-Marek G. et al. // Oncotarget. 2014. V. 5 (16). P. 6633. https://doi.org/10.18632/oncotarget.1955
Mezyk-Kopeć R., Bzowska M., Stalińska K. et al. // Cytokine. 2009. V. 46 (3). P. 309. https://doi.org/10.1016/j.cyto.2009.03.002
Anders A., Gilbert S., Garten W. et al. // FASEB J. 2001. V. 15 (10). P. 1837. https://doi.org/10.1096/fj.01-0007fje
Moss M.L., Bomar M., Liu Q. et al. // J. Biol. Chem. 2007. V. 282 (49). P. 35712. https://doi.org/10.1074/jbc.M703231200
Vassar R. // Neuron. 2013. V. 80 (2). P. 250. https://doi.org/10.1016/j.neuron.2013.09.031
Fujii Y., Okuda D., Fujimoto Z. et al. // J. Mol. Biol. 2003. V. 332 (5). P. 1115. https://doi.org/10.1016/S0022-2836(03)00991-4
Senn H., Klaus W. // J. Mol. Biol. 1993. V. 232 (3). P. 907. https://doi.org/10.1006/jmbi.1993.1439
Seegar T.C.M., Killingsworth L.B., Saha N. et al. // Cell. 2017. V. 171 (7). P. 1638. https://doi.org/10.1016/j.cell.2017.11.014
Fahrenholz F., Gilbert S., Kojro E. et al. // Ann. N. Y. Acad. Sci. 2000. V. 920. P. 215.
Atapattu L., Saha N., Chheang C. et al. // J. Exp. Med. 2016. V. 213 (9). P. 1741. https://doi.org/10.1084/jem.20151095
Ahmed R.R., Holler C.J., Webb R.L. et al. // J. Neurochem. 2010. V. 112 (4). P. 1045. https://doi.org/10.1111/j.1471-4159.2009.06528.x
Wang Z., Xu Q., Cai F. et al. // JCI Insight. 2019. V. 4. № 1. P. e123431. https://doi.org/10.1172/jci.insight.123431
Willem M., Garratt A.N., Novak B. et al. // Science. 2006. V. 314 (5799). P. 664. https://doi.org/10.1126/science.1132341
Hu X., Hicks C.W., He W. et al. // Nat. Neurosci. 2006. V. 9 (12). P. 1520. https://doi.org/10.1038/nn1797
Hu X., He W., Luo X. et al. // Cell Rep. 2013. V. 4 (1). P. 40. https://doi.org/10.1016/j.celrep.2013.06.005
Kim D.Y., Carey B.W., Wang H. et al. // Nat. Cell. Biol. 2007. V. 9 (7). P. 755. https://doi.org/10.1038/ncb1602
Sachse C.C., Kim Y.H., Agsten M. et al. // FASEB J. 2013. V. 27 (6). P. 2458. https://doi.org/10.1096/fj.12-214056
Kuhn P.-H., Koroniak K., Hogl S. et al. // EMBO J. 2012. V. 31 (14). P. 3157. https://doi.org/10.1038/emboj.2012.173
Hemming M.L., Elias J.E., Gygi S.P., Selkoe D.J. // PloS One. 2009. V. 4 (12). P. e8477. https://doi.org/10.1371/journal.pone.0008477
Venugopal C., Demos C.M., Rao K.S.J. et al. // CNS Neurol. Disord. Drug. Targets. 2008. V. 7 (3). P. 278.
Shi X.-P., Chen E., Yin K.-C. et al. // J. Biol. Chem. 2001. V. 276 (13). P. 10366. https://doi.org/10.1074/jbc.M009200200
Yan R., Han P., Miao H. et al. // J. Biol. Chem. 2001. V. 276 (39). P. 36788. https://doi.org/10.1074/jbc.M104350200
Hong L. // Science. 2000. V. 290 (5489). P. 150. https://doi.org/10.1126/science.290.5489.150
Ostermann N., Eder J., Eidhoff U. et al. // J. Mol. Biol. 2006. V. 355 (2). P. 249. https://doi.org/10.1016/j.jmb.2005.10.027
Hong L., Tang J. // Biochemistry. 2004. V. 43 (16). P. 4689. https://doi.org/10.1021/bi0498252
Lin X., Koelsch G., Wu S. et al. // Proc. Natl. Acad. Sci. 2000. V. 97 (4). P. 1456. https://doi.org/10.1073/pnas.97.4.1456
Bittner H.J., Guixà-González R., Hildebrand P.W. // Biochim. Biophys. Acta BBA – Biomembr. 2018. V. 1860 (5). P. 1105. https://doi.org/10.1016/j.bbamem.2018.01.019
Zhang X., Li Y., Xu H., Zhang Y. // Front. Cell. Neurosci. 2014. V. 8. P. 427. https://doi.org/10.3389/fncel.2014.00427
Kopan R., Ilagan M.X.G. // Nat. Rev. Mol. Cell. Biol. 2004. V. 5 (6). P. 499. https://doi.org/10.1038/nrm1406
Haapasalo A., Kovacs D.M. // J. Alzheimers Dis. JAD. 2011. V. 25 (1). P. 3. https://doi.org/10.3233/JAD-2011-101065
Xu X. // J. Alzheimers Dis. JAD. 2009. V. 16 (2). P. 211. https://doi.org/10.3233/JAD-2009-0957
Shah S., Lee S.-F., Tabuchi K. et al. // Cell. 2005. V. 122 (3). P. 435. https://doi.org/10.1016/j.cell.2005.05.022
Shea Y.-F., Chu L.-W., Chan A.O.-K. et al. // J. Formos. Med. Assoc. 2016. V. 115 (2). P. 67. https://doi.org/10.1016/j.jfma.2015.08.004
De Strooper B. // Neuron. 2003. V. 38 (1). P. 9. https://doi.org/10.1016/s0896-6273(03)00205-8
Shah S., Lee S.-F., Tabuchi K. et al. // Cell. 2005. V. 122 (3). P. 435. https://doi.org/10.1016/j.cell.2005.05.022
Dries D.R., Yu G. // Curr. Alzheimer Res. 2008. V. 5 (2). P. 132. https://doi.org/10.2174/156720508783954695
Alattia J.-R., Matasci M., Dimitrov M. et al. // Biotechnol. Bioeng. 2013. V. 110 (7). P. 1995. https://doi.org/10.1002/bit.24851
Sun L., Zhao L., Yang G. et al. // Proc. Natl. Acad. Sci. 2015. V. 112 (19). P. 6003. https://doi.org/10.1073/pnas.1506242112
Bai X., Yan C., Yang G. et al. // Nature. 2015. V. 525 (7568). P. 212. https://doi.org/10.1038/nature14892
Sato C., Takagi S., Tomita T., Iwatsubo T. // J. Neurosci. 2008. V. 28. № 24. P. 6264. https://doi.org/10.1523/JNEUROSCI.1163-08.2008
Zhou R., Yang G., Guo X. et al. // Science. 2019. V. 363 (6428). P. eaaw0930. https://doi.org/10.1126/science.aaw0930
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография