

Микробиология, 2019, T. 88, № 2, стр. 240-244
Новый бактериофаг порядка Caudovirales, обнаруженный в метагеноме грунта в районе подземного горения угля
В. В. Кадников a, А. В. Марданов a, Д. А. Ивасенко b, Д. В. Анциферов b, А. В. Белецкий a, О. В. Карначук b, Н. В. Равин a, *
a Институт биоинженерии, ФИЦ Биотехнологии РАН
119071 Москва, Россия
b Лаборатория биохимии и молекулярной биологии,
Томский государственный университет
634050 Томск, Россия
* E-mail: nravin@biengi.ac.ru
Поступила в редакцию 10.12.2018
После доработки 26.12.2018
Принята к публикации 30.12.2018
Вирусы микроорганизмов, число частиц которых в биосфере оценивается в 1031, являются наиболее многочисленной формой жизни на Земле (Paez-Espino et al., 2016). За счет взаимодействия со своими хозяевами они играют важную роль в преобразовании микробной биомассы и тем самым влияют на ключевые биогеохимические циклы и состояние экосистем (Paez-Espino et al., 2016). Например, в морской воде отношение вирусных частиц к числу микроорганизмов оценивается в среднем как 10 : 1 и по некоторым оценкам ежедневно около 20% микроорганизмов погибает из-за инфекции бактериофагами (Suttle, 2007; Breitbart et al., 2018). Хотя первые работы по метагеномному анализу “некультивируемых” бактериофагов были опубликованы более 15 лет назад (Breitbart et al., 2002), развитие методов метагеномики в последние годы существенно расширилo наши знания о разнообразии бактериофагов и их распространении в природе, в первую очередь в морских экосистемах и в составе микробиома человека (Paez-Espino et al., 2016; Dutilh et al., 2017; López-Pérez et al., 2017). Тем не менее, для многих экосистем, в первую очередь с экстремальными физико-химическими условиями, разнообразие вирусов и их возможная экологическая роль исследованы мало.
Ранее с использованием метагеномных методов мы исследовали микробное сообщество приповерхностного слоя грунта в зоне подземного горения угля на территории Горного Алтая (Kadnikov et al., 2018). Явления подземного горения угольных пластов известны в различных регионах Земли, и могут продолжаться на протяжении десятков и, в отдельных случаях, тысяч лет (Stracher, Taylor, 2004). Нагрев грунта в местах горения в сочетании с доступностью газов, образующихся при термическом разложении угля, обуслoвливает формирование специфических сообществ термофильных микроорганизмов. Метагеномный анализ показал, что более 98% микробного сообщества составляли бактерии филума Firmicutes, среди которых преобладали четыре вида, – новый представитель порядка Bacillales, названный “Candidatus Carbobacillus altaicus” (47% от всех последовательностей генов 16S рРНК), Brockia lithotrophica (15%), Hydrogenibacillus schlegelii (4.9%) и новый вид рода Thermoanaerobacter (5.8%) (Kadnikov et al., 2018). Также были обнаружены Kyrpidia tusciae, Bacillus methanolicus и различные линии фирмикут, филогенетически удаленные от известных видов. Целью данной работы является идентификация бактериофагов и определение их геномных последовательностей.
Выделение ДНК из образца грунта проводили с помощью набора MO BIO Power Soil DNA Kit (MO BIO Laboratories, Qiagen Inc., Valencia, CA). Секвенирование метагеномной ДНК на Illumina HiSeq2500 (Illumina Inc., USA) описано нами ранее (Kadnikov et al., 2018). В результате секвенирования в формате одиночных чтений длиной 250 нт. были получены последовательности суммарной длиной около 2.9 млрд нт., после удаления адапторов и фильтрации по качеству (Q > 33). Сборку полученных последовательностей в контиги проводили с помощью программы metaSPAdes v. 3.7.1 (Bankevich et al., 2012), в результате чего было получено 25249 контигов длиной более 500 нт. (Kadnikov et al., 2018).
Один из полученных контигов имел среднюю кратность прочтения 1970, что существенно выше, чем средняя кратность прочтения геномов доминирующих микроорганизмов, “Candidatus Carbobacillus altaicus” (439), B. lithotrophica (236) и H. schlegelii (102). Сравнение последовательности этого контига с базой NCBI GenBank по протоколу BLASTX выявило ее сходство с геномами фагов порядка Caudovirales. При визуализации графа сборки метагенома с помощью программы Bandage (Wick et al., 2015) для данного контига был определен дополнительный короткий участок с такой же высокой кратностью прочтения, который замыкал его в кольцо. В результате была получена полная последовательность генома бактериофага, названного Altai3, имеющая длину 33 044 нт. (GenBank PRJNA399827). Высокая кратность прочтения генома фага Altai3 указываeт на то, что он не интегрирован в хромосому, а присутствует в исследованном образце грунта в форме вирусных частиц и/или активно реплицируется в клетках бактерии-хозяина. Кольцевая топология геномного контига не означает, что фаговая ДНК в вирионах имеет кольцевую форму, и может объясняться наличием длинных концевых повторов в линейной фаговой ДНК (концевая избыточность), если ее упаковка в капсиды происходит до их полного заполнения. Другим объяснением может быть присутствие значительной часть фаговой ДНК в инфицированных клетках в виде конкатемеров и/или кольцевой репликативной формы. Других контигов длиной более 5000 нт. с высоким покрытием, которые могли бы представлять геномы других литических фагов, обнаружено не было.
По результатам анализа генома фага Altai3 было идентифицировано 46 потенциальных белок-кодирующих генов (табл. 1). Поиск генов и их аннотацию проводили с использованием сервера RAST (Brettin et al., 2015) с последующей проверкой аннотации в результате сравнения последовательностей предсказанных белков с базами данных NCBI. Геном включает два противоположно направленных кластера генов, – с 1 по 5 и с 6 по 46 (рис. 1). Первый кластер включает гены, кодирующие два белка с helix-turn-helix (HTH) мотивами, которые могут обеспечивать связывание с ДНК, гомолог белка ParA системы сегрегационной стабильности бактериальных плазмид и два белка с неизвестными функциями. Структурная организация и набор генов во втором кластере сходны с таковыми у различных фагов фирмикут, в частности, у хорошо охарактеризованного фага SPP1 семейства Siphoviridae (Godinho et al., 2018), хотя заметная гомология аминокислотных последовательностей белков Altai3 и SPP1 наблюдается лишь для нескольких предсказанных белков. В начале этого кластера расположены гены белков, функции которых связаны с процессами репликации фаговой ДНК, в том числе 5'-3' эндонуклеазы (гомолог gp34.1 фага SPP1), RecT-подобной рекомбиназы (гомолог gp35), репликационных белков DnaD и DnaC/DnaI, RecU резолвазы структур Холлидея, сигма фактора РНК полимеразы и ДНК-модифицирующей метилазы. Далее следуют гены, кодирующие терминазу и структурные компоненты вириона. Первый из них, ген 19, предшествует гену большой субъединицы терминазы и кодирует 163 а.о. белок, содержащий НТН мотив. Возможно, он является малой субъединицей терминазы. Семь следующих генов кодируют вероятные компоненты капсида, четыре из которых сходны с соответствующими белками фага SPP1, – портальным белком gp6, портал-связывающим белком gp7, белком скаффолда (procapsid scaffolding protein) gp11 и основным капсидным белком gp13. Далее следует ген, кодирующий коннектор головки и хвоста и 11 генов, кодирующих компоненты хвоста фага (32−35 и 37−42). Ближайшие гомологи большинства белков капсида и хвоста фага Altai3 были найдены у фагов семейства Siphoviridae, что указывает на принадлежность Altai3 к этому семейству. Среди генов хвоста расположен также ген 36, аннотированный как фенилаланин рацемаза. Единственный гомолог этого белка у бактериофагов имеется у Streptococcus phage 315.5 (47% идентичности) и, вероятно, этот “орфанный” ген был получен в результате горизонтального переноса. Последними во втором кластере являются два гена, кодирующие белки с неизвестными функциями, и два гена, продукты которых могут обеспечивать лизис клетки, – гликозил-гидролаза семейства GH18 и N-acetylmuramoyl-L-alanine amidase. Гены, связанные с лизогенным путем развития, такие как интеграза и репрессор профага, в геноме Altai3 обнаружены не были.
Рис. 1.
Организация генома бактериофага Altai3. Гены показаны прямоугольниками со стрелками, указывающими направление их транскрипции, номера генов указаны цифрами. Прямоугольники с заливкой скошенным штрихом, – гены белков системы репликации и рекомбинации; серые прямоугольники – гены терминазы и капсидных белков; прямоугольники с точечной заливкой, – гены белков хвостового отростка; светлые прямоугольники, – гены белков с другими предсказанными функциями; черные прямоугольники, – гены гипотетических белков. Шкала координат приведена в т.п.н.
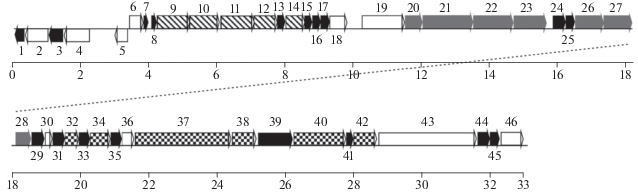
Поиск близких к фагу Altai3 нуклеотидных последовательностей в GenBank выявил, что около 87% его генома (от 4065 до 33 040 нт, гены с 8 по 46), включая почти весь ориентированный направо кластер генов, имеет 96% идентичность с профагом алкалофильной гетеротрофной бактерии Bacillus halodurans C-125 (Takami et al., 2000). В исследованном нами микробном сообществе этот вид бацилл не был обнаружен. Интересно, что этот профаг содержит также участок длиной около 3.5 т.п.н., содержащий гены интегразы и репрессора профага. У фага Altai3 этот район отсутствует и на его месте расположен первый, ориентированный справа налево, кластер генов (рис. 1) . По-видимому, именно отличие этими участками генома определяют вирулентный или умеренный характер этих фагов. Подобная мозаичная структура характерна для фаговых геномов, образование которых часто включает события горизонтального переноса не только отдельных “орфанных” генов, но и крупных функциональных модулей (Hatfull, Hendrix, 2011).
В целом, доля фага Altai3 во всем метагеноме, определяемая как доля соответствующих ему просеквенированных последовательностей от общего числа прочтений, составляет 2.3%. С учетом то, что размер генома Altai3 примерно в 100 раз меньше размеров геномов доминирующих в сообществе бактерий (1.7–3.1 млн нт.), число геномных копий фага Altai3 в микробном сообществе превышает суммарное число всех бактериальных геномов. Хотя фаговая ДНК может присутствовать не только в составе вирионов, но и в виде репликативных форм внутри клеток, эти результаты указывают на то, что число фаговых частиц в исследованной экосистеме может превышать число бактерий.
Исследованная нами экосистема грунта в зоне подземного горения угля в районе Горного Алтая является относительно молодой термальной экосистемой, образовавшейся лишь несколько лет или десятилетий назад (Kadnikov et al., 2018). По-видимому, термофильные фирмикуты, споры которых могут распространяться на большие расстояния от их исходных геотермальных местообитаний (Bonjour et al., 1988), являются первыми колонизаторами этой новой термальной экологической ниши (Kadnikov et al., 2018). Как минимум два механизма дисперсии бактериофагов могут объяснять присутствие фага Altai3 в исследованной экосистеме. Во-первых, геномы фагов могут включаться в состав эндоспор и таким образом распространяться вместе с их хозяевами (Sonenshein, 2005). Альтернативным объяснением может быть образование генома фага Altai3, предком которого мог быть умеренный фаг, подобный профагу B. halodurans C-125, в результате замещения специфичной для лизогении области генома близким по размеру фрагментом ДНК из другого фага или плазмиды, уже в новой термальной экосистеме.
Работа поддержана Российским научным фондом (грант 14-14-01016).
Таблица 1.
Гены бактериофага Altai3
| Номер гена | Начало | Конец | Цепь ДНК | Предсказанная функция | Гомолог у фага SPP1 |
|---|---|---|---|---|---|
| 1 | 331 | 14 | – | Hypothetical protein | |
| 2 | 1104 | 328 | – | ParA family protein | |
| 3 | 1403 | 1116 | – | Hypothetical protein | |
| 4 | 2319 | 1429 | – | Helix-turn-helix domain protein | |
| 5 | 3430 | 3047 | – | Xre family transcriptional regulator | |
| 6 | 3455 | 3688 | + | Helix-turn-helix domain protein, Xre family | |
| 7 | 3691 | 3891 | + | Hypothetical protein | |
| 8 | 4093 | 4293 | + | Hypothetical protein | |
| 9 | 4280 | 5248 | + | Phage-related protein, predicted 5'–3' endonuclease | 34.1 |
| 10 | 5248 | 6144 | + | Recombinational protein RecT | 35 |
| 11 | 6201 | 7106 | + | DNA replication protein DnaD | |
| 12 | 7186 | 7785 | + | DNA replication helicase loader DnaC/DnaI | |
| 13 | 7798 | 8007 | + | Hypothetical protein | |
| 14 | 8076 | 8582 | + | Holliday junction resolvase RecU | 44 |
| 15 | 8587 | 8760 | + | Hypothetical protein | |
| 16 | 8852 | 9043 | + | Hypothetical protein | |
| 17 | 9040 | 9279 | + | Hypothetical protein | |
| 18 | 9305 | 9766 | + | RNA polymerase sigma factor | |
| 19 | 10 327 | 11 490 | + | DNA modification methylase | |
| 20 | 11 507 | 11 998 | + | Helix-turn-helix domain protein | |
| 21 | 11 991 | 13 397 | + | Terminase large subunit | |
| 22 | 13 398 | 14 753 | + | Phage portal protein | 6 |
| 23 | 14 734 | 15 711 | + | Phage head morphogenesis protein | 7 |
| 24 | 15 883 | 16 206 | + | Hypothetical protein | |
| 25 | 16 196 | 16 381 | + | Hypothetical protein | |
| 26 | 16 476 | 17 249 | + | Scaffold protein, DUF4355 domain-containing | 11 |
| 27 | 17 266 | 18 255 | + | Phage capsid protein | 13 |
| 28 | 18 272 | 18 481 | + | Сapsid accessory protein | |
| 29 | 18 643 | 18 885 | + | Hypothetical protein | |
| 30 | 18 898 | 19 221 | + | Phage head-tail connector protein | |
| 31 | 19 218 | 19 526 | + | Hypothetical protein | |
| 32 | 19 523 | 19 900 | + | Phage tail component, HK97 gp10 family | |
| 33 | 19 907 | 20 317 | + | DUF5072 domain-containing protein | |
| 34 | 20 332 | 20 862 | + | Phage major tail protein, TP901-1 family | |
| 35 | 20 891 | 21 193 | + | Hypothetical protein | |
| 36 | 21 259 | 21 648 | + | Phenylalanine racemase | |
| 37 | 21 649 | 24 471 | + | Phage tail tape measure protein | |
| 38 | 24 471 | 25 217 | + | Phage tail protein | 19.1 |
| 39 | 25 231 | 26 268 | + | Hypothetical protein | |
| 40 | 26 274 | 27 752 | + | Phage endopeptidase tail | |
| 41 | 27 764 | 27 964 | + | Hypothetical protein | |
| 42 | 27 971 | 28 774 | + | Phage distal tail protein | |
| 43 | 28 771 | 31 644 | + | Glycoside hydrolase | |
| 44 | 31 697 | 31 948 | + | Hypothetical protein | |
| 45 | 31 949 | 32 197 | + | Hypothetical protein | |
| 46 | 32 199 | 33 044 | + | N-acetylmuramoyl-L-alanine amidase | 25 |
Список литературы
Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., Pyshkin A.V., Sirotkin A.V., Vyahhi N., Tesler G., Alekseyev M.A., Pevzner P.A. SPAdes: a new genome assem-bly algorithm and its applications to single-cell sequencing // J. Comput. Biol. 2012. V. 19. P. 455–477.
Bonjour F., Graber A., Aragno M. Isolation of Bacillus schlegelii, a thermophilic, hydrogen oxidizing, aerobic autotroph, from geothermal and nongeothermal environments // Microb. Ecol. 1988. V. 16. P. 331–337.
Breitbart M., Bonnain C., Malki K., Sawaya N.A. Phage puppet masters of the marine microbial realm // Nat. Microbiol. 2018. V. 3. P. 754–766.
Breitbart M., Salamon P., Andresen B., Mahaffy J.M., Segall A.M., Mead D., Azam F, Rohwer F. Genomic analysis of uncultured marine viral communities // Proc. Natl. Acad. Sci. U.S.A. 2002. V. 99. P. 14250–14255.
Brettin T., Davis J.J., Disz T., Edwards R.A., Gerdes S., Olsen G.J., Olson R., Overbeek R., Parrello B., Pusch G.D., Shukla M., Thomason J.A. 3rd, Stevens R., Vonstein V., Wattam A.R., Xia F. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes // Sci. Rep. 2015. V. 5. P. 8365.
Dutilh B.E., Reyes A., Hall R.J., Whiteson K.L. Editorial: Virus discovery by metagenomics: the (im)possibilities // Front. Microbiol. 2017. V. 8. P. 1710.
Godinho L.M., El Sadek Fadel M., Monniot C., Jakutyte L., Auzat I., Labarde A., Djacem K., Oliveira L., Carballido-Lopez R., Ayora S., Tavares P. The revisited genome of Bacillus subtilis bacteriophage SPP1 // Viruses. 2018. V. 10(12). pii E705.
Hatfull G.F., Hendrix R.W. Bacteriophages and their genomes // Curr. Opin. Virol. 2011. V. 1. P. 298–303.
Kadnikov V.V., Mardanov A.V., Ivasenko D.A., Antsiferov D.V., Beletsky A.V., Karnachuk O.V., Ravin N.V. Lignite coal burning seam in the remote Altai Mountains harbors a hydrogen-driven thermophilic microbial community // Sci. Rep. 2018. V. 8(1). P. 6730.
López-Pérez M., Haro-Moreno J.M., Gonzalez-Serrano R., Parras-Moltó M., Rodriguez-Valera F. Genome diversity of marine phages recovered from Mediterranean metagenomes: Size matters // PLoS Genet. 2017. V. 13(9). e1007018.
Paez-Espino D., Eloe-Fadrosh E.A., Pavlopoulos G.A., Thomas A.D., Huntemann M., Mikhailova N., Rubin E., Ivanova N.N., Kyrpides N.C. Uncovering Earth’s virome // Nature. 2016. V. 536(7617). P. 425–430.
Sonenshein A.L. Bacteriophages: how bacterial spores capture and protect phage DNA // Curr. Biol. 2005. V. 16. P. R14–R16.
Stracher G.B., Taylor T.P. Coal fires burning out of control around the world: thermodynamic recipe for environmental catastrophe // Int. J. Coal Geol. 2004. V. 59. P. 7–17.
Suttle C.A. Marine viruses-major players in the global ecosystem // Nat. Rev. Microbiol. 2007. V. 5(10). P. 801–812.
Takami H., Nakasone K., Takaki Y., Maeno G., Sasaki R., Masui N., Fuji F., Hirama C., Nakamura Y., Ogasawara N., Kuhara S., Horikoshi K. Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis // Nucleic Acids Res. 2000. V. 28. P. 4317–4331.
Wick R.R., Schultz M.B., Zobel J., Holt K.E. Bandage: interactive visualisation of de novo genome assemblies // Bioinformatics. 2015. V. 31. P. 3350–3352.
Дополнительные материалы отсутствуют.
Инструменты
Микробиология