

Микробиология, 2019, T. 88, № 4, стр. 470-478
Полный геном некультивируемой бактерии кандидатного филума Bipolaricaulota
В. В. Кадников a, b, *, А. В. Марданов a, А. В. Белецкий a, Ю. А. Франк c, О. В. Карначук c, Н. В. Равин a, b
a Институт биоинженерии, ФИЦ Биотехнологии РАН
119071 Москва, Россия
b Биологический факультет МГУ им. М.В. Ломоносова
119192 Москва, Россия
c Лаборатория биохимии и молекулярной биологии, ТГУ
634050 Томск, Россия
* E-mail: vkadnikov@bk.ru
Поступила в редакцию 25.12.2018
После доработки 06.01.2019
Принята к публикации 07.01.2019
Аннотация
Бактерии кандидатного филума Bipolaricaulota, ранее известного как OP1 и Acetothermia, были идентифицированы в различных наземных и морских экосистемах в результате молекулярного анализа микробных сообществ. До настоящего времени ни один из представителей Bipolaricaulota не был выделен в чистую культуру. Мы определили полную замкнутую последовательность генома бактерии этого филума из метагенома подземного резервуара термальных вод и использовали геномные данные для анализа филогенетического положения этой бактерии и особенностей ее метаболизма. Анализ генома этой бактерии, названной Ch78, выявил различные транспортеры сахаров и пептидов и ограниченные возможности внеклеточного гидролиза полисахаридов. Реконструкция путей центрального метаболизма позволила предположить, что бактерия Ch78 является анаэробным органотрофом, способным сбраживать сахара и белковые субстраты. Известные пути аэробного и анаэробного дыхания, а также автотрофной фиксации углерода у Ch78 отсутствуют. В подземном водоносном горизонте представители Bipolaricaulota, вероятно, потребляют низкомолекулярные органические соединения, образуя водород и ацетат. На основе результатов филогенетического и геномного анализа новую бактерию предлагается классифицировать как “Candidatus Bipolaricaulis sibiricus”.
Последовательности генов 16S рибосомной РНК (рРНК) представителей кандидатного филума OP1 были впервые обнаружены в осадках горячего источника (75–95°С) Obsidian Pool в Йеллоустонском национальном парке США (Hugenholtz et al., 1998). Это один из первых 12 кандидатных филумов, описанных в результате анализа микробного разнообразия молекулярными методами. В последующие годы последовательности генов 16S рРНК, отнесенные к этому кандидатному филуму, были обнаружены в различных наземных и морских экосистемах, в том числе в глубоководных гидротермах (Teske et al., 2002; Stott et al., 2008; Kato et al., 2009), горячих источниках (Costa et al., 2009; Tobler, Benning, 2011), анаэробных биореакторах (Nobu et al., 2015) и подземных водах (Hirayama et al., 2005). Несмотря на широкое распространение в природе, в большинстве местообитаний доля представителей ОР1 в сообществах не превышала 5%. В настоящее время база данных SILVA (Quast et al., 2013) содержит более тысячи последовательностей генов 16S рРНК, отнесенных к кандидатному филуму ОР1.
Хотя на сегодняшний день в составе OP1 нет культивируемых представителей, некоторая информация о свойствах бактерий этого кандидатного филума была получена в результате секвенирования метагеномов и геномов единичных клеток. В настоящее время в Genome Taxоnomy Database (GTDB, Parks et al., 2018) доступно 24 генома представителей ОР1, но только один из них является полным. Первый неполный геном (MAG, metagenome-assembled genome) организма этой группы был получен из метагенома микробного мата в горячем источнике (Takami et al., 2012). Анализ этого генома показал, что бактерия имеет фолат-зависимый путь фиксации СО2 Вуда–Льюнгдаля и является ацетогеном. Соответственно, для этой бактерии было предложено название “Candidatus Acetothermum autotrophicum” (Takami et al., 2012), а для кандидатного филума ОР1 – Acetothermia (Rinke et al., 2013). Другой MAG (Acetothermia bacterium 64_32) был получен из метагенома осадков в морском шельфовом месторождении нефти (Hu et al., 2016). В этом геноме отсутствовали ключевые гены, кодирующие пути автотрофной фиксации углерода, что указывает на гетеротрофный образ жизни. Единственный полный (одна кольцевая хромосома) геном представителя ОР1 был получен из метагенома анаэробного биореактора по переработке органических отходов (Hao et al., 2018). Аннотация генома и реконструкция путей метаболизма указывают на анаэробный хемогетеротрофный метаболизм, при котором бактерия получает энергию и углерод посредством ферментации пептидов, аминокислот и простых сахаров до ацетата, формиата и водорода. Для новой бактерии предложено название “Candidatus Bipolaricaulis anaerobius”, отражающее необычную морфологию клеток, а филум ОР1 было предложено переименовать в “Candidatus Bipolaricaulota” из-за номенклатурных проблем с ранее использованным именем (Hao et al., 2018).
Глубинные подземные экосистемы представляют собой экстремальные местообитания, характеризующиеся высокими температурой и давлением. Микробные сообщества таких экосистем обычно включают представителей различных некультивируемых групп бактерий и архей. Для многих из них с помощью метагеномики или секвенирования геномов единичных клеток были определены частичные или полные геномы (Anantharaman et al., 2016; Magnabosco et al., 2016; Hernsdorf et al., 2017; Probst et al., 2017). Ранее мы исследовали микробные сообщества глубинных подземных вод Западно-Сибирского региона. Эти подземные термальные воды, залегающие на глубинах от 1 до 3 км в осадочных породах мелового периода, доступны для исследования благодаря большому числу пробуренных нефтепоисковых скважин, через которые вода вытекает под естественным давлением. Мы проанализировали состав микробных сообществ (Кадников и соавт., 2017a, 2017b) и секвенировали метагеном подземных вод, вскрываемых скважиной 5Р в Томской области (Kadnikov et al., 2017). В этом сообществе около половины микроорганизмов составляют метаногенные археи, а вторую половину – бактерии различных некультивируемых линий (Кадников и соавт., 2017a). В частности, анализ состава сообщества по последовательностям генов 16S рРНК показал, что в нем присутствуют бактерии филума Bipolaricaulota, и их доля составляет около 1.5%.
Целью настоящей работы было, используя результаты метагеномного анализа, определилить полный, собранный на уровне одной хромосомы, геном нового представителя Bipolaricaulota и охарактеризовать особенности его метаболизма и глобальное распространение.
МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Характеристика скважины, отбор проб воды и выделение метагеномной ДНК. Отбор воды проводили из нефтепоисковой скважины 5Р, пробуренной в 1950-х годах до глубины 2.8 км в районе пос. Чажемто Томской области. Пробу воды объемом 20 л отобрали в апреле 2016 г. (Kadnikov et al., 2017с). Вода имела температуру около 20°С, нейтральную реакцию (pH 7.43–7.6) и отрицательный окислительно-восстановительный потенциал (от –304 до –338 mV). Концентрация сульфатов в воде была невысока (90.4 мг/л; Кадников и соавт., 2017b).
Для сбора биомассы микроорганизмов образец воды (объемом 20 л) пропускали через фильтры с диаметром пор 0.22 мкм. Фильтры гомогенизировали, растирая с жидким азотом, препарат метагеномной ДНК выделяли с помощью Power Soil DNA Isolation Kit (“MO BIO Laboratories Inc.”, Carlsbad, США). Всего было выделено около 1 мкг ДНК.
Секвенирование метагеномной ДНК на платформе Illumina и сборка геномов микроорганизмов – членов сообщества. Секвенирование метагеномной ДНК на Illumina HiSeq2500 (“Illumina Inc.”, США) описано нами ранее (Kadnikov et al., 2017с). В результате секвенирования (в формате парных чтений 2 × 250 нт) с последующей фильтрацией по качеству (Q > 33) были получены последовательности суммарной ДНК длиной около 16.9 млрд нт. Сборку полученных последовательностей в контиги проводили с помощью программы metaSPAdes v. 3.7.1. Для разбиения полученных контигов на кластеры, соответствующие геномам отдельных микроорганизмов – членов сообщества (MAG), использовали программу CONCOСT (Alneberg et al., 2014). Полноту собранных MAG и их “загрязнение” (т.е. наличие в MAG контигов, представляющих другие микроорганизмы), оценивали с помощью программы CheckM (Parks et al., 2015).
Таксономическую принадлежность собранных геномов определяли по базе GTDB с использованием программы GTDB-Tk v. 0.1.3 (Parks et al., 2018). Один из геномов, обозначенный как Ch78, был идентифицирован как принадлежащий представителю Bipolaricaulota.
Секвенирование метагеномной ДНК с помощью системы MinION и сборка генома представителя Bipolaricaulota. Дополнительно метагеномную ДНК секвенировали на MinION (“Oxford Nanopore”, Великобритания), используя Ligation Sequencing kit 1D согласно рекомендациям производителя. В результате секвенирования было получено 1 418 419 чтений суммарной длиной около 1.54 млрд нт. Эти длинные чтения использовали для объединения контигов, составляющих геном Ch78, в более длинные последовательности. Для этого c помощью программы BWA v. 0.7.15 (Li, Durbin, 2009) отбирали полученные на MinION прочтения, имеющие высокую гомологию с последовательностями контигов Ch78. Контиги объединяли с использованием программы npScarf (Cao, 2017). Возможные ошибки в полученной консенсусной последовательности корректировали с помощью Pilon 1.22 (Walker et al., 2014) на основании наложения чтений Illumina, выполненного программой Bowtie 2 (Langmead, Salzberg, 2012). Последовательность генома бактерии Ch78 депонирована в NCBI GenBank под номером CP034928.
Определение уровней сходства геномных последовательностей. Средний уровень идентичности аминокислотных последовательностей (average amino acid identity, AAI) между геномами определяли с помощью скрипта aai.rb из Enveomics Collection (Rodriguez-R, Konstantinidis, 2016). Значения ДНК–ДНК гибридизации in silico вычисляли с помощью сервиса GGDC2 (Meier-Kolthoff et al., 2013), доступного на сайте http://ggdc.dsmz.de/, используя рекомендованную формулу 2.
Аннотация и анализ генома представителя Bipolaricaulota. Поиск генов и их аннотацию проводили с использованием сервера RAST, с последующей проверкой аннотации в результате сравнения последовательностей предсказанных белков с базами данных NCBI. Для предсказания N-концевых сигнальных пептидов использовали программы Signal P v. 4.1 (http://www.cbs.dtu.dk/services/SignalP/) и PRED-TAT (http://www.compgen.org/tools/PRED-TAT/), трансмембранные домены идентифицировали с помощью TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/). Программа iRep была использована для расчета индекса репликации геномной ДНК (Brown et al., 2016). Для классификации гидрогеназ использовали онлайн сервис HydDB (https://services.birc.au.dk/hyddb/) (Søndergaard et al., 2016).
Филогенетический анализ. Для проведения филогенетического анализа на основе полногеномных данных была определена выборка, включающая геном Ch78, геномы 21 других представителей Bipolaricaulota (табл. 1), геномы представителей Synergistetes, Deinococcus-Thermus, Thermotogae, кандидатных филумов Aerophobetes и Calescamantes, а также геном Aquifex aeolicus. Для этой выборки с помощью CheckM в каждом геноме были идентифицированы 43 консервативных однокопийных маркерных гена и построено множественное выравнивание конкатенированных последовательностей этих маркерных генов. Полученное множественное выравнивание было использовано для построения филогенетического дерева методом Maximum Likelihood с помощью программы PhyML v. 3.3 (Guindon et al., 2010) с параметрами по умолчанию.
Таблица 1.
Основные характеристики геномов представителей Bipolaricaulota
| Организм/геном | GenBank | Суммарная длина скаффолдов, нт | Полнота генома, % | Число скаффолдов |
|---|---|---|---|---|
| Candidatus Bipolaricaulis anaerobius Ran1 | LS483254 | 1 324 338 | 100 | 1 |
| Candidatus Bipolaricaulis sibiricus Ch78 | CP034928 | 1 701 655 | 100 | 1 |
| Acetothermia bacterium 64_32 | GCA_001508155 | 1 810 203 | 89.83 | 33 |
| Candidatus Acetothermum autotrophicum | AP011800–AP011803 | 1 970 005 | 77.97 | 4 |
| Candidatus Fraserbacteria bacterium RBG_16_55_9 | GCA_001778455 | 1 955 059 | 76.27 | 133 |
| Acetothermia bacterium UBA7950 | GCA_003535305 | 1 040 075 | 75.42 | 165 |
| Acetothermia bacterium UBA9294 | GCA_003545155 | 1 055 817 | 77.12 | 144 |
| Candidatus Acetothermia bacterium JdFR-49 | GCA_002010355 | 1 698 648 | 68.41 | 312 |
| Candidatus Acetothermia bacterium JdFR-50 | GCA_002010365 | 1 292 279 | 58.63 | 255 |
| Candidatus Acetothermia bacterium JdFR-52 | GCA_002010385 | 1 710 598 | 86.44 | 155 |
| Candidatus Acetothermia bacterium JdFR-46 | GCA_002011365 | 1 088 071 | 54.01 | 254 |
| Candidatus Acetothermia bacterium JdFR-47 | GCA_002011415 | 1 622 290 | 82.33 | 230 |
| Candidatus Acetothermia bacterium JdFR-48 | GCA_002011425 | 1 893 129 | 91.53 | 62 |
| Candidatus Acetothermia bacterium JdFR-51 | GCA_002011435 | 1 763 844 | 88.14 | 185 |
| Acetothermia bacterium UBA3574 | GCA_002375935 | 1 576 779 | 91.53 | 83 |
| Acetothermia bacterium UBA3571 | GCA_002375995 | 1 842 568 | 91.53 | 46 |
| Acetothermia bacterium UBA3565 | GCA_002376075 | 1 833 941 | 88.14 | 96 |
| Acetothermia bacterium UBA3564 | GCA_002376085 | 1 497 836 | 83.9 | 72 |
| Acetothermia bacterium UBA3563 | GCA_002376125 | 1 519 271 | 80.35 | 162 |
| Acetothermia bacterium UBA3560 | GCA_002376485 | 1 872 529 | 88.14 | 51 |
| Acetothermia bacterium UBA4801 | GCA_002402715 | 1 727 801 | 83.05 | 125 |
| Acetothermia bacterium UBA4777 | GCA_002403185 | 1 150 656 | 82.2 | 70 |
| Acetothermia bacterium UBA7521 | GCA_002479455 | 1 790 708 | 86.44 | 68 |
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Сборка генома представителя кандидатного филума Bipolaricaulota. В результате секвенирования метагенома микробного сообщества подземных вод скважины 5Р по технологии Illumina было прочтено и собрано в контиги около 16.9 млрд нт (Kadnikov et al., 2017). Кластеризацию контигов проводили с помощью программы CONCOCT. Один из полученных MAG, обозначенный Ch78, был представлен 6 контигами общей длиной 1.7 млн нт, средняя кратность их прочтения составляла 55. Относительная доля этого генотипа в микробном сообществе, определяемая по доле MAG Ch78 во всем метагеноме, составляла 0.6%. Анализ таксономической принадлежности этого генома с помощью Genome Taxonomy database показал, что он относится к кандидатному филуму Bipolaricaulota (ОР1).
Для сборки полного генома бактерии Ch78 были использованы 1.4 млн длинных чтений (общим объемом около 1.5 млрд нт), полученных в результате мономолекулярного нанопорового секвенирования. Это позволило объединить 6 контигов в одну кольцевую последовательность длиной 1 701 655 нт. По оценке сервиса CheckM этот геном имеет полноту 100% при отсутствии возможного загрязнения. Это второй известный полный геном представителя Bipolaricaulota.
В результате анализа генома Ch78 был обнаружен оперон генов 5S–23S рРНК, отдельно расположенный ген 16S рРНК, и 47 генов транспортных РНК. По результатам аннотации предсказано 1668 потенциальных белок-кодирующих генов, функции лишь 808 из которых могут быть предсказаны в результате сравнения с базами данных NCBI. Геном Ch78 содержит единственный CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) локус, включающий 50 единиц повтор–спейсер, и набор генов, кодирующих CRISPR систему типа 1-C. Размер генома и количество генов у Ch78 близки к таковым у других Bipolaricaulota (табл. 1).
Для оценки скорости репликации ДНК бактерии Ch78 in situ, в подземных водах, мы использовали программу iRep (Brown et al., 2016). Значение индекса iRep для генома Ch78 составляло 1.15, что указывает на медленный рост этих бактерий (около 15% клеток активно реплицировались во время отбора проб), но свидетельствует о том, что они представляют метаболически активную часть микробного сообщества.
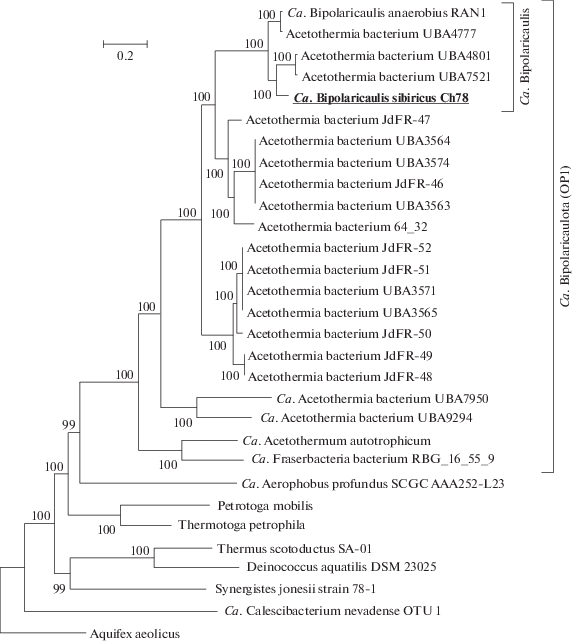
Филогенетическое положение Ch78. Поиск филогенетически родственных микроорганизмов на основе сходства геномов показал, что ближайшим родственником Ch78 является “Candidatus Bipolaricaulis anaerobius” Ran1 (Hao et al., 2018) со средней идентичностью аминокислотных последовательностей (AAI) 76.08%. Значения AAI между Ch78 и другими представителями Bipolaricaulota не превышали 60.1%. В соответствии с критериями, предложенными в работе Konstantinidis et al. (2017) для определения филогенетического положения некультивируемых микроорганизмов, Ch78 и “Ca. Bipolaricaulis anaerobius” являются разными видами одного рода. На принадлежность этих микроорганизмов к разным видам указывает и значение их ДНК–ДНК гибридизации in silico, оцениваемое в 18.4–23.0%. Уровень идентичности нуклеотидных последовательностей генов 16S рРНК Ch78 и “Ca. Bipolaricaulis anaerobius” Ran1 составляет 97%.
Для анализа филогении Bipolaricaulota было построено филогенетическое дерево на основе конкатенированных последовательностей 43 консервативных маркерных генов, включающее Ch78 и другие доступные геномы Bipolaricaulota. Полученные результаты (рис. 1) подтверждают, что ближайшим родственником Ch78 является “Ca. Bipolaricaulis anaerobius”. Ранее охарактеризованный на геномном уровне автотрофный представитель этого филума, “Ca. Acetothermum autotrophicum”, кластеризуется вместе с “Ca. Fraserbacteria bacterium” RBG_16_55_9 и образует отдельную удаленную ветвь в кандидатном филуме Bipolaricaulota.
Рис. 1.
Положение “Candidatus Bipolaricaulis sibiricus” Ch78 на филогенетическом дереве, построенном по конкатенированным последовательностям 43 консервативных маркерных генов.
Анализ путей метаболизма бактерии Ch78. Анализ генома Ch78 выявил гены транспортеров АВС типа, отвечающих за импорт сахаров, аминокислот и пептидов в клетку. Поиск генов гликозилгидролаз, содержащих характерные для секретируемых белков N-концевые сигнальные последовательности, выявил лишь две альфа-амилазы, которые могут участвовать во внеклеточном гидролизе крахмала и его производных. У Ch78 также имеются транспортеры АВС-типа для импорта мальтозы/мальтодекстрина и внутриклеточные ферменты для утилизации этих углеводов (мальтoдекстринглюкозидаза, альфа-амилаза, амиломальтаза и альфа-глюкозидаза). Гены других гидролитических ферментов, участвующих в гидролизе полисахаридов, в геноме не обнаружены. Импортированные в клетку сахара могут метаболизироваться в пути гликолиза Эмбдена–Мейергофа (рис. 2). При анализе генома были выявлены гены всех ферментов гликолиза, глюконеогенеза и неокислительной части пентозофосфатного пути. В качестве запасного полисахарида Ch78 может использовать гликоген, пути синтеза и расщепления которого кодируются в геноме. Пируват, образованный в результате гликолиза, может быть конвертирован в ацетил-КоА с помощью пируват-ферредоксиноксидоредуктазы. Окисление ацетил-КоА с образованием ацетата и генерацией АТФ может осуществляться AДФ-образующей ацетил-КоА синтетазой. Цикл трикарбоновых кислот незамкнут (отсутствует сукцинатдегидрогеназа) и, вероятно, используется только для биосинтетических целей.
Рис. 2.
Основные пути метаболизма “Candidatus Bipolaricaulis sibiricus” Ch78. Обозначения ферментов: Atr – аминотрансферазы; POR – пируват ферредоксин оксидоредуктаза; OOR – ферредоксин-зависимые оксидоредуктазы 2-кетокислот; ACD – ацетил-КоА синтетаза (ADP-образующая); Hdr/Mvh – комплекс гетеродисульфид редуктазы и гидрогеназы группы 3с; Mbh – гидрогеназа группы 4d; Hox – гидрогеназа группы 3d; PPase – пирофосфатаза. Другие обозначения: EMP – путь Эмбдена–Мейергофа; PEP – фосфоэнолпируват; Fd(ox) – окисленный ферредоксин; Fd(red) – восстановленный ферредоксин; CoA – коэнзим А; PP – пирофосфат; Р – фосфат.
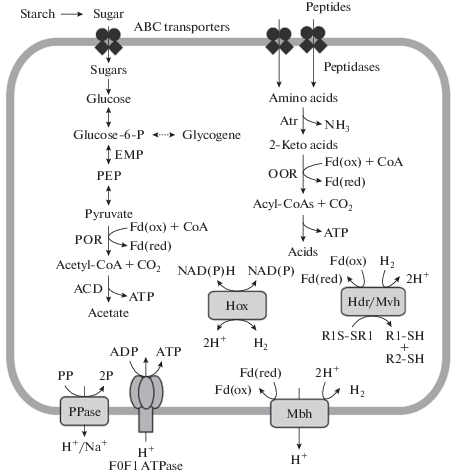
Источником углерода и энергии могут быть также аминокислоты и пептиды, импорт которых к клетку может обеспечиваться транспортерами АВС-типа, специфичными в отношении аминокислот, олиго- и дипептидов. Гены, кодирующие потенциально секретируемые протеолитические ферменты, не обнаружены. Импортированные или образовавшиеся в результате внутриклеточного гидролиза пептидов аминокислоты могут дезаминироваться и конвертироваться в 2-кетокислоты (пируват, 2-оксоглутарат и др.). 2-Кетокислоты могут окисляться с помощью ферредоксин-зависимых оксидоредуктаз различной специфичности до ацил-КоА производных, которые затем расщепляются с продукцией АТФ и образованием соответствующих кислот (рис. 2).
Геном первого охарактеризованного представителя ОР1, “Ca. Acetothermum autotrophicum”, кодировал полный путь фиксации СО2 Вуда–Льюнгдаля, на основании чего было сделано предположение о возможности его автотрофного метаболизма (Takami et al., 2012). В геноме Ch78 ключевые ферменты этого пути, в частности комплекс СО-дегидрогензы и ацетил-КоА синтетазы, отсутствуют. Анализ геномов других представителей ОР1 показал, что путь Вуда–Льюнгдаля имеется у ближайшего родственника “Ca. Acetothermum autotrophicum”, ”Ca. Fraserbacteria” bacterium RBG_16_55_9, и отсутствует у остальных представителей ОР1. По-видимому, метаболические возможности бактерии Ch78 ограничиваются ферментацией низкомолекулярных органических соединений, а известные пути аэробного или анаэробного дыхания отсутствуют.
О важной роли водорода в метаболизме бактерии Ch78 свидетельствует наличие у нее нескольких гидрогеназ (NiFe) класса (рис. 2). Первая из них, относящаяся к группе 3с, входит в состав локализованного в цитоплазме комплекса с гетеродисульфидредуктазой (Hdr-Mvh комплекс), который может окислять Н2, восстанавливая гетеродисульфид и ферредоксин за счет бифуркации электронов (Greening et al., 2016). Вторая гидрогеназа (Mbh), группы 4d, является мультисубъединичным комплексом, связанным с цитоплазматической мембраной. Гидрогеназы этой группы окисляют восстановленный ферредоксин и образуют молекулярный водород, при этом транспортируют ионы H+ или Na+ через мембрану, генерируя трансмембранный ионный градиент (Greening et al., 2016). Третий фермент (Hox) относится к группе 3d. В зависимости от редокс статуса клетки эти цитоплазматические гидрогеназы могут окислять NADH и восстанавливать протоны до Н2 или осуществлять обратную реакцию (Greening et al., 2016). В целом, активность гидрогеназ может не только обеспечивать повторное окисление NADH и восстановленного ферредоксина, образующихся в ходе ферментации, но также генерировать трансмембранный ионный градиент, который может использоваться мембранной АТФ-синтазой типа F0F1 для синтеза АТФ. Наряду с гидрогеназой группы 4d, в формировании трансмембранного ионного градиента может также участвовать Na+-транспортирующая мембранная пирофосфатаза. В целом, бактерия Ch78 обладает ограниченными возможностями генерации трансмембранного ионного градиента, поскольку у нее отсутствует NADH дегидрогеназа и другие ионные помпы.
Интересно отметить, что в геноме “Ca. Bipolaricaulis anaerobius” имеется (NiFe)-гидрогеназа группы 4d, способная генерировать трансмембранный ионный градиент, но отсутствует АТФ-синтаза. Гомологи АТФ-синтазы бактерии Ch78 обнаружены у многих других представителей ОР1, что указывает на ее потерю у “Ca. Bipolaricaulis anaerobius”. Сравнение участков генома, фланкирующих кластер генов АТФ-синтазы у Ch78 и “Ca. Bipolaricaulis anaerobius” выявило их сходство и делецию этого локуса у “Ca. Bipolaricaulis anaerobius”. Вероятно, у “Ca. Bipolaricaulis anaerobius” трансмембранный градиент используется исключительно для активного транспорта, а образование АТФ происходит только в реакциях субстратного фосфорилирования.
Глобальное распространение Bipolaricaulota и их экологическая роль. В базе данных SILVA (Quast et al., 2013) имеется 1054 последовательности генов 16S рРНК, относящихся к филуму Bipolaricaulota, но большая их часть имеет длину лишь порядка 300–700 нт. Из 366 практически полноразмерных последовательностей (длина более 1200 нуклеотидов) большинство были обнаружены в глубинных осадках морей и озер, горячих источниках, нефтяных резервуарах, осадках соленых озер. Все эти местообитания объединяет богатая органикой среда и анаэробные условия, благоприятные для роста органторофов-бродильщиков филума Bipolaricaulota.
Микробное сообщество подземного водоносного горизонта, в котором была обнаружена бактерия Ch78, примерно наполовину состоит из метаногенных архей, а вторую половину составляют различные “некультивируемые” линии бактерий, филогенетически удаленные от известных видов и в основном относящиеся к филумам Firmicutes, Chloroflexi, Proteobacteria, Bacteroidetes и Ignavibacteriae (Кадников и соавт., 2017a). Среди бактерий этих филумов известны организмы, способные гидролизовать сложные полимеры. Так, представители филумов Ignavibacteriae и Chloroflexi (сем. Anaerolineaceae) , встречающиеся в подземных водах Западно-Сибирского региона, могут участвовать в деградации полисахаридов и белков (Kadnikov et al., 2018). Вероятно, бактерии Bipolaricaulota в этих экосистемах играют роль “мусорщиков”, потребляя образованные гидролитическими микроорганизмами низкомолекулярные органические соединения, при этом образуя водород и ацетат, являющиеся субстратами для метаногенных архей.
Описание “Candidatus Bipolaricaulis sibiricus” Ch78. На основании проведенного анализа полного генома мы предлагаем описать новый вид как “Candidatus Bipolaricaulis sibiricus”. Bipolaricaulis sibiricus sp. nov. (si.bi’ri.cus N.L. masc. adj. sibiricus, originating from Siberia). Не культивирован. Обнаружен в подземном водоносном горизонте в Западной Сибири. Предположительно является анаэробом, способным к ферментации сахаров и белковых субстратов. Содержание Г + Ц в ДНК 67.23 мол. %. Представлен геномом (GenBank CP034928), полученным из метагенома подземных термальных вод скважины 5Р (п. Чажемто, Томская область, Россия).
Список литературы
Кадников В.В., Франк Ю.А, Марданов А.В., Белецкий А.В., Ивасенко Д.А., Пименов Н.В., Карначук О.В., Равин Н.В. Некультивируемые бактерии и метаногенные археи доминируют в микробном сообществе подземных вод Западной Сибири // Микробиология. 2017a. Т. 86. № 3. С. 383–386.
Kadnikov V.V., Frank Y.A., Mardanov A.V., Beletskii A.V., Ivasenko D.A., Pimenov N.V., Karnachuk O.V., Ravin N.V. Uncultured bacteria and methanogenic archaea predominate in the microbial community of Western Siberian deep subsurface aquifer // Microbiology (Moscow). 2017a. V. 86. P. 412–415.
Кадников В.В., Франк Ю.А, Марданов А.В., Белецкий А.В., Ивасенко Д.А., Пименов Н.В., Карначук О.В., Равин Н.В. Вариабельность состава микробного сообщества резервуара подземных термальных вод в Западной Сибири // Микробиология. 2017b. Т. 86. № 6. С. 739–747.
Kadnikov V.V., Frank Y.A., Mardanov A.V., Beletsky A.V., Ivasenko D.A., Pimenov N.V., Karnachuk O.V., Ravin N.V. Variability of the composition of the microbial community of the deep subsurface thermal aquifer in Western Siberia // Microbiology (Moscow). 2017b. V. 86. P. 765–772.
Alneberg J., Bjarnason B.S., De Bruijn I., Schirmer M., Quick J., Ijaz U.Z., Lahti L., Loman N.J., Andersson A.F., Quince C. Binning metagenomic contigs by coverage and composition // Nat. Methods. 2014. V. 11. P. 1144–1146.
Anantharaman K., Brown C.T., Hug L.A., Sharon I., Castelle C.J., Probst A.J., Thomas B.C., Singh A., Wilkins M.J., Karaoz U., Brodie E.L., Williams K.H., Hubbard S.S., Banfield J.F. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system // Nat. Commun. 2016. V. 7. P. 13219.
Brown C.T., Olm M.R., Thomas B.C., Banfield J.F. Measurement of bacterial replication rates in microbial communities // Nat. Biotechnol. 2016. V. 34. P. 1256–1263.
Cao M.D., Nguyen S.H., Ganesamoorthy D., Elliott A.G., Cooper M.A., Coin L.J. Scaffolding and completing genome assemblies in real-time with nanopore sequencing // Nat. Commun. 2017. V. 8. P. 14515.
Costa K.C., Navarro J.B., Shock E.L., Zhang C.L., Soukup D., Hedlund B.P. Microbiology and geochemistry of great boiling and mud hot springs in the United States Great Basin // Extremophiles. 2009. V. 13. P. 447–459.
Greening C., Biswas A., Carere C.R., Jackson C.J., Taylor M.C., Stott M.B., Cook G.M., Morales S.E. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival // ISME J. 2016. V. 10. P. 761–777.
Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0 // Syst. Biol. 2010. V. 59. P. 307–321.
Hao L., McIlroy S.J., Kirkegaard R.H., Karst S.M., Fernando W.E.Y., Aslan H., Meyer R.L., Albertsen M., Nielsen P.H., Dueholm M.S. Novel prosthecate bacteria from the candidate phylum Acetothermia // ISME J. 2018. V. 12. P. 2225–2237.
Hernsdorf A.W., Amano Y., Miyakawa K., Ise K., Suzuki Y., Anantharaman K., Probst A., Burstein D., Thomas B.C., Banfield J. F. Potential for microbial H2 and metal transformations associated with novel bacteria and archaea in deep terrestrial subsurface sediments // ISME J. 2017. V. 11. P. 1915–1929.
Hirayama H., Takai K., Inagaki F., Yamato Y., Suzuki M., Nealson K.H., Horikoshi K. Bacterial community shift along a subsurface geothermal water stream in a Japanese gold mine // Extremophiles. 2005. V. 9. P. 169–184.
Hu P., Tom L., Singh A., Thomas B.C., Baker B.J., Piceno Y.M., Andersen G.L., Banfield J.F. Genome-resolved metagenomic analysis reveals roles for candidate phyla and other microbial community members in biogeochemical transformations in oil reservoirs // MBio. 2016. V. 7. P. e01669-15.
Hugenholtz P., Pitulle C., Hershberger K.L., Pace N.R. Novel division level bacterial diversity in a Yellowstone hot spring // J. Bacteriol. 1998. V. 180. P. 366–376.
Kadnikov V.V., Frank Y.A., Mardanov A.V., Beletsky A.V., Karnachuk O.V., Ravin N.V. Metagenome of the Siberian underground water reservoir // Genome Announc. 2017. V. 5. P. e01317-17.
Kadnikov V.V., Mardanov A.V., Beletsky A.V., Banks D., Pimenov N.V., Frank Y.A., Karnachuk O.V., Ravin N.V. A metagenomic window into the 2-km-deep terrestrial subsurface aquifer revealed multiple pathways of organic matter decomposition // FEMS Microbiol. Ecol. 2018. V. 94. P. fiy152.
Kato S., Kobayashi C., Kakegawa T., Yamagishi A. Microbial communities in iron silica rich microbial mats at deep sea hydrothermal fields of the Southern Mariana Trough // Environ. Microbiol. 2009. V. 11. P. 2094–2111.
Konstantinidis K.T., Rosselló-Móra R., Amann R. Uncultivated microbes in need of their own taxonomy // ISME J. 2017. V. 11. P. 2399–2406.
Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2 // Nat. Methods. 2012. V. 9. P. 357–359.
Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform // Bioinformatics. 2009. V. 25. P. 1754–1760.
Magnabosco C., Ryan K., Lau M.C., Kuloyo O., Lollar B.S., Kieft T.L., van Heerden E., Onstott T.C. A metagenomic window into carbon metabolism at 3 km depth in Precambrian continental crust // ISME J. 2016. V. 10. P. 730–741.
Meier-Kolthoff J.P., Auch A.F., Klenk H.-P., Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions // BMC Bioinform. 2013. V. 14. P. 60.
Nobu M.K., Narihiro T., Rinke C., Kamagata Y., Tringe S.G., Woyke T., Liu W.T. Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor // ISME J. 2015. V. 9. P. 1710–1722.
Parks D.H., Chuvochina M., Waite D.W., Rinke C., Skarshewski A., Chaumeil P.A., Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life // Nat. Biotechnol. 2018. V. 36. P. 996–1004.
Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes // Genome Res. 2015. V. 25. P. 1043–1055.
Probst A.J., Castelle C.J., Singh A., Brown C.T., Anantharaman K., Sharon I., Hug L.A., Burstein D., Emerson J.B., Thomas B.C., Banfield J.F. Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations // Environ. Microbiol. 2017. V. 19. P. 459–474.
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools // Nucleic Acids Res. 2013. V. 41. P. D590–D596.
Rinke C., Schwientek P., Sczyrba A., Ivanova N.N., Anderson I.J., Cheng J.F., Darling A., Malfatti S., Swan B.K., Gies E.A., Dodsworth J.A., Hedlund B.P., Tsiamis G., Sievert S.M., Liu W.T., Eisen J.A., Hallam S.J., Kyrpides N.C., Stepanauskas R., Rubin E.M., Hugenholtz P., Woyke T. Insights into the phylogeny and coding potential of microbial dark matter // Nature. 2013. V. 499. P. 431–437.
Rodriguez-R L.M., Konstantinidis K.T. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes // PeerJ Preprints. 2016. V. 4. P. e1900v1.
Søndergaard D, Pedersen CN, Greening C. HydDB: A web tool for hydrogenase classification and analysis // Sci. Rep. 2016. V. 6. P. 34212.
Stott M.B., Saito J.A., Crowe M.A., Dunfield P.F., Hou S., Nakasone E., Daughney C.J., Smirnova A.V., Mountain B.W., Takai K., Alam M. Culture-independent characterization of a novel microbial community at a hydrothermal vent at Brothers volcano, Kermadec arc, New Zealand // J. Geophys. Res. 2008. V. 113. P. B08S06.
Takami H., Noguchi H., Takaki Y., Uchiyama I., Toyoda A., Nishi S., Chee G.J., Arai W., Nunoura T., Itoh T., Hattori M., Takai K. A deeply branching thermophilic bacterium with an ancient acetyl-CoA pathway dominates a subsurface ecosystem // PLoS One. 2012. V. 7. e30559.
Teske A., Hinrichs K.U., Edgcomb V., de Vera Gomez A., Kysela D., Sylva S.P., Sogin M.L., Jannasch H.W. Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities // Appl. Environ. Microbiol. 2002. V. 68. P. 1994–2007.
Tobler D.J., Benning L.G. Bacterial diversity in five Icelandic geothermal waters: temperature and sinter growth rate effects // Extremophiles. 2011. V. 15. P. 473–485.
Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement // PLoS One. 2014. V. 9. P. e112963.
Дополнительные материалы отсутствуют.