

Микробиология, 2019, T. 88, № 3, стр. 336-344
Геном археи кандидатного филума Verstraetearchaeota из подземных термальных вод выявил пути метил-редуцирующего метаногенеза и ферментативного метаболизма
В. В. Кадников a, b, *, А. В. Марданов a, А. В. Белецкий a, Ю. А. Франк c, О. В. Карначук c, Н. В. Равин a, b
a Институт биоинженерии, ФИЦ Биотехнологии РАН
119071 Москва, Россия
b Биологический факультет МГУ им. М.В. Ломоносова
119192 Москва, Россия
c Лаборатория биохимии и молекулярной биологии,
Томский государственный университет
634050 Томск, Россия
* E-mail: vkadnikov@bk.ru
Поступила в редакцию 20.12.2018
После доработки 24.12.2018
Принята к публикации 26.12.2018
Аннотация
Процессы метаногенеза являются основным источником биогенного метана в атмосфере и играют важную роль в изменении климата. Все известные до последнего времени метаногены относили к филуму Euryarchaeota, но недавно потенциальные метаногены были обнаружены среди двух некультивируемых филумов архей ‒ Bathyarchaeota и Verstraetearchaeota. Анализ геномов нескольких представителей Verstraetearchaeota показал, что они используют для метаногенеза метилированные соединения, однако все эти геномы являются неполными, что не позволяет провести однозначную реконструкцию путей метаболизма. В этой работе мы определили полный геном нового представителя Verstraetearchaeota из метагенома микробного сообщества глубинного подземного резервуара термальных вод в Западной Сибири. Филогенетический анализ показал, что новая архея представляет новый вид рода ‘Candidatus Methanosuratus’, названный нами ‘Candidatus Methanosuratus subterraneum’. О возможности метил-редуцирующего метаногенеза свидетельствует наличие метил-коэнзим М редуктазного комплекса и генов, необходимых для продукции метана с использованием в качестве донора метильной группы метанола при отсутствии генов, кодирующих ферменты окисления метильной группы до СО2. Анализ генома показал, что ‘Ca. Methanosuratus subterraneum’ имеет пути метаболизма, необходимые для роста за счет ферментации белковых субстратов. Анализ глобального распространения ‘Ca. Methanosuratus’ показал, что последовательности генов 16S рРНК, отнесенные к этому роду, обнаруживают в горячих источниках, подземных водах и нефтяных резервуарах, что позволяет отнести этот род к представителям подземной биосферы.
Глубинная подземная биосфера является одной из наименее изученных экосистем на Земле, содержащей функционально-активные сообщества экстремофильных микроорганизмов, развивающихся в условиях высокого давления и температуры (Ollivier et al., 2007). Развитие микроорганизмов ограничивается лишь ростом температуры с глубиной и возможно на глубинах до 3‒5 км. Материковые подземные экосистемы содержат большую часть органического углерода на Земле и, по последним оценкам, от 12 до 20% всей биомассы микроорганизмов (Magnobosco et al., 2018). Культивирование микроорганизмов подземной биосферы является сложной задачей, поскольку в естественных условиях они, как правило, развиваются в условиях низкой доступности субстратов источников энергии, вследствие чего время генерации может достигать сотен‒тысяч лет (Lomstein et al., 2012; Braun et al., 2017). Поэтому наши знания о разнообразии микробных сообществ подземной биосферы в первую очередь основаны на результатах молекулярного анализа последовательностей генов 16S рРНК, а в последние годы также и метагеномных исследований (Schloss et al., 2016; Magnobosco et al., 2018).
В отличие от поверхностных местообитаний, для большинства из которых характерно присутствие кислорода, подземные экосистемы являются преимущественно анаэробными, вследствие чего микроорганизмы используют альтернативные акцепторы электрона. Микроорганизмы подземной биосферы могут осуществлять такие процессы как метаногенез (Kotelnikova, 2002; Nyyssönen et al., 2014), анаэробное окисление метана (Ino et al., 2018), ацетогенез (Lever, 2012), восстановление сульфата (Chivian et al., 2008), сопряженное с денитрификацией окисление серы (Lau et al., 2016), восстановление железа (Haveman et al., 1999).
В отсутствие альтернативных акцепторов электронов в большинстве исследованных глубинных подземных экосистем основным микробиологическим процессом является метаногенез. Метаногенные археи широко распространены в природе и обычно обнаруживаются в местообитаниях с ограниченным содержанием кислорода – почвах, болотах, донных осадках водоемов, желудочно-кишечном тракте жвачных животных и термитов и т.п. (Hedderich, Whitman, 2013). Субстратами для метаногенеза могут являться H2/CO2 (гидрогенотрофный), ацетат (ацетокластический) или метилированные соединения (метилотрофный метаногенез). Известные до последнего времени метаногены относились к филуму Euryarchaeota, но недавно с использованием геномных методов потенциальные метаногены были обнаружены среди двух некультивируемых филумов архей ‒ Bathyarchaeota (Miscellaneous Crenarchaeota Group) (Evans et al., 2015; Borrel et al., 2016) и Verstraetearchaeota (Terrestrial Miscellaneous Crenarchaeota Group) (Vanwonterghem et al., 2016). Представители кандидатного филума Verstraetearchaeota были обнаружены в различных анаэробных местообитаниях с высоким содержанием метана, в том числе анаэробных биореакторах, в которых осуществляется деградация органических соединений, осадках озер, термальных источниках, подземных водах (Vanwonterghem et al., 2016). Vanwonterghem et al. (2016) на геномном уровне описали 3 кандидатных вида – ‘Ca. Methanomethylicus mesodigestum’, ‘Ca. Methanomethylicus oleusabulum’ и ‘Ca. Methanosuratus petracarbonis’. Их геномы были получены, соответственно, из метагеномов анаэробного биореактора, накопительной культуры, деградирующей изо-алканы, и скважины по добыче сланцевого газа в бассейне Сурат в Австралии. Однако все эти геномы являются неполными, что не позволяет провести однозначную реконструкцию путей метаболизма этих некультивируемых архей.
Ранее мы исследовали микробные сообщества глубинных подземных вод Западно-Сибирского региона, залегающих на глубинах от 1 до 3 км в осадочных породах Мелового периода (Кадников и соавт., 2017a, 2017b), и просеквенировали метагеном подземных вод, вскрываемых скважиной 5Р в Томской области (Kadnikov et al., 2017с). Археи составляли около половины микробного сообщества и, в основном, относились к метаногенам родов Methanothermobacter и Methanosaeta, небольшую долю составляли представители Methanomassiliicoccales и Verstraetearchaeota. Это указывает на протекание in situ всех трех типов метаногенеза ‒ гидрогенотрофного, ацетокластического и метилотрофного.
Целью работы было, используя результаты метагеномного анализа, реконструировать геном нового представителя Verstraetearchaeota, охарактеризовать возможности его метаболизма и глобального распространения.
МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Характеристика скважины, отбор проб воды и выделение метагеномной ДНК. Отбор воды проводили из нефтепоисковой скважины 5Р, пробуренной в 1950-х годах до глубины 2.8 км в районе пос. Чажемто Томской области. Пробу воды объемом 20 л отобрали в апреле 2016 г. (Kadnikov et al., 2017с). Вода имела температуру 19.8‒19.9°С, нейтральный pH (7.43‒7.6) и низкий окислительно-восстановительный потенциал (от –304 до ‒338 mV). Концентрация сульфатов в воде была невысока (90.4 мг/л), а содержание растворенного сероводорода составляло около 16 мг/л (Кадников и соавт., 2017b). В газовой фазе, в основном, присутствовал метан, изотопный состав которого (δ13С/CH4 – 59.87‒60.03‰) указывает на преимущественно биогенное происхождение (Кадников и соавт., 2017b).
Для сбора биомассы микроорганизмов образец воды (объемом 20 л) пропускали через фильтры с диаметром пор 0.22 мкм. Фильтры с биомассой гомогенизировали, растирая с жидким азотом, препарат метагеномной ДНК выделяли с помощью Power Soil DNA Isolation Kit (“MO BIO Laboratories Inc.”, Carlsbad, США). Всего было выделено около 1 мкг ДНК.
Секвенирование метагеномной ДНК на платформе Illumina и сборка геномов микроорганизмов – членов сообщества. Секвенирование метагеномной ДНК на Illumina HiSeq2500 (“Illumina Inc.”, США) описано нами ранее (Kadnikov et al., 2017с). В результате секвенирования (в формате парных чтений 2 × 250 нт) были получены последовательности суммарной длиной около 16.9 млрд нт, после фильтрации по качеству (Q > 33). Сборку полученных последовательностей в контиги проводили с помощью программы metaSPAdes v. 3.7.1, в результате чего было получено 185 598 контигов длиной более 500 нт (Kadnikov et al., 2017с). Для разбиения полученных контигов на кластеры, соответствующие геномам отдельных микроорганизмов – членов сообщества, использовали программу CO-NCOСT (Alneberg et al., 2014). Полноту собранных геномов, их “загрязнение” (т.е. наличие в кластере контигов, представляющих другие микроорганизмы) и гетерогенность (наличие в кластере последовательностей из нескольких близких штаммов), оценивали с помощью программы CheckM (Parks et al., 2015). Всего в результате кластеризации метагеномных контигов было получено 59 геномов (metagenome-assembled genomes), имеющих полноту свыше 90%.
Таксономическую принадлежность собранных геномов определяли по базе Genome Taxanomy database (GTDB) с использованием программы GTDB-Tk v. 0.1.3 (Parks et al., 2018). Один из геномов, обозначенный как Ch88, был идентифицирован как представитель Verstraetearchaeota.
Нанопоровое секвенирование метагеномной ДНК и сборка генома представителя Verstraetearchaeota. Дополнительно метагеномную ДНК секвенировали с помощью системы MinION (“Oxford Nanopore”, Великобритания), используя 1D Genomic DNA by ligation протокол. В результате секвенирования было получено 1 418 419 чтений суммарной длиной около 1.54 млрд нт. Эти длинные чтения использовали для объединения контигов, составляющих геном Ch88, в более длинные последовательности. Для этого c помощью программы BWA v. 0.7.15 (Li, Durbin, 2009) отбирали полученные на MinION прочтения, имеющие высокую гомологию с последовательностями контигов Ch88. Контиги объединяли с использованием программы npScarf (Cao, 2017). Возможные ошибки в полученной консенсусной последовательности корректировали с помощью Pilon 1.22 (Walker et al., 2014) на основании наложения чтений Illumina, выполненного программой Bowtie 2 (Langmead, Salzberg, 2012). Последовательность генома бактерии Ch88 депонирована в NCBI GenBank под номером RXGA00000000.
Определение уровней сходства геномных последовательностей. Средний уровень идентичности аминокислотных последовательностей (average amino acid identity, AAI) между геномами определяли с помощью скрипта aai.rb из Enveomics Collection (Rodriguez-R, Konstantinidis, 2016). Значения ДНК‒ДНК гибридизации in silico вычисляли с помощью сервиса GGDC2 (Meier-Kolthoff et al., 2013), доступного на сайте http://ggdc.dsmz. de/, используя рекомендованную формулу 2.
Аннотация и анализ генома представителя Verstraetearchaeota. Поиск генов и их аннотацию проводили с использованием сервера RAST, с последующей проверкой аннотации в результате сравнения последовательностей предсказанных белков с базами данных NCBI. Для предсказания N-концевых сигнальных пептидов использовали программы Signal P v. 4.1 (http://www.cbs.dtu.dk/ services/SignalP/) и PRED-TAT (http://www.compgen.org/tools/PRED-TAT/), трансмембранные домены идентифицировали с помощью TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/ TMHMM/).
Филогенетический анализ. Для проведения филогенетического анализа на основе полногеномных данных была определена выборка, включающая геном Ch88, 7 других известных геномов Verstraetearchaeota (табл. 1), 2 генома представителей кандидатного филума Marsarchaeota и геном кренархеи Vulcanisaeta thermophila. Для этой выборки с помощью CheckM в каждом геноме были идентифицированы 43 консервативных однокопийных маркерных гена, и построено множественное выравнивание конкатенированных последовательностей этих маркерных генов. Полученное множественное выравнивание было использовано для построения филогенетического дерева методом Maximum Likelihood с помощью программы PhyML v. 3.3 (Guindon et al., 2010) с параметрами по умолчанию.
Таблица 1.
Основные характеристики геномов представителей Verstraetearchaeota
| Организм/геном | GenBank | Суммарная длина скаффолдов, нт | Полнота генома, % | Число скаффолдов | Медианная длина скаффолда (N50) | Белок- кодирующие гены |
|---|---|---|---|---|---|---|
| ‘Ca. Methanomethylicus mesodigestum’ V1 | MAGS00000000 | 1 428 061 | 90.3 | 88 | 34 148 | 1579 |
| ‘Ca. Methanomethylicus mesodigestum’ V2 | MAGT00000000 | 1 637 224 | 99.1 | 11 | 256 139 | 1757 |
| ‘Ca. Methanomethylicus mesodigestum’ UBA76 | DAZQ00000000 | 1 164 921 | 87.8 | 10 | 125 122 | 1264 |
| ‘Ca. Methanomethylicus oleusabulum’ V3 | MAGU00000000 | 1 374 477 | 99.1 | 86 | 24 139 | 1550 |
| ‘Ca. Methanomethylicus oleusabulum’ UBA156 | DAUC00000000 | 1 178 906 | 88.8 | 15 | 113 809 | 1292 |
| ‘Ca. Methanosuratus petracarbonis’ V4 | MAGV00000000 | 1 268 545 | 91.6 | 84 | 20 171 | 1424 |
| ‘Ca. Methanosuratus petracarbonis’ V5 | MAGW00000000 | 735 081 | 60.1 | 295 | 2926 | 1248 |
| ‘Ca. Methanosuratus subterraneum’ Ch88 | RXGA00000000 | 1 512 112 | 100 | 8 | 371 187 | 1658 |
Последовательности генов 16S рРНК выравнивали с помощью программы MUSCLE, включенной в пакет MEGA 7 (Kumar et al., 2016). Предварительно из алаймента удаляли неоднозначно выравнивающиеся участки с помощью Gblocks (Talavera, Castresana, 2007). Филогенетические деревья строили методом Maximum Likelihood с помощью MEGA 7, используя модели Tamura-Nei. Достоверность ветвлений оценивали с помощью бутстрепа по 100 итерациям.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Сборка генома представителя кандидатного филума Verstraetearchaeota. В результате секвенирования метагенома микробного сообщества подземных вод, поступающих через скважину 5Р, на платформе Illumina были получены последовательности суммарной длиной около 16.9 млрд нт, которые были собраны в 185 598 контигов длиной более 500 нт (Kadnikov et al., 2017с). Кластеризацию контигов на основе их кратности прочтения и особенностей нуклеотидных последовательностей проводили с помощью программы CONCOCT. Один из полученных геномов, обозначенный Ch88, был представлен 14 контигами с общей длиной 1.5 млн нт, со средней кратностью прочтения 19. Относительная доля этого генотипа в микробном сообществе, определяемая по доле генома Ch88 во всем метагеноме, составляла лишь 0.2%. Анализ таксономической принадлежности этого генома с помощью Genome Taxonomy database (GTDB) показал, что он относится к кандидатному филуму Verstraetearchaeota. Отметим, что в соответствии с предложением Vanwonterghem et al. (2016), Verstraetearchaeota рассматривается в качестве филума в таксономии NCBI (Federhen, 2012), а таксономия GTDB определяет эту линию как класс Methanomethylicia в филуме Crenarchaeota.
Для улучшения сборки генома Ch88 были использованы 1.4 млн длинных чтений (общим объемом около 1.5 млрд нт), полученных в результате мономолекулярного секвенирования метагенома на MinION. Это позволило объединить 14 контигов в 8, суммарная длина которых составила 1 512 112 нт. По оценке сервиса CheckM этот геном имеет полноту 100% при отсутствии возможного загрязнения и гетерогенности популяции.
В результате анализа генома Ch88 был обнаружен оперон генов 16S‒23S рРНК, отдельно расположенный ген 5S рРНК и 44 гена транспортных РНК (тРНК), кодирующих все 20 аминокислот. По результатам аннотации предсказано 1658 потенциальных белок-кодирующих генов, функции лишь 636 из которых могут быть предсказаны в результате сравнения с базами данных NCBI. Геном Ch88 содержит единственный CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) локус, включающий 27 единиц повтор‒спейсер, и набор генов, кодирующих CRI-SPR систему типа 1-А.
Размер генома и количество предсказанных генов у Ch88 близки к соответствующим показателям для других Verstraetearchaeota (табл. 1). Полученный нами геном является наиболее полным и представлен наименьшим числом контигов, по сравнению со всеми другими известными геномами Verstraetearchaeota.
Филогенетическое положение Ch88. Поиск родственных Ch88 микроорганизмов на основе сходства геномов показал, что ближайшим к нему является ‘Candidatus Methanosuratus petracarbonis’ V4 (Vanwonterghem et al., 2016) со средней идентичностью аминокислотных последовательностей (AAI) 88.7%. Геном ‘Ca. Methanosuratus petracarbonis’ V4 был получен из метагенома подземных вод бассейна Сурат в Австралии. Полнота этого генома оценивается в 91.6%; он собран в виде 84 скаффолдов и включает неполную последовательность гена 16S рРНК (около 600 нт). Значения AAI между Ch88 и двумя другими представителями Verstraetearchaeota, ‘Ca. Methanomethylicus oleusabulum’ и ‘Ca. Methanomethylicus mesodigestum’, составляли 61.1 и 60.8% соответственно. В соответствии с критериями, предложенными в работе Konstantinidis et al. (2017) для определения филогенетического положения некультивируемых микроорганизмов, Ch88 и ‘Ca. Methanosuratus petracarbonis’ являются разными видами одного рода. На принадлежность этих микроорганизмов к разным видам указывает и значение их ДНК‒ДНК гибридизации in silico, оцениваемое в 33.3‒38.2%.
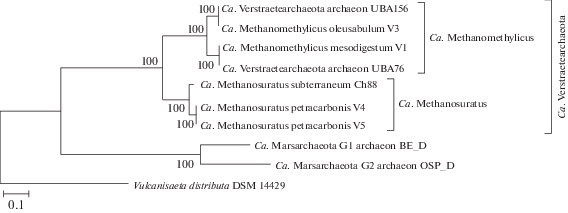
Для анализа филогении Verstraetearchaeota было построено филогенетическое дерево на основе конкатенированных последовательностей 43 консервативных маркерных генов, включающее Ch88 и 7 других известных геномов Verstraetearchaeota. Полученные результаты (рис. 1) подтверждают, что ближайшим родственником Ch88 является ‘Ca. Methanosuratus petracarbonis’, а представители двух видов рода ‘Ca. Methanomethylicus’ кластеризуются совместно и образуют сестринскую ветвь.
Рис. 1.
Положение ‘Ca. Methanosuratus subterraneum’ Ch88 на филогенетическом дереве, построенном по конкатенированным последовательностям 43 консервативных маркерных генов.
Анализ путей метаболизма археи Ch88. Ключевой особенностью метаболизма всех представителей Verstraetearchaeota, предсказанной на основе анализа их геномов, является способность к метил-редуцирующему метаногенезу (Vanwonterghem et al., 2016). В подтверждение этого в геноме Ch88 присутствуют гены метил-коэнзим М редуктазного комплекса (McrABG и вспомогательные компоненты С и А2) и гены метилтрансфераз, необходимых для продукции метана с использованием метильной группы метанола (рис. 2). Наличие генов других метилтрансфераз, которые могут переносить метильные группы на CoM-HS, указывает на возможность использования других метилированных соединений, но специфичность этих ферментов не может быть точно предсказана на основе аминокислотных последовательностей. Гены, необходимые для окисления метильной группы до CO2, у Ch88 отсутствуют, что указывает на метил-редуцирующий путь метаногенеза и неспособность диспропорционировать метанол. Восстановление гетеродисульфида (CoM-SS-CoB), образующегося в реакциях метаногенеза, до HS-CoM и HS-CoB может осуществляться комплексом гетеродисульфидредуктазы (HdrABC) и гидрогеназы (Ni–Fe) типа. Vanwonterghem et al. (2016) на основе анализа геномов Verstraetearchaeota сделали заключение, что гетеродисульфидредуктаза в этих археях действует в комплексе с цитоплазматической (Ni–Fe) гидрогеназой группы 3 (MvhADG и MvhB), окисляющей Н2 и восстанавливающей ферредоксин с бифуркацией электронов. У Ch88 ген hdrA входит в состав оперона с генами цитоплазматической гидрогеназы, а гены hdrCB кластеризуются с генами другой гидрогеназы, (Ni–Fe) гидрогеназы группы 4. Один из генов этой гидрогеназы кодирует NuoH-подобный мембранный белок, что указывает на связь этого комплекса с цитоплазматической мембраной.
Рис. 2.
Основные пути метаболимзма ‘Ca. Methanosuratus subterraneum’ Ch88. Обозначения ферментов: Atr ‒ аминотрансфераза, POR ‒ пируватферредоксиноксидоредуктаза; OOR ‒ ферредоксин-зависимые оксидоредуктазы 2-кетокислот; ACD ‒ ацетил-КоА-синтетаза (ADP-образующая); ACS ‒ ацетил-КоА-синтетаза (ацетат-КоА-лигаза); AOR ‒ альдегидферредоксиноксидоредуктаза; Mta ‒ метанол-СоМ-метилтрансфераза; Mcr ‒ метил-CoМ-редуктаза; Hdr ‒ гетеродисульфидредуктаза; Hdr/Mvh ‒ комплекс гетеродисульфидредуктазы и гидрогеназы; NDH ‒ мембранный комплекс, сходный с NADH-дегидрогеназой (без субъединиц NuoEFG); Ehb ‒ мультисубъединичная гидрогеназа Ehb-типа; Mnh ‒ натрий/протон антипортер; Hyd-4 ‒ гидрогеназа группы 4; PPase ‒ пирофосфатаза. Другие обозначения: EMP ‒ путь Эмбдена‒Мейергофа; PEP ‒ фосфоэнолпируват; Fd(ox) ‒ окисленный ферредоксин; Fd(red) ‒ восстановленный ферредоксин; Co ‒ коэнзим (А, В или М); PP ‒ пирофосфат; Р ‒ фосфат.
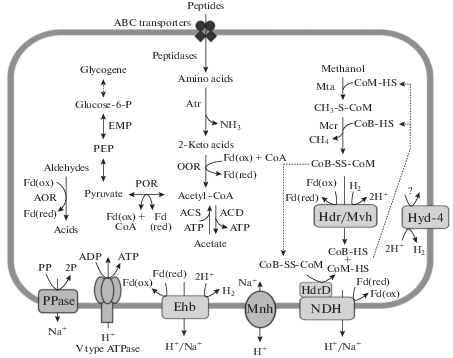
Генерация трансмембранного протонного градиента, который может использоваться АТФазой V-типа для синтеза АТФ, может осуществляться несколькими ионными помпами. Первый комплекс по структуре схож с NADH-дегидрогеназой, но отсутствие субъединиц NuoEFG, взаимодействующих с NADH, указывает, что вероятным донором электронов для него является восстановленный ферредоксин, генерируемый за счет работы комплекса Mvh-Hdr. Восстановленный ферредоксин может также окисляться мембранно-связанной мультисубъединичной (Ni–Fe) гидрогеназой Ehb-типа, создающей трансмембранный градиент H+ или Na+ и образующей H2, который затем может использоваться в качестве донора для восстановления метильной группы в метан. В формировании трансмембранного ионного градиента может также участвовать Na+-транспортирующая мембранная пирофосфатаза, а мультисубъединичный Na+/H+ антипортер обеспечивает связь градиентов протонов и натрия.
Также, как и у представителей Methanomassiliicoccales и других Verstraetearchaeota, в геноме у Ch88 кодируется субъединица HdrD гетеродисульфидредуктазы, а мембранно-связанная субъединица HdrE отсутствует. Вероятно, HdrD взаимодействует с NADH дегидрогеназа-подобным комплексом, образуя протон-транспортирующую ферредоксин : гетеродисульфид оксидоредуктазу, как это было предложено для ‘Ca. Methanoplasma termitum’ (Lang et al., 2015).
Анализ геномов Verstraetearchaeota позволил предположить, что помимо осуществления метаногенеза, эти археи могут расти и ферментативно, используя сахара в качестве субстратов и сбраживая их в пути Эмбдена‒Мейергофа (Vanwonterghem et al., 2016), хотя два ключевых фермента, энолаза и фосфоглицератмутаза, в неполном геноме ‘Ca. Methanosuratus petracarbonis’ V4 отсутствовали. Оба эти фермента обнаружены у Ch88, что указывает на возможность ферментативного метаболизма. Однако у Ch88 не обнаружены системы транспорта сахаров в клетку, а потенциальные сахаролитические ферменты не содержат N-концевые сигнальные пептиды и, вероятно, участвуют лишь во внутриклеточном метаболизме сахаров (запасание гликогена и т.п.). Как и у ‘Ca. Methanosuratus petracarbonis’ V4, у Ch88 были найдены гены, кодирующие транспортеры пептидов, а также ферменты, обеспечивающие их внутриклеточный метаболизм, заканчивающийся образованием 2-кето-кислот ‒ внутриклеточные пептидазы, глутаматдегидрогеназа и аминотрансферазы. Кетокислоты могут окисляться с образованием ацетил-КоА с помощью ферредоксин-зависимых оксидоредуктаз различной специфичности. Ацетил-КоА синтетаза (ADP-образующая) может затем окислять ацетил-КоА с образованием ацетата и генерацией АТФ, что позволяет Ch88 получать энергию за счет субстратного фосфорилирования (рис. 2).
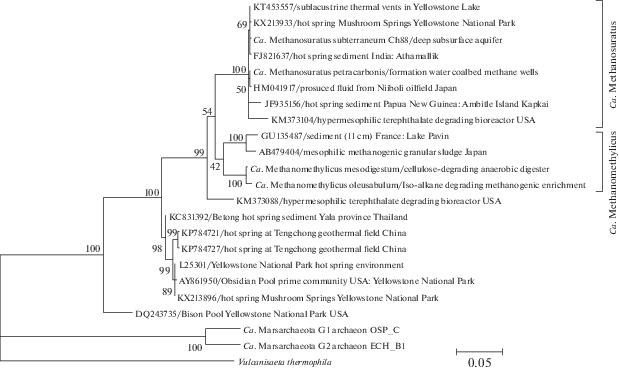
Глобальное распространение Verstraetearchaeota. В базе данных SILVA (Quast et al., 2013) имеется 261 последовательность генов 16S рРНК, относящихся к археям филума Verstraetearchaeota, но большая их часть имеет длину лишь порядка 300‒700 нт. Из 19 полноразмерных последовательностей большинство были обнаружены в горячих источниках США (Йеллоустонский парк), Таиланда, Индии, Китая и Папуа‒Новой Гвинеи (10 клонов), а также в биореакторах, осадках меромиктического озера Павин (Франция) и нефтяном месторождении в Японии. Филогенетический анализ последовательностей генов 16S рРНК показал, что разнообразие Verstraetearchaeota не ограничивается родами ‘Ca. Methanomethylicus’ и ‘Ca. Methanosuratus’, но включает еще несколько монофилетических линий уровня рода и выше (рис. 2).
Все последовательности генов 16S рРНК, отнесенные к ‘Ca. Methanosuratus’, были детектированы в экосистемах, связанных с подземными термальными местообитаниями, ‒ горячих источниках, подземных водах и нефтяных резервуарах (рис. 3), что позволяет отнести это род к представителям подземной биосферы. По-видимому, представители ‘Ca. Methanosuratus’ в этих экосистемах могут участвовать в анаэробной деградации низкомолекулярных органических соединений с образованием метана.
Рис. 3.
Филогения кандидатного филума Verstraetearchaeota, основанная на анализе последовательностей генов 16S рРНК. Указаны номера последовательностей в GenBank и местообитания, в которых они были детектированы.
Описание ‘Candidatus Methanosuratus subterraneum’ Ch88. На основе определенного полного генома мы предлагаем описать новый вид как ‘Candidatus Methanosuratus subterraneum’ Ch88.
Methanosuratus subterraneum sp. nov. (Sub.terr.a’ne.um. L. neutr. adj. subterraneum, underground, subterranean). Не культивирован. Обнаружен в подземном водоносном горизонте в Западной Сибири. Наиболее вероятно является анаэробом, способным к метаногенезу за счет метилредукции и, возможно, так же способен к ферментации белковых субстратов. Представлен геномом (GenBank RXGA00000000), полученным из метагенома подземных термальных вод скважины 5Р (п. Чажемто, Томская область, Россия).
БЛАГОДАРНОСТИ
Мы благодарим Д.Ю. Сорокина за помощь в анализе путей метаболизма.
Работа выполнена с использованием научного оборудования ЦКП “Биоинженерия”.
ФИНАНСИРОВАНИЕ РАБОТЫ
Работа выполнена при финансовой поддержке РФФИ (грант № 16-34-60124) и Министерства науки и высшего образования Российской Федерации.
Список литературы
Кадников В.В., Франк Ю.А., Марданов А.В., Белецкий А.В., Ивасенко Д.А., Пименов Н.В., Карначук О.В., Равин Н.В. Некультивируемые бактерии и метаногенные археи доминируют в микробном сообществе подземных вод Западной Сибири // Микробиология. 2017a. Т. 86. № 3. С. 383‒386.
Kadnikov V.V., Frank Y.A., Mardanov A.V., Beletskii A.V., Ivasenko D.A., Pimenov N.V., Karnachuk O.V., Ravin N.V. Uncultured bacteria and methanogenic archaea predominate in the microbial community of Western Siberian deep subsurface aquifer // Microbiology (Moscow). 2017a. V. 86. P. 412‒415.
Кадников В.В., Франк Ю.А., Марданов А.В., Белецкий А.В., Ивасенко Д.А., Пименов Н.В., Карначук О.В., Равин Н.В. Вариабельность состава микробного сообщества резервуара подземных термальных вод в Западной Сибири // Микробиология. 2017b. Т. 86. № 6. С. 739‒747.
Kadnikov V.V., Frank Y.A., Mardanov A.V., Beletsky A.V., Ivasenko D.A., Pimenov N.V., Karnachuk O.V., Ravin N.V. Variability of the composition of the microbial community of the deep subsurface thermal aquifer in Western Siberia // Microbiology (Moscow). 2017b. V. 86. P. 765‒772.
Alneberg J., Bjarnason B.S., De Bruijn I., Schirmer M., Quick J., Ijaz U.Z., Lahti L., Loman N.J., Andersson A.F., Quince C. Binning metagenomic contigs by coverage and composition // Nat. Methods. 2014. V. 11. P. 1144‒1146.
Borrel G., Adam P.S., Gribaldo S. Methanogenesis and the Wood–Ljungdahl pathway: an ancient, versatile, and fragile association // Genome Biol. Evol. 2016. V. 8. P. 1706‒1711.
Braun S., Mhatre S.S., Jaussi M., Røy H., Kjeldsen K.U., Pearce C., Seidenkrantz M.S., Jørgensen B.B., Lomstein B.A. Microbial turnover times in the deep seabed studied by amino acid racemization modelling // Sci. Rep. 2017. V. 7. Article 5680.
Cao M.D., Nguyen S.H., Ganesamoorthy D., Elliott A.G., Cooper M.A., Coin L.J. Scaffolding and completing genome assemblies in real-time with nanopore sequencing // Nat. Commun. 2017. V. 8. P. 14515.
Chivian D., Brodie E.L., Alm E.J., Culley D.E., Dehal P.S., DeSantis T.Z., Gihring T.M., Lapidus A., Lin L.-H., Lowry S.R., Moser D.P., Richardson P.M., Southam G., Wanger G., Pratt L.M., Andersen G.L., Hazen T.C., Brockman F.J., Arkin A.P., Onstott T.C. Environmental genomics reveals a single-species ecosystem deep within Earth // Science. 2008. V. 322. № 5899. P. 275‒278.
Evans P.N., Parks D.H., Chadwick G.L., Robbins S.J., Orphan V.J., Golding S.D., Tyson G.W. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics // Science. 2015. V. 350. № 6259. P. 434‒438.
Federhen S. The NCBI taxonomy database // Nucl. Acids Res. 2011. V. 40. P. D136‒D143.
Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0 // Syst. Biol. 2010. V. 59. P. 307‒321.
Haveman S.A., Pedersen K., Ruotsalainen P. Distribution and metabolic diversity of microorganisms in deep igneous rock aquifers of Finland // Geomicrobiol. J. 1999. V. 16. P. 277‒294.
Hedderich R., Whitman W.B. Physiology and biochemistry of the methane-producing archaea // The Prokaryotes. Berlin‒Heidelberg: Springer, 2013. P. 635‒662.
Ino K., Hernsdorf A.W., Konno U., Kouduka M., Yanagawa K., Kato S., Sunamura M., Hirota A., Togo Y.S., Ito K., Fukuda A., Iwatsuki T., Mizuno T., Komatsu D.D., Tsunogai U., Ishimura T., Amano Y., Thomas B.C., Banfield J.F., Suzuki Y. Ecological and genomic profiling of anaerobic methane-oxidizing archaea in a deep granitic environment // ISME J. 2017. V. 12. P. 31‒47.
Kadnikov V.V., Frank Y.A., Mardanov A.V., Beletsky A.V., Karnachuk O.V., Ravin N.V. Metagenome of the Siberian underground water reservoir // Genome Announc. 2017c. V. 5. № 47. P. e01317-17.
Konstantinidis K.T., Rosselló-Móra R., Amann R. Uncultivated microbes in need of their own taxonomy // ISME J. 2017. V. 11. P. 2399‒2406.
Kotelnikova S. Microbial production and oxidation of methane in deep subsurface // Earth-Sci. Rev. 2002. V. 58. P. 367‒395.
Kumar S., Stecher G., Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets // Mol. Biol. Evol. 2016. V. 33. P. 1870‒1874.
Lang K., Schuldes J., Klingl A., Poehlein A., Daniel R., Brunea A. New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of “Candidatus Methanoplasma termitum” // A-ppl. Environ. Microbiol. 2015. V. 81. P. 1338‒1352.
Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2 // Nat. Methods. 2012. V. 9. P. 357‒359.
Lau M.C., Kieft T.L., Kuloyo O., Linage-Alvarez B., van Heerden E., Lindsay M.R., Magnabosco C., Wang W., Wiggins J.B., Guo L., Perlman D.H., Kyin S., Shwe H.H., Harris R.L., Oh Y., Yi M.J., Purtschert R., Slater G.F., Ono S., Wei S., Li L., Lollar B.S., Onstott T.C. An oligotrophic deep-subsurface community dependent on syntrophy is dominated by sulfur-driven autotrophic denitrifiers // Proc. Natl. Acad. Sci. USA. 2016. V. 113. P. E7927‒E7936.
Lever M.A. Acetogenesis in the energy-starved deep biosphere–a paradox? // Front. Microbiol. 2012. V. 2. P. 284.
Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform // Bioinformatics. 2009. V. 25. P. 1754‒1760.
Lomstein B.A., Langerhuus A.T., D’Hondt S., Jørgensen B.B., Spivack A.J. Endospore abundance, microbial growth and necromass turnover in deep sub-seafloor sediment // Nature. 2012. V. 484. № 7392. P. 101‒104.
Magnabosco C., Lin L.H., Dong H., Bomberg M., Ghiorse W., Stan-Lotter H., Pedersen K., Kieft T.L., van Heerden E., Onstott, T.C. The biomass and biodiversity of the continental subsurface // Nat. Geosci. 2018. V. 1. P. 707‒717.
Meier-Kolthoff J.P., Auch A.F., Klenk H.-P., Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions // BMC Bioinformatics. 2013. V. 14. P. 60.
Nyyssönen M., Hultman J., Ahonen L., Kukkonen I., Paulin L., Laine P., Itävaara M., Auvinen P. Taxonomically and functionally diverse microbial communities in deep crystalline rocks of the Fennoscandian shield // ISME J. 2014. V. 8. P. 126‒138.
Ollivier B., Cayol J.-L., Fauque G. Sulphate-reducing bacteria from oil field environments and deep-sea hydrothermal vents, in Sulphate-Reducing Bacteria // Environmental and Engineered Systems / Eds. Barton L.L., Hamilton W.A. Cambridge: Cambridge University Press, 2007. P. 305–328.
Parks D.H., Chuvochina M., Waite D.W., Rinke C., Skarshewski A., Chaumeil P.A., Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life // Nat. Biotechnol. 2018. V. 36. P. 996‒1004.
Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes // Genome Res. 2015. V. 25. P. 1043‒1055.
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools // Nucl. Acids Res. 2013. V. 41. P. D590‒D596.
Rodriguez-R L.M., Konstantinidis K.T. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes // Peer J. 2016. V. 4. e1900v1.
Schloss P.D., Girard R.A., Martin T., Edwards J., Thrash J.C. Status of the archaeal and bacterial census: an update // MBio. 2016. V. 7. P. e00201–e00216.
Talavera G., Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments // Syst. Biol. 2007. V. 56. P. 564‒577.
Vanwonterghem I., Evans P.N., Parks D.H., Jensen P.D., Woodcroft B.J., Hugenholtz P., Tyson G.W. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota // Nat. Microbiol. 2016. V. 1. P. 16170.
Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement // PLoS One. 2014. V. 9. P. e112963.
Дополнительные материалы отсутствуют.