

Микробиология, 2020, T. 89, № 4, стр. 381-389
Хитиназы, кодируемые в геномах ацидобактерий: происхождение и эволюция
Д. Г. Наумов a, *, С. Н. Дедыш a
a Институт микробиологии им. С.Н. Виноградского, ФИЦ Биотехнологии РАН
119071 Москва, Россия
* E-mail: daniil_naumoff@yahoo.com
Поступила в редакцию 09.03.2020
После доработки 27.03.2020
Принята к публикации 30.03.2020
Аннотация
Ацидобактерии – одна из широко распространенных и численно доминирующих в почвах и торфяниках групп прокариот. Функциональный потенциал этих микроорганизмов остается не вполне раскрытым из-за ограниченного числа полученных в культурах и охарактеризованных представителей. Acidisarcina polymorpha SBC82 является единственной ацидобактерией, для которой экспериментально доказана способность к использованию хитина как источника азота и углерода. Геном штамма SBC82 кодирует шесть белков из семейства GH18 гликозил-гидролаз, каждый из которых может рассматриваться как представитель самостоятельного подсемейства. В настоящей работе методами биоинформатики показано, что одно из этих подсемейств содержит обширную группу гипотетических хитиназ ацидобактерий. Такая функциональная аннотация сделана на основе близкого родства с экспериментально охарактеризованным ферментом из протеобактерии Xylella fastidiosa. Анализ филогении показал, что исследованная группа ацидобактериальных белков появилась за счет горизонтального переноса предкового гена от фирмикут. В дальнейшем ацидобактерии послужили источником для множественных горизонтальных переносов в бактерии целого ряда фил, включая протеобактерии. Анализ метагеномных последовательностей выявил широкую распространенность генов хитиназ у ацидобактерий из болот и вечной мерзлоты умеренного и субарктического климатических поясов.
Гликозил-гидролазы – широко распространенная группа ферментов, отвечающих за расщепление углеводов и их производных. В соответствии с субстратной специфичностью выделяют (McDonald et al., 2009) более двухсот вариантов их энзиматических активностей (К.Ф. 3.2.1.1–К.Ф. 3.2.1.213). На основании гомологии каталитических доменов, большинство известных гликозил-гидролаз объединены в базе данных CAZy (Lombard et al., 2014; Terrapon et al., 2017; Garron, Henrissat, 2019) в 161 семейство: GH1–GH167 (кроме GH21, GH40, GH41, GH60, GH61 и GH69). При этом в пределах одного семейства обычно бывают представлены ферменты с различающимися энзиматическими активностями, а одна и та же субстратная специфичность может быть обнаружена у белков сразу в нескольких семействах. Семейства принято разбивать на основе филогенетического анализа на более-менее монофункциональные подсемейства для более надежного предсказания биологической функции у экспериментально не охарактеризованных белков. Спектр гликозил-гидролаз у конкретного микроорганизма зависит от его таксономического положения и занимаемой им экологической ниши (Наумов, 2011). Большую роль в эволюции генов этих ферментов играют горизонтальные переносы, дупликации и элиминации генов. Для многих гликозил-гидролаз характерна многодоменная структура, разные домены одного белка часто имеют независимую эволюционную историю (Nguyen et al., 2019).
Хитиназы или 1,4-β-поли-N-ацетилглюкозаминидазы (К.Ф. 3.2.1.14) являются эндо-гликозил-гидролазами, катализирующими расщепление хитина и хитодекстринов (Talamantes et al., 2016; Nguyen et al., 2018; Oyeleye, Normi, 2018; Tamadoni Jahromi, Barzkar, 2018; Куличевская и соавт., 2019; Le, Yang, 2019). Ферменты с данной активностью отнесены в базе данных CAZy к четырем семействам: GH18, GH19, GH23 и GH48. Однако каждое из этих семейств помимо белков с хитинолитической активностью также содержит ферменты с иной субстратной специфичностью. Подавляющее большинство экспериментально охарактеризованных прокариотических хитиназ принадлежит семейству GH18. Это семейство содержит около пятисот функционально изученных белков из архей, бактерий, эукариот и вирусов (Lombard et al., 2014). Среди них есть хитиназы, лизоцимы (К.Ф. 3.2.1.17), эндо-β-N-ацетилглюкозаминидазы (К.Ф. 3.2.1.96), пептидогликан гидролазы, гидролазы Nod-фактора и ряд белков, не обладающих энзиматической активностью (конканавалин B, нарбонин, ингибитор ксиланазы и др.). Исследование филогенетических взаимоотношений между белками этого семейства позволило раскрыть их сложную эволюционную историю и показать огромную роль горизонтальных переносов генов (Karlsson, Stenlid, 2009).
Acidobacteria – это филогенетическая группа прокариот, которые широко распространены в различных местообитаниях, но сложны в культивировании и изучены сравнительно слабо (Ludwig et al., 1997; Kielak et al., 2016; Dedysh, Sinninghe Damsté, 2018). По данным молекулярных исследований, ацидобактерии являются одним из основных компонентов почвенных микробных сообществ, причем их численность наиболее высока в кислых почвах и торфяниках (Lipson, Schmidt, 2004; Janssen, 2006; Lee et al., 2008; Jones et al., 2009; Lauber et al., 2009; Serkebaeva et al., 2013; Foesel et al., 2014; Kielak et al., 2016; Belova et al., 2018; Eichorst et al., 2018). Подавляющее большинство охарактеризованных ацидобактерий – это гетеротрофные микроорганизмы с большим гликолитическим геномным потенциалом и широким арсеналом кодируемых в их геномах гидролитических ферментов (Ward et al., 2009; Naumoff, Dedysh, 2012; Rawat et al., 2012; Kielak et al., 2016; Belova et al., 2018; Oshkin et al., 2019; Eichorst et al., 2020). Известные ныне функции ацидобактерий в природных экосистемах – деструкция ряда биополимеров и участие в глобальных циклах углерода, железа и водорода (Kielak et al., 2016; Dedysh, Sinninghe Damsté, 2018; Eichorst et al., 2018), однако этот перечень сформирован на основе изучения ограниченного числа представителей группы и далеко не полон. Способность к росту на хитине упоминается в таксономических описаниях ряда ацидобактерий (Foesel et al., 2013; Huber et al., 2014; García-Fraile et al., 2015; Lladó et al., 2016), однако экспериментальные доказательства наличия хитинолитического потенциала были опубликованы только для Acidisarcina polymorpha SBC82 (Belova et al., 2018). Эта ацидобактерия была выделена из верхнего горизонта покрытой лишайниками родов Cladonia и Cetraria почвы субарктической зоны России. Согласно базе данных CAZy, геном штамма SBC82 содержит 191 ген, кодирующий белки из 51 различного семейства гликозил-гидролаз. В их числе шесть белков из семейства GH18: ACPOL_1623, ACPOL_2565, ACPOL_3555, ACPOL_3848, ACPOL_5649 и ACPOL_6017.
Анализ филогении этих белков послужил отправной точкой для детального исследования происхождения и эволюции хитиназ, кодируемых в геномах ацидобактерий, что явилось целью настоящей работы. При этом акцент был сделан на выявлении подсемейства хитиназ, которое содержало бы многочисленных ацидобактериальных представителей. Им оказалась группа близких гомологов гипотетической хитиназы ACPOL_5649 из A. polymorpha.
МЕТОДЫ ИССЛЕДОВАНИЯ
Выбор подсемейства белков в пределах семейства. Шесть закодированных в геноме Acidisarcina polymorpha SBC82 GH18-содержащих белков (согласно списку в базе данных CAZy) служили в качестве запросов (query) для скрининга базы данных NCBI. При этом были использованы полноразмерные аминокислотные последовательности, кроме случая многодоменного белка ACPOL_3555, из которого брали фрагмент, соответствующий GH18-домену (первые 450 аминокислотных остатков). В качестве запросов также служили все 432 энзиматически как-либо охарактеризованных прокариотических белка семейства GH18 (согласно CAZy, по состоянию на август 2019 г.), использовались полноразмерные аминокислотные последовательности. В обоих случаях поиск вели в базе данных GenPept (раздел “non-redundant protein sequences”) только среди белков, отнесенных к филе Acidobacteria.
Поиск гомологов белка ACPOL_5649 в NCBI. Полноразмерную аминокислотную последовательность использовали для поиска близких гомологов в базе данных GenPept (разделы “non-redundant protein sequences” и “metagenomic proteins”) с помощью программы blastp (http:// www.ncbi.nlm.nih.gov/). Для построения множественного выравнивания и дальнейшего филогенетического анализа были отобраны 189 ближайших полноразмерных гомологов (в т.ч. только один белок – CBI00159.1 – из раздела “metagenomic proteins”), при этом близкородственные белки из штаммов одного вида и видов одного рода, как правило, не использовали.
Поиск гомологов белка ACPOL_5649 в базе данных Integrated Microbial Genomes and Microbiomes (https://img.jgi.doe.gov). Полноразмерную аминокислотную последовательность использовали для поиска близких гомологов среди белков, закодированных в метагеномах из болот и вечной мерзлоты Канады и севера США (умеренный и субарктический климатические пояса). На основании значений E-value было отобрано 119 полноразмерных или почти полноразмерных аминокислотных последовательностей белков изучаемого подсемейства, для удобства им были присвоены номера ENV_01–ENV_119 (см. список в он-лайн табл. 1). Далее проводили проверку на идентичность этих белков на участке, используемом для филогенетического анализа.
Филогенетический анализ. Множественное выравнивание аминокислотных последовательностей проводили вручную в программе-редакторе BioEdit (http://www.mbio.ncsu.edu/BioEdit/bioedit.html), при этом учитывали результаты попарных выравниваний с помощью программы blastp (Наумов, 2006). Результаты множественного выравнивания (после удаления наиболее вариабельных участков последовательностей) использовали для построения филогенетических деревьев с помощью программ PROTPARS (метод максимальной экономии, Protein Sequence Parsimony method, MP) и NEIGHBOR (метод ближайших соседей, Neighbor-Joining method, NJ) из пакета PHYLIP (http:// evolution.gs.washington.edu/phylip.html). Статистическую надежность узлов оценивали с использованием бутстреп-анализа (по 1000 псевдореплик для каждого древа). Программу TreeView Win32 (http:// taxonomy.zoology.gla.ac.uk/rod/treeview.html) применяли для получения графических изображений деревьев.
РЕЗУЛЬТАТЫ
Выбор подсемейства белков для филогенетического анализа. Для выявления широко распространенной моно- или парафилетической группы наиболее вероятных кандидатов на роль хитиназы среди GH18-белков ацидобактерий проводились скрининги базы данных NCBI с использованием двух разных стратегий. В первом случае в качестве запросов были последовательно взяты все шесть GH18-белков, закодированных в геноме Acidisarcina polymorpha SBC82 (GenBank, CP030840.1). Во втором случае в качестве запросов служили все энзиматически как-либо охарактеризованные прокариотические белки семейства GH18.
Попарное сравнение аминокислотных последовательностей шести белков A. polymorpha SBC82 показало, что их гены не являются продуктами эволюционно недавних дупликаций исходного гена, т.к. максимальный уровень сходства составил лишь 35% (для пары ACPOL_1623/ACPOL_3848). Каждый из этих белков может рассматриваться как представитель самостоятельного подсемейства в составе семейства GH18 гликозил-гидролаз.
Скрининг базы данных NCBI с использованием белков из A. polymorpha SBC82 показал, что соответствующие им подсемейства существенно различаются по содержанию белков из ацидобактерий. Так скрининг с помощью белков ACPOL_1623, ACPOL_2565 и ACPOL_6017 в каждом из случаев выявлял менее 20 гомологов из ацидобактерий с порогом ≥40% идентичности аминокислотных последовательностей. Поэтому эти три подсемейства рассматривались нами как неперспективные для дальнейшего анализа.
Скрининги базы данных с помощью всех энзиматически охарактеризованных прокариотических белков семейства GH18 с дальнейшей сортировкой обнаруженных ацидобактериальных белков по минимальному значению E-value (среди результатов всех скринингов) позволили нам отобрать 19 лучших белков. Предсказание энзиматической активности для них представляется нам наиболее надежным среди всех ацидобактериальных белков изучаемого семейства. Для каждого из этих 19 белков было определено по два ближайших энзиматически охарактеризованных гомолога (на основании E-value). При этом для 17 из 19 ацидобактериальных белков этими лучшими гомологами оказались протеобактериальные белки: хитиназа из Xylella fastidiosa (GenPept, AAO29659.1) и эндо-β-N-ацетилглюкозаминидаза из Xanthomonas campestris (AAM42161.1). Данные две энзиматические активности являются очень сходными и, вероятно, не могут быть различены биоинформатическими методами. Эти два протеобактериальных белка имеют 74% идентичности аминокислотных последовательностей и существенно различаются только в лидерной области. Оба белка принадлежат подсемейству, содержащему белок ACPOL_5649 (GenPept, AXC14897.1) из A. polymorpha SBC82 и имеют с ним 47–48% идентичности.
Остальными двумя ацидобактериальными белками (из 19) оказались белки из Terriglobus saanensis SP1PR4 (GenPept, ADV82022.1) и Luteitalea pratensis DSM100886 (AMY07034.1). Первый из них в качестве ближайших энзиматически охарактеризованных гомологов имел хитиназ из фирмикута Lachnoclostridium phytofermentans (ABX42168.1) и актинобактерии Streptomyces coelicolor (CAB92596.1), а второй – хитиназ из бактероидетеса Rhodothermus marinus (AAU11838.1) и фирмикута Laceyella putida (BAO37115.1). Эти два ацидобактериальных белка принадлежат подсемействам, первое из которых не содержит белков из A. polymorpha SBC82, а второе включает в себя белок ACPOL_3848.
По итогам этой части работы нами было выбрано для проведения дальнейшего исследования подсемейство, содержащее белок ACPOL_5649 из A. polymorpha SBC82 и два энзиматически охарактеризованных представителя филы Proteobacteria.
Построение множественного выравнивания. Для определения взаимного расположения на филогенетическом древе ацидобактериальных белков исследуемого подсемейства и его экспериментально охарактеризованных протеобактериальных представителей было построено множественное выравнивание соответствующих аминокислотных последовательностей. Белки для анализа отбирали из базы данных NCBI на основании максимального сходства с ACPOL_5649 из A. polymorpha SBC82 (см. раздел “Методы исследования”).
Для выявления степени распространенности и уровня разнообразия ацидобактериальных хитиназ данного подсемейства в природных экосистемах был проведен скрининг метагеномных последовательностей в базе данных Integrated Microbial Genomes and Microbiomes (https://img.jgi.doe.gov) с помощью аминокислотной последовательности белка ACPOL_5649. Было обнаружено 119 (почти) полноразмерных белков изучаемого подсемейства. Их попарное сравнение позволило отобрать 76 неидентичных метагеномных последовательностей, которые были добавлены во множественное выравнивание.
Анализ полученного множественного выравнивания показал наличие инвариантного мотива GxxxDxE во всех последовательностях (всего было лишь четыре инвариантных остатка). При этом остаток Glu является каталитически важным, играющим роль донора протона в активном центре фермента, как было показано для хитиназы Serratia marcescens (van Aalten et al., 2000, 2001). Единственным исключением оказались две идентичные метагеномные последовательности ENV_95 и ENV_96, в которых на соответствующем участке имеется протяженная делеция длиной 45 аминокислотных остатков, что указывает на отсутствие у этого белка энзиматической активности.
Филогенетический анализ хитиназ. Построение двумя различными методами – максимальной экономии (MP) и ближайших соседей (NJ) – филогенетического древа (рис. 1) изучаемого пула из 265 белков (189 из NCBI и 76 из метагеномной базы данных) выявило шесть основных кластеров ветвей (I–VI). Четыре из них почти эксклюзивно содержали представителей только какой-то одной филы бактерий: Acidobacteria (кластер V), Chloroflexi (IV), Firmicutes (III) и Proteobacteria (VI). При этом кластеры V и VI формировали общий стабильный суперкластер, в то время как кластеры III и IV хотя и проявляли тенденцию к кокластеризации, но их объединение не имело высокой статистической поддержки. Кластер I содержал белки из бактериальных фил Bacteroidetes, Balneolaeota, Calditrichaeota, Gemmatimonadetes, Ignavibacteria, Candidatus Kapabacteria и Proteobacteria. Кластер II оказался сформированным исключительно белками бактерий из фил-кандидатов: Candidatus Azambacteria, Candidatus Colwellbacteria, Candidatus Giovannonibacteria, Candidatus Kaiserbacteria, Candidatus Nomurabacteria, Candidatus Staskawiczbacteria, Candidatus Sungbacteria, Candidatus Taylorbacteria и Candidatus Vogelbacteria, а также из “Parcubacteria group”. Все они принадлежат так называемой кладе Candidate Phyla Radiation (Brown et al., 2015), которая на основании филогеномного анализа была недавно классифицирована в качестве единой филы-кандидата Patescibacteria (Parks et al., 2018).
Рис. 1.
Схема филогенетического древа исследуемого подсемейства семейства GH18 гликозил-гидролаз, построенного методами максимальной экономии (а) и ближайших соседей (б). Статистическую надежность узлов древа оценивали с помощью бутстреп-анализа, около каждого узла указано число подтверждающих псевдореплик из 1000. Обозначены шесть кластеров (I–VI), для каждого из них внутри треугольника указана бутстреп-поддержка. Число белков в этих кластерах: 46, 19, 47, 21, 71 и 51 соответственно. Желтым цветом выделен кластер V и шесть одиночных аминокислотных последовательностей, соответствующих ацидобактериям (названия организмов подписаны). Синим цветом выделен кластер VI, содержащий белки протеобактерий (также он содержит по одному белку из фирмикут и растений, см. текст). Для каждого кластера указано число относящихся к нему неидентичных метагеномных последовательностей из базы данных Integrated Microbial Genomes and Microbiomes (список см. в он-лайн табл. 1). Строение кластера V приведено на рис. 2.
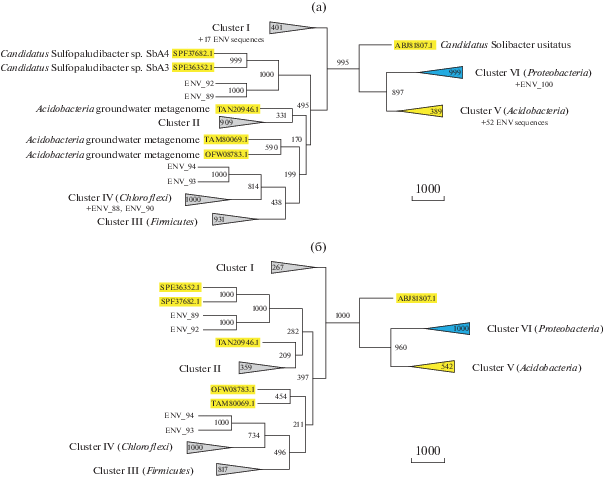
Исключительно высокая статистическая поддержка кластера VI (более 99% при использовании обоих алгоритмов) и слабая поддержка кластера V (54 и 39% на NJ- и MP-древе соответственно), а также более чем 99%-я поддержка суперкластера, сформированного дивергентным белком ацидобактерии Candidatus Solibacter usitatus Ellin6076 (GenPept, ABJ81807.1) и кластерами V и VI, указывают на имевший место горизонтальный перенос гена хитиназы от ацидобактерий к протеобактериям (рис. 1). В кластере VI оказались представлены только 11 родов протеобактерий, относящиеся к трем классам: Alphaproteobacteria (3 рода из 2 порядков), Betaproteobacteria (1 род) и Gammaproteobacteria (7 родов из 2 порядков). Такая ограниченная и мозаичная таксономическая представленность указывает на эволюционно поздний перенос от ацидобактерий, за которым последовали множественные горизонтальные переносы гена между разными группами протеобактерий. В состав кластера VI также попали два уникальных белка, один из которых проаннотирован как фирмикутный (RAN76990.1), а второй – эукариотический (XP_015870934.1). По всей видимости, аннотации этих двух белков являются ошибочными и обусловлены контаминацией образцов нуклеиновых кислот, взятых для секвенирования.
Добавление в множественное выравнивание белков архей (GenPept: RLF05149.1, RLE66198.1, RLE92050.1 и др.) и ряда близкородственных им бактериальных белков (ACB07477.1, OGF96134.1, OGY24845.1 и др.) приводило к их попаданию на древе внутрь фирмикутного кластера, однако его стабильность при этом существенно снижалась (рисунок не приводится). Это указывает на то, что кластер III может рассматриваться в качестве аутгруппы. Однако добавленная группа архейных и бактериальных белков представляет собой сильно дивергированных представителей изучаемого подсемейства, выравнивание аминокислотных последовательностей которых не является однозначным на некоторых участках. Поэтому мы исключили эту группу белков из филогенетического анализа.
Анализ распределения метагеномных последовательностей из базы данных Integrated Microbial Genomes and Microbiomes показал, что подавляющее большинство из них (52 из 76) попали в кластер V на филогенетическом древе (рис. 1, 2). Еще две последовательности (ENV_89 и ENV_92) оказались близки к двум ацидобактериальным белкам (SPE36352.1 и SPF37682.1), не вошедшим ни в один из шести основных кластеров. Одна последовательность (ENV_100) попала в кластер VI, 17 – в кластер I, две – в кластер IV и еще две образовали общий суперкластер с кластером IV.
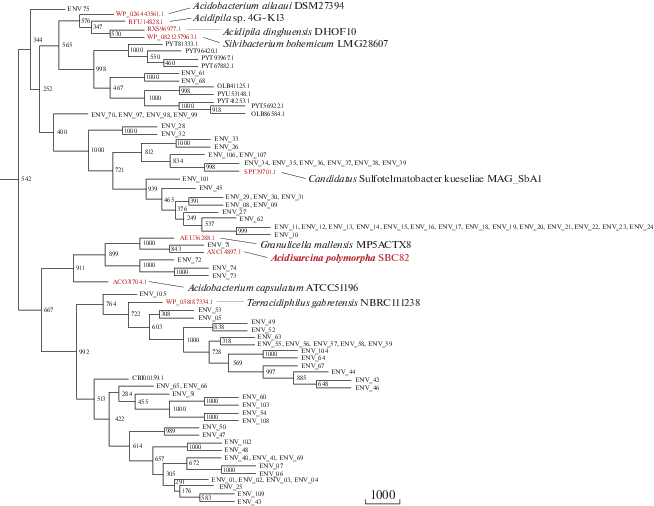
Рис. 2.
Фрагмент филогенетического древа (кластер V), схема которого представлена на рис. 1б. Подписаны видовые названия организмов, если информация о них имеется в базе данных NCBI. Для белков из метагеномов через запятую указаны номера-синонимы идентичных последовательностей.
ОБСУЖДЕНИЕ
Подавляющее большинство известных прокариотических хитиназ принадлежит семейству GH18 гликозил-гидролаз (Lombard et al., 2014). Хотя это семейство и содержит белки с рядом других функций, но хитиназы являются безусловными лидерами среди его экспериментально охарактеризованных представителей. В этом семействе нам удалось выявить подсемейство белков, которое содержит многочисленных ацидобактериальных представителей, при этом они находятся в близком эволюционном родстве с экспериментально охарактеризованной хитиназой из Xylella fastidiosa (Killiny et al., 2010). Точную субстратную специфичность исследованных белков предсказать не удалось, т.к. согласно базе данных CAZy для близкого гомолога этой хитиназы была продемонстрирована эндо-β-N-ацетилглюкозаминидазная активность. Однако, с одной стороны, эти две активности являются очень близкими, а с другой стороны, в соответствующей статье (Boulanger et al., 2010) нам не удалось найти результатов энзиматических тестов, которые исключали бы наличие хитиназной активности у фермента из Xanthomonas campestris. Поэтому мы склонны рассматривать все белки изученного подсемейства как потенциальные хитиназы.
Ферменты с хитиназными активностями широко распространены у прокариот (Lombard et al., 2014). При этом явными лидерами (в порядке убывания) по числу известных генов потенциальных хитиназ являются филы Proteobacteria, Actinobacteria, Firmicutes и Bacteroidetes (Le, Yang, 2019), что не удивительно, т.к. именно эти четыре группы бактерий имеют наибольшее число секвенированных геномов (Наумов, Дедыш, 2018). Однако исследованное нами подсемейство семейства GH18 гликозил-гидролаз имеет существенно иную распространенность среди бактериальных фил: оно совсем не представлено у актинобактерий и редко встречается у протеобактерий и бактероидетесов. При этом подавляющее число генов протеобактериальных белков этого подсемейства восходят к эволюционно позднему горизонтальному переносу от ацидобактерий (кластер VI, рис. 1).
Непропорционально широко относительно числа секвенированных геномов (Наумов, Дедыш, 2018) в изучаемом подсемействе белков представлены ацидобактерии, однако это не удивительно, т.к. обусловлено тем алгоритмом, который мы использовали для выбора этого подсемейства. Большинство его ацидобактериальных белков являются близкородственными (формируют кластер V) и принадлежат представителям порядка Acidobacteriales. Несколько более дивергентным является белок из Candidatus Solibacter usitatus Ellin6076 (GenPept, ABJ81807.1). Эта ацидобактерия принадлежит к порядку Bryobacterales и характеризуется существенно более крупным геномом (9.9 Мб). Ее гипотетическая хитиназа была использована в раннем исследовании филогении семейства GH18 (Karlsson, Stenlid, 2009) и была там отнесена (под устаревшим номером ZP_00526542) к группе бактериальных белков номер VII в составе кластера A. Это позволяет определить примерное положение всего изучаемого нами подсемейства белков относительно глобальной классификации хитиназ семейства GH18. Следует отметить, что из 12 белков группы VII девять принадлежали фирмикутам и по одному – актинобактериям, ацидобактериям и протеобактериям. При этом белок Ca. Solibacter usitatus был наиболее дивергентным представителем этой группы (см. Fig. 2 в работе Karlsson, Stenlid, 2009).
Расположение кластера VI внутри суперкластера, образованного белками ацидобактерий (кластер V и белок из Ca. Solibacter usitatus), и представленность в нем только 11 родов трех классов протеобактерий указывают на происхождение генов соответствующих протеобактериальных белков посредством горизонтального переноса предкового гена от ацидобактерий и дальнейшее его распространение между протеобактериями множественными горизонтальными переносами. Необычно эволюционно поздний перенос гена от ацидобактерий именно к протеобактериям сделал возможным довольно надежное предсказание функции обширной группы ацидобактериальных белков на основе имеющихся экспериментальных данных о субстратной специфичности протеобактериальных гомологов. Наличие пяти дополнительных ацидобактериальных белков (GenPept: OFW08783.1, SPE36352.1, SPF37682.1, TAM80069.1 и TAN20946.1), расположенных за пределами этого суперкластера, делает ацидобактерий наиболее равномерно распределенной филой бактерий на филогенетическом древе изучаемого подсемейства белков (рис. 1). Это позволяет предположить, что ацидобактерии были источником горизонтальных переносов генов хитиназ не только для протеобактерий, но и для многих других таксонов бактерий (кластеры I и II, а возможно и IV). Использование более дальних гомологов в качестве аутгруппы (данные не приводятся) показало, что корень филогенетического древа лежит в кластере III, что указывает на исходно фирмикутное происхождение всего пула белков, проанализированных в настоящей работе. Следует отметить, что шесть основных кластеров ветвей (I–VI) многократно воспроизводились при филогенетическом анализе при использовании нами разных субпулов белков (данные не приводятся), что дополнительно указывает на их стабильность.
Для проверки наличия энзиматической активности у исследованной группы белков было изучено расположение высококонсервативных аминокислотных остатков во множественном выравнивании. Среди них был обнаружен инвариантный остаток Glu, который является ключевым компонентом активного центра фермента, играющим там роль донора протона (van Aalten et al., 2000, 2001). Окружающий сайт оказался консервативным у всего подсемейства, что позволяет сделать вывод о наличии гликозидазных активностей у всех белков. Единственным исключением является последовательность ENV_95/ENV_96, которая имеет делецию в этом сайте. Соответствующий белок (если он экспрессируется), по всей видимости, лишен какой-либо энзиматической активности. На филогенетическом древе (рис. 1) он находится в кластере I и имеет 53% идентичности с белком из Gemmatimonas phototrophica (GenPept, AMW04476.1).
Анализ распределения метагеномных последовательностей из болот и вечной мерзлоты Канады и севера США на филогенетическом древе подсемейства показал, что подавляющее большинство из них попали в кластер V и соответствуют ацидобактериям (рис. 1, 2). При этом большинство из них оказались в очень близком родстве с гипотетическими хитиназами Terracidiphilus gabretensis (GenPept, WP_058187334.1) или Candidatus Sulfotelmatobacter kueseliae (SPF39701.1). Две другие последовательности оказались близки к белкам из ацидобактерии Candidatus Sulfopaludibacter (SPE36352.1 и SPF37682.1), не вошедшим ни в один из шести основных кластеров (рис. 1). В отношении остальных метагеномных последовательностей можно предполагать их принадлежность другим филам бактерий. Примечательно, что лишь одна последовательность (ENV_100) попала в протобактериальный кластер VI и еще одна (ENV_91) оказалась близка белку протеобактерии Aquicella lusitana (RDI39838.1; 42% идентичности) из кластера I. Результаты этого анализа свидетельствуют о широком распространении ацидобактерий, обладающих хитиназами исследованного нами подсемейства, в мерзлотных почвах, торфяниках и болотах севера Америки.
Представленные в настоящей работе результаты базируются на основе анализа аминокислотных последовательностей, доступных в базах данных по состоянию на конец 2019 года. Однако 12 января 2020 г. в NCBI появились 5 новых белков ацидобактерий изучаемого подсемейства (GenPept: WP_158789196.1, WP_158793234.1, WP_158820483.1, WP_158910956.1 и WP_158941762.1), принадлежащих пяти штаммам Granulicella sp. Все они принадлежат кластеру V и находятся в близком родстве с гипотетическими хитиназами Acidisarcina polymorpha (AXC14897.1) и Granulicella mallensis (AEU36288.1), имея с ними 56–85% идентичности аминокислотных последовательностей.
Он-лайн табл. 1. Метагеномные последовательности из базы данных Integrated Microbial Genomes and Microbiomes (https://img.jgi.doe.gov), использованные в работе.
Список литературы
Куличевская И.С., Наумов Д.Г., Иванова А.А., Ракитин А.Л., Дедыш С.Н. Выявление хитинолитического потенциала у пресноводного планктомицета Planctomicrobium piriforme // Микробиология. 2019. Т. 88. С. 426–437.
Kulichevskaya I.S., Naumoff D.G., Ivanova A.A., Rakitin A.L., Dedysh S.N. Detection of chitinolytic capabilities in the freshwater planctomycete Planctomicrobium piriforme // Microbiology (Moscow). 2019. V. 88. P. 423–432.
Наумов Д.Г. Иерархическая классификация гликозил-гидролаз // Биохимия. 2011. Т. 76. С. 764–780.
Naumoff D.G. Hierarchical classification of glycoside hydrolases // Biochemistry (Moscow). 2011. V. 76. P. 622–635.
Наумов Д.Г. Филогенетический анализ семейства белков-гомологов // Zbio. 2006. V. 1. Art. 3. http://zbio.net/bio/001/003.html.
Наумов Д.Г., Дедыш С.Н. Малоизученные группы бактерий – источник новых ферментов: β-галактозидазы из планктомицетов и веррукомикробий // Микробиология. 2018. Т. 87. С. 695–705.
Naumoff D.G., Dedysh S.N. Bacteria from poorly studied phyla as a potential source of new enzymes: β-galactosidases from Planctomycetes and Verrucomicrobia // Microbiology (Moscow). 2018. V. 87. P. 796–805.
Belova S.E., Ravin N.V., Pankratov T.A., Rakitin A.L., Ivanova A.A., Beletsky A.V., Mardanov A.V., Sinninghe Damsté J.S., Dedysh S.N. Hydrolytic capabilities as a key to environmental success: chitinolytic and cellulolytic Acidobacteria from acidic Sub-arctic soils and boreal peatlands // Front. Microbiol. 2018. V. 9. Art. 2775.
Boulanger A., Déjean G., Lautier M., Glories M., Zischek C., Arlat M., Lauber E. Identification and regulation of the N-acetylglucosamine utilization pathway of the plant pathogenic bacterium Xanthomonas campestris pv. campestris // J. Bacteriol. 2010. V. 192. P. 1487–1497.
Brown C.T., Hug L.A., Thomas B.C., Sharon I., Castelle C.J., Singh A., Wilkins M.J., Wrighton K.C., Williams K.H., Banfield J.F. Unusual biology across a group comprising more than 15% of domain Bacteria // Nature. 2015. V. 523. P. 208–211.
Dedysh S.N., Sinninghe Damsté J.S. Acidobacteria // eLS. Chichester, UK: John Wiley & Sons, Ltd; 2018. P. 1–10. https://doi.org/10.1002/9780470015902.a0027685
Eichorst S.A., Trojan D., Huntemann M., Clum A., Pillay M., Palaniappan K., Varghese N., Mikhailova N., Stamatis D., Reddy T.B.K., Daum C., Goodwin L.A., Shapiro N., Ivanova N., Kyrpides N., Woyke T., Woebken D. One complete and seven draft genome sequences of subdivision 1 and 3 Acidobacteria isolated from soil // Microbiol. Resour. Announc. 2020. V. 9. Art. e01087-19.
Eichorst S.A., Trojan D., Roux S., Herbold C., Rattei T., Woebken D. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments // Environ. Microbiol. 2018. V. 20. P. 1041–1063.
Foesel B.U., Nägele V., Naether A., Wüst P.K., Weinert J., Bonkowski M., Lohaus G., Polle A., Alt F., Oelmann Y., Fischer M., Friedrich M.W., Overmann J. Determinants of Acidobacteria activity inferred from the relative abundances of 16S rRNA transcripts in German grassland and forest soils // Environ. Microbiol. 2014. V. 16. P. 658–675.
Foesel B.U., Rohde M., Overmann J. Blastocatella fastidiosa gen. nov., sp. nov., isolated from semiarid savanna soil – the first described species of Acidobacteria subdivision 4 // Syst. Appl. Microbiol. 2013. V. 36. P. 82–89.
García-Fraile P., Benada O., Cajthaml T., Baldrian P., Lladó S. Terracidiphilus gabretensis gen. nov., sp. nov.: an abundant and active forest soil Acidobacteria important in organic matter transformation // Appl. Environ. Microbiol. 2015. V. 82. P. 560–569.
Garron M.L., Henrissat B. The continuing expansion of CAZymes and their families // Curr. Opin. Chem. Biol. 2019. V. 53. P. 82–87.
Huber K.J., Wust P.K., Rohde M., Overmann J., Foesel B.U. Aridibacter famidurans gen. nov., sp. nov. and Aridibacter kavangonensis sp. nov., two novel members of subdivision 4 of the Acidobacteria isolated from semiarid savannah soil // Int. J. Syst. Evol. Microbiol. 2014. V. 64. P. 1866–1875.
Janssen P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes // Appl. Environ. Microbiol. 2006. V. 72. P. 1719–1728.
Jones R.T., Robeson M.S., Lauber C.L., Hamady M., Knight R., Fierer N. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses // ISME J. 2009. V. 3. P. 442–453.
Karlsson M., Stenlid J. Evolution of family 18 glycoside hydrolases: diversity, domain structures and phylogenetic relationships // J. Mol. Microbiol. Biotechnol. 2009. V. 16. P. 208–223.
Kielak A.M., Barreto C.C., Kowalchuk G.A., van Veen J.A., Kuramae E.E. The ecology of Acidobacteria: moving beyond genes and genomes // Front. Microbiol. 2016. V. 7. Art. 744.
Killiny N., Prado S.S., Almeida R.P.P. Chitin utilization by the insect-transmitted bacterium Xylella fastidiosa // Appl. Environ. Microbiol. 2010. V. 76. P. 6134–6140.
Lauber C.L., Hamady M., Knight R., Fierer N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale // Appl. Environ. Microbiol. 2009. V. 75. P. 5111–5120.
Le B., Yang S.H. Microbial chitinases: properties, current state and biotechnological applications // World J. Microbiol. Biotechnol. 2019. V. 35. Art. 144.
Lee S.H., Ka J.O., Cho J.C. Members of the phylum Acidobacteria are dominant and metabolically active in rhizosphere soil // FEMS Microbiol. Lett. 2008. V. 285. P. 263–269.
Lipson D.A., Schmidt S.K. Seasonal changes in an alpine soil bacterial community in the Colorado Rocky Mountains // Appl. Environ. Microbiol. 2004. V. 70. P. 2867–2879.
Lladó S., Benada O., Cajthaml T., Baldrian P., García-Fraile P. Silvibacterium bohemicum gen. nov. sp. nov., an acidobacterium isolated from coniferous soil in the Bohemian Forest National Park // Syst. Appl. Microbiol. 2016. V. 39. P. 14–19.
Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013 // Nucleic Acids Res. 2014. V. 42 (Database issue). P. D490–D495.
Ludwig W., Bauer S.H., Bauer M., Held I., Kirchhof G., Schulze R., Huber I., Spring S., Hartmann A., Schleifer K.H. Detection and in situ identification of representatives of a widely distributed new bacterial phylum // FEMS Microbiol. Lett. 1997. V. 153. P. 181–190.
McDonald A.G., Boyce S., Tipton K.F. ExplorEnz: the primary source of the IUBMB enzyme list // Nucleic Acids Res. 2009. V. 37 (Database issue). P. D593–D597.
Naumoff D.G., Dedysh S.N. Lateral gene transfer between the Bacteroidetes and Acidobacteria: the case of α-L-rhamnosidases // FEBS Lett. 2012. V. 586. P. 3843–3851.
Nguyen S.N., Flores A., Talamantes D., Dar F., Valdez A., Schwans J., Berlemont R. GeneHunt for rapid domain-specific annotation of glycoside hydrolases // Sci. Rep. 2019. V. 9. Art. 10137.
Nguyen S.T.C., Freund H.L., Kasanjian J., Berlemont R. Function, distribution, and annotation of characterized cellulases, xylanases, and chitinases from CAZy // Appl. Microbiol. Biotechnol. 2018. V. 102. P. 1629–1637.
Oshkin I.Y., Kulichevskaya I.S., Rijpstra W., Sinninghe Damsté J.S., Rakitin A.L., Ravin N.V., Dedysh S.N. Granulicella sibirica sp. nov., a psychrotolerant acidobacterium isolated from an organic soil layer in forested tundra, West Siberia // Int. J. Syst. Evol. Microbiol. 2019. V. 69. P. 1195–1201.
Oyeleye A., Normi Y.M. Chitinase: diversity, limitations, and trends in engineering for suitable applications // Biosci. Rep. 2018. V. 38. Art. BSR2018032300.
Parks D.H., Chuvochina M., Waite D.W., Rinke C., Skarshewski A., Chaumeil P.A., Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life // Nat. Biotechnol. 2018. V. 36. P. 996–1004.
Rawat S.R., Männistö M.K., Bromberg Y., Häggblom M.M. Comparative genomic and physiological analysis provides insights into the role of Acidobacteria in organic carbon utilization in Arctic tundra soils // FEMS Microbiol. Ecol. 2012. V. 82. P. 341–355.
Serkebaeva Yu.M., Kim Y., Liesack W., Dedysh S.N. Pyrosequencing-based assessment of the Bacteria diversity in surface and subsurface peat layers of a northern wetland, with focus on poorly studied phyla and candidate divisions // PLoS One. 2013. V. 8. Art. e63994.
Talamantes D., Biabini N., Dang H., Abdoun K., Berlemont R. Natural diversity of cellulases, xylanases, and chitinases in bacteria // Biotechnol. Biofuels. 2016. V. 9. Art. 133.
Tamadoni Jahromi S., Barzkar N. Marine bacterial chitinase as sources of energy, eco-friendly agent, and industrial biocatalyst // Int. J. Biol. Macromol. 2018. V. 120. Pt. B. P. 2147–2154.
Terrapon N., Lombard V., Drula E., Coutinho P.M., Henrissat B. Chapter 6. The CAZy database/the Carbohydrate-Active Enzyme (CAZy) database: principles and usage guidelines // A Practical Guide to Using Glycomics Databases / Ed. Aoki-Kinoshita K.F. Tokyo: Springer. 2017. P. 117–131.
van Aalten D.M.F., Komander D., Synstad B., Gåseidnes S., Peter M.G., Eijsink V.G.H. Structural insights into the catalytic mechanism of a family 18 exo-chitinase // Proc. Natl. Acad. Sci. USA. 2001. V. 98. P. 8979–8984.
van Aalten D.M.F., Synstad B., Brurberg M.B., Hough E., Riise B.W., Eijsink V.G.H., Wierenga R.K. Structure of a two-domain chitotriosidase from Serratia marcescens at 1.9-Å resolution // Proc. Natl. Acad. Sci. USA. 2000. V. 97. P. 5842–5847.
Ward N.L., Challacombe J.F., Janssen P.H., Henrissat B., Coutinho P.M., Wu M., Haft D.H., Sait M., Badger J., Barabote R.D., Bradley B., Brettin T.S., Brinkac L.M., Bruce D., Creasy T., Daugherty S.C., Davidsen T.M., DeBoy R.T., Detter J.C., Dodson R.J., Durkin A.S., Ganapathy A., Gwinn-Giglio M., Han C.S., Khouri H., Kiss H., Kothari S.P., Madupu R, Nelson K.E., Nelson W.C., Paulsen I., Penn K., Ren Q., Rosovitz M.J., Selengut J.D., Shrivastava S., Sullivan S.A., Tapia R., Thompson L.S., Watkins K.L., Yang Q., Yu C., Zafar N., Zhou L., Kuske C.R. Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils // Appl. Environ. Microbiol. 2009. V. 75. P. 2046–2056.
Дополнительные материалы
- скачать ESM_1.docx
- Таблица S1. Метагеномные последовательности из базы данных Integrated Microbial Genomes and Microbiomes (https://img.jgi.doe.gov), использованные в работе
Инструменты
Микробиология