

Микробиология, 2021, T. 90, № 5, стр. 543-552
Метагеномный анализ микробного сообщества в районе подземного горения угля в Кемеровской области выявил доминирование термофильных бактерий филумов Deinococcus-Thermus, Aquificae и Firmicutes
В. В. Кадников a, *, А. В. Марданов a, А. В. Белецкий a, О. В. Карначук b, Н. В. Равин a
a Институт биоинженерии, ФИЦ Биотехнологии РАН
119071 Москва, Россия
b Лаборатория биохимии и молекулярной биологии, Томский государственный университет
634050 Томск, Россия
* E-mail: vkadnikov@bk.ru
Поступила в редакцию 12.05.2021
После доработки 13.05.2021
Принята к публикации 14.05.2021
Аннотация
Подземное горение угольных пластов, сопровождающееся образованием газов, приводит к формированию локальных термальных экосистем. Мы исследовали микробное сообщество нагретого до 72°С грунта в районе выхода на поверхность горячих газов, образующихся при подземном горении отходов добычи угля на угольном месторождении Бунгурский-Северный в Кемеровской области России. Анализ состава микробного сообщества по 16S рРНК показал, что в нем доминируют термофильные бактерии филумов Deinococcus-Thermus, Aquificae и Firmicutes. В результате секвенирования метагенома получено 18 геномов основных членов микробного сообщества, в том числе полные геномы Hydrogenobacter thermophiles, Thermoflexus hugenholtzii, Thermus antranikianii и представителя кандидатного рода UBA11096 филума Aquificae (RBS10-58). Анализ генома RBS10-58 указывает, что эта бактерия может автотрофно фиксировать углерод в восстановительном цикле трикарбоновых кислот и получать энергию за счет окисления водорода и соединений серы с использованием в качестве акцептора электронов кислорода или нитрата. Анализ геномов двух доминирующих представителей Firmicutes, Hydrogenibacillus schlegelii и некультивируемой линии класса Thermaerobacteria, показал, что эти бактерии могут расти аэробно, окисляя водород и монооксид углерода. В целом в сообществе преобладали аэробные бактерии, способные расти автотрофно и получать энергию за счет окисления основных компонентов угольных газов, водорода и монооксида углерода. Thermus antranikianii, составляющий около половины микробного сообщества, вероятно, использует органические вещества, образуемые автотрофными представителями Firmicutes и Aquificae.
Исследования термофильных микроорганизмов расширили наши представления о разнообразии микроорганизмов и их эволюции, механизмах адаптации к экстремальным условиям окружающей среды (Urbieta et al., 2015; Counts et al., 2017). Большинство исследований термофильных микроорганизмов было сосредоточено на термальных экосистемах, связанных с вулканической активностью, таких как наземные горячие источники и глубоководные гидротермы, или на техногенных биотопах (высокотемпературные биореакторы и т.п.). Помимо вулканической активности, к образованию локальных термальных экологических ниш могут приводить процессы естественного горения ископаемых углеводородов и угля. Характеристика микробных сообществ таких экосистем расширяет наши знания о разнообразии термофильных микроорганизмов и осуществляемых ими процессах.
Явления подземного горения угольных пластов распространены в природе и встречаются в Австралии, Германии, США, Китае, России, Индии и других странах (Stracher, Taylor, 2004). Такие подземные пожары могут длиться веками, например, угольный пласт в Дудвайлере (Саар, Германия) горит, начиная с 1668 г. Примером длительного естественного подземного горения угля является Пылающая гора (Burning Mountain) в Австралии, продолжительность горения которой оценивается примерно в 6000 лет (Rattigan, 1967).
Горение угля в условиях недостатка кислорода в присутствии воды приводит к образованию угольных газов в реакциях: 3C + O2 + H2O → H2 + + 3CO и CO + H2O → CO2 + H2 (Shafirovich, Varma, 2009). Подземное горение угля является естественным аналогом процесса получения синтез-газа при газификации угля. Помимо СО2, угольные газы содержат в основном водород, СО и газообразные углеводороды (Stracher, Taylor, 2004; Engle et al., 2012). Эти газы также могут содержать сероводород, оксиды серы, другие токсичные соединения, такие как бензол, ксилол, алифатические и галогенированные соединения (Engle et al., 2012; Zhang et al., 2013). С потоком газа могут переноситься на поверхность сера и другие элементы, присутствующие в угольных пластах (Pone et al., 2007).
В районах, где горячие угольные газы выходят на поверхность, могут образовываться локальные экстремальные экосистемы, для которых характерны высокие температуры (>50°C) и присутствие токсичных веществ (Tammy et al., 2005). Содержащиеся в угольных газах высокоэнергетические соединения, такие как водород и СО, могут использоваться микроорганизмами в качестве субстратов, а кислород – в качестве акцептора электронов, что определяет возможность развития специфических сообществ термофильных микроорганизмов. Однако сравнительно немного известно о составе микробных сообществ таких экосистем и генетическом потенциале входящих в них микроорганизмов.
Явления подземного горения угля встречаются и на угольных месторождениях в России. Кузнецкий угольный бассейн (Кузбасс), расположенный на юге Западной Сибири, является одним из крупнейших районов добычи угля в мире. В настоящее время добыча угля в этом регионе в основном ведется открытым способом, что приводит к образованию больших количеств отходов, включающих вскрышные породы. Породы, содержащие достаточное количество остаточного угля, складируют в виде отвалов непосредственно в местах добычи, где природные и техногенные причины вызывают воспламенение и длительное горение. Широко известным примером экологической катастрофы является горение отвалов угля в непосредственной близости шахтерского города Киселевск в Кузбассе (Kadnikov et al., 2021). В этой работе мы изучили микробное сообщество грунта, ассоциированное с зоной подземного горения угля и выходами горячих угольных газов, на отвалах месторождения Бунгурский-Северный в Новокузнецком районе Кемеровской области.
Целью исследования было изучение состава и генетического потенциала этого микробного сообщества. Мы приводим данные о составе сообщества, полученные с помощью высокопроизводительного секвенировании ампликонов гена 16S рРНК, и метагенома грунта в районе выхода горячих угольных газов на поверхность. В результате метагеномного анализа были получены высококачественные геномы (metagenome-assembled genomes, MAG) большинства членов сообщества, что позволило охарактеризовать метаболический потенциал входящих в него микроорганизмов.
МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Отбор проб и выделение ДНК. Пробы были отобраны в месте складирования отходов добычи угля на месторождении угля Бунгурский-Северный в районе поселка Апанас Новокузнецкого района, Кемеровской области (53.542314 N, 86.862370 E). Отобранный на глубине 5–10 см от поверхности образец грунта, обозначенный как RBS10, представлял собой влажный осадок на склоне отвала отходов добычи вблизи места выхода на поверхность горячего пара и газов. Отобранный образец представлял собой мелкодисперсную горную породу, содержащую уголь. Препарат метагеномной ДНК выделяли с использованием набора MO BIO Power Soil DNA Kit (MO BIO Laboratories, “Qiagen Inc.”, Valencia, CШA).
Секвенирование и анализ фрагментов генов 16S рРНК. ПЦР-амплификацию фрагментов гена 16S рибосомной РНК, включающих гипервариабельные области V3–V6, проводили с использованием универсальных праймеров 341F (5'-CCTAYGGGDBGCWSCAG-3') и 806R (5'-GGACTACNVGGGTHTCTAAT-3') (Frey et al., 2016). ПЦР фрагменты баркодировали, используя набор Nextera XT Index Kit v.2 (“Illumina”, СШA). Очистку ПЦР фрагментов проводили с использованием Agencourt AMPure beads (“Beckman Coulter”, Brea, CA, СШA), количество ДНК определяли с помощью Qubit dsDNA HS Assay Kit (“Invitrogen”, Carlsbad, CA, СШA). Затем ампликоны секвенировали на Illumina MiSeq (парные чтения, 2 × 300 нт). Пересекающиеся чтения объединяли с помощью FLASH v.1.2.11 (Magoc, Salzberg, 2011). Фильтрацию по качеству и кластеризацию последовательностей в оперативные таксономические единицы (ОТЕ) на уровне 97% идентичности последовательностей проводили с помощью программы Usearch (Edgar, 2010). Химерные последовательности и синглтоны удаляли при кластеризации алгоритмом Usearch. Для расчета относительной численности OTЕ все чтения (включая синглтоны и низкокачественные) с помощью Usearch были картированы на последовательности OTЕ с порогом идентичности 97%.
Таксономическую идентификацию OTЕ проводили в результате поиска по базе данных последовательностей рРНК SILVA v.132 с использованием алгоритма VSEARCH (Rognes et al., 2016).
Секвенирование метагеномной ДНК, сборка контигов и их кластеризация для получения MAG. Метагеномную ДНК секвенировали с использованием Illumina HiSeq2500 в соответствии с инструкциями производителя (“Illumina Inc.”, США). В результате секвенирования библиотеки ДНК TruSeq (парные чтения, 2 × 150 нт) было получено 232 885 794 пар чтений. Удаление адапторов и исключение низкокачественных последовательностей (Q < 30) выполнялись с использованием Cutadapt v.1.8.3 (Martin, 2011) и Sickle v.1.33 (https:// github.com/najoshi/sickle) соответственно. Обработанные парные чтения были объединены с помощью FLASH v.1.2.11 (Magoc, Salzberg, 2011).
Метагеномную ДНК дополнительно секвенировали на приборе MinION (“Oxford Nanopore”, Великобритания) с использованием набора 1D Genomic DNA by ligation kit (SQK-LSK108). В результате секвенирования этой библиотеки на MinION в проточной ячейке R9.4 (FLO-MIN106) было получено 8 2802 28 прочтений общей длиной 16.12 млрд нт.
Все полученные чтения Illumina (всего около 25 млрд нт) и Nanopore были de novo собраны в контиги с помощью metaSPAdes hybrid assembler v.3.13.0 (Nurk et al., 2017). Контиги длиной более 1500 п.н. были объединены в кластеры, представляющие MAG, с использованием MetaBAT v.2.12.1 (Kang et al., 2015). Для улучшения сборки MAG, чтения MinION были картированы на входящие в MAG контиги с помощью BWA v.0.7.15 (Li, Durbin, 2010). Затем программа Npscarf v.1.0 (Cao et al., 2017) была использована для формирования цепочек контигов (скаффолдов) и заполнения пробелов между контигами с использованием консенсусных последовательностей Illumina из графа сборки metaSPAdes.
Кроме того, чтения MinION собирали в контиги de novo с использованием Flye v. 2.7 (Kolmogorov et al., 2019). Последовательности контигов были скорректированы с помощью Pilon v.1.2.2 (Walker et al., 2014) в результате двух итераций картирования чтений Illumina на собранные последовательности контигов с использованием Bowtie 2 (Langmead, Salzberg, 2012). Полученные контиги были кластеризованы в MAG с помощью MetaBAT v.2.12.1 (Kang et al., 2015).
Аннотация и анализ геномов (MAG). Полноту MAG и их возможное загрязнение (т.е. возможное наличие в них контигов, представляющих другие геномы вследствие неправильной кластеризации) оценивали с помощью CheckM v.1.05 (Parks et al., 2015). Собранные MAG были таксономически классифицированы с использованием набора инструментов Genome Taxonomy Database Toolkit (GTDB-Tk) v.0.3.2 (Chaumeil et al., 2020) и Genome Taxonomy Database (GTDB) (Parks et al., 2018).
Поиск генов и аннотацию MAG выполняли с использованием NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al., 2016) или RAST server 2.0 (Brettin et al., 2015) с последующей корректировкой аннотации путем сравнения предсказанных последовательностей белков с базами данных National Center for Biotechnology Information (NCBI). N-концевые сигнальные пептиды были предсказаны с помощью Signal P v.5.0, а присутствие трансмембранных доменов – с помощью TMHMM v.2.0 (http://www.cbs.dtu.dk/services/TMHMM/).
Определениe уровня сходства между геномами и филогенетический анализ на основе полногеномных данных. Средние уровни идентичности нуклеотидных (average nucleotide identity, ANI) и аминокислотных (average amino acid identity, AAI) последовательностей между выбранными геномами были рассчитаны с использованием скриптов из Enveomics Collection (Rodriguez-R, Konstantinidis, 2016).
GTDB-Tk v.0.3.2 был использован для поиска однокопийных маркерных генов в MAG и для построения множественного выравнивания конкатенированных последовательностей однокопийных маркерных генов из данного MAG и всех видов из GTDB. Часть множественного выравнивания, созданного в GTDB-Tk, была использована для построения филогенетического дерева с помощью PhyML v.3.3 (Guindon et al., 2010) с использованием параметров по умолчанию. Уровень поддержки внутренних ветвей оценивался с помощью байесовского теста в PhyML.
Депонирование нуклеотидных последовательностей. Первичные данные, полученные в результате секвенирования фрагментов гена 16S рРНК и секвенирования метагенома, были депонированы в NCBI Sequence Read Archive (SRA) под номерами SRX10881305, SRX10881306 и SRX10881307. Аннотированные последовательности MAG депонированы в базе данных GenBank и доступны через BioProject PRJNA728906.
РЕЗУЛЬТАТЫ
Состав микробного сообщества по результатам анализа ПЦР фрагментов генов 16S рРНК. Образец грунта был отобран из горящего отвала добычи угля с многочисленными выходами продуктов горения на поверхность. Температура грунта в месте отбора составляла 72°C.
Для характеристики состава микробного сообщества были использованы 57 213 последовательностей фрагментов гена 16S рРНК. В результате кластеризации этих последовательностей было идентифицировано 29 OTЕ на уровне 97% идентичности. Все выявленные ОТЕ представляли бактерии, архей обнаружено не было. Результаты таксономической классификации OTЕ представлены на рис. 1.
Рис. 1.
Состав микробного сообщества по результатам анализа генов 16S рРНК и метагеномного секвенирования. Категория others (прочие) в случае метагенома включает последовательности, не вошедшие в MAG, отнесенные к указанным группам.
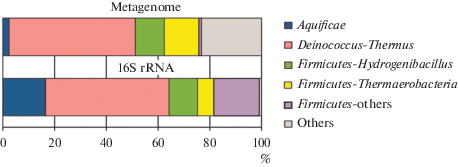
В сообществе доминировали представители трех филумов: Deinococcus-Thermus (47.7% всех последовательностей генов 16S рРНК), Firmicutes (34.9%) и Aquificae (16.5%). В минорных количествах были обнаружены представители филумов Bacteroidetes (0.48%), Proteobacteria (0.27%), Chloroflexi (0.02%) и Actinobacteria (0.01%).
Филум Deinococcus-Thermus был представлен двумя ОТЕ, одна из которых доминировала в сообществе, составляя 47.4% всех последовательностей 16S рРНК. Эта ОТЕ относится к роду Thermus со сходством последовательности 98.7% с термофильной гетеротрофной бактерии Thermus antranikianii, выделенной из горячих источников в Исландии (Chung et al., 2000).
12 ОТЕ представляли филум Firmicutes. Наиболее многочисленная из них (14.1%) представляла род Brevibacillus и имела идентичность 98.73% по 16S рРНК с Brevibacillus borstelensis, умеренно термофильной гетеротрофной спорообразующей бактерией, встречающейся в почвах и гидротермальных местообитаниях (Shida et al., 1995; Khalil et al., 2018). Около 11% последовательностей представляли одну ОТЕ, отнесенную к роду Hydrogenibacillus (порядок Thermicanales, семейство Thermicanaceae в соответствии с таксономической системой Genome Taxonomy Database). Культивируемый представитель этого рода, Hydrogenibacillus schlegelii, – факультативно хемолитотрофная аэробная термофильная бактерия, которая способна окислять водород и, предположительно, СО (Schenk, Aragno, 1979; Kämpfer et al., 2013). Около 6.3% сообщества представляла ОТЕ, филогенетически удаленная от культивируемых представителей филума Firmicutes (<87% идентичности по 16S рРНК).
Филум Aquificae был представлен одной ОТЕ, филогенетически близкой к роду Hydrogenobacter семейства Aquificaceae. Представители Hydrogenobacter – типичные обитатели высокотемпературных горячих источников, хемолитоавтотрофные термофилы, способные аэробно окислять водород (Reysenbach et al., 2000; Takacs-Verbach et al., 2013).
Секвенирование метагенома и сборка MAG. Для получения геномов представителей микробного сообщества мы просеквенировали метагеном образца RBS10, используя комбинацию технологий Illumina и Oxford Nanopore. Собранные контиги были кластеризованы в 18 MAG, имеющих полноту свыше 80% и загрязнение (избыточность) менее 10% по оценке CheckM на основе анализа присутствия набора консервативных однокопийных маркерных генов (табл. 1). В сумме эти MAG представляли около 80% всего метагенома сообщества. Таксономическая принадлежность полученных MAG была определена на основе филогенетического анализа по конкатенированным последовательностями консервативных маркерных генов по Genome Taxonomy Database (Parks et al., 2018).
Таблица 1.
Основные характеристики MAG
| MAG ID | Полнота/ загрязнение, % | Размер генома, нт | Число контигов | GC, % | Доля в метагеноме, % | Таксономическая принадлежность* |
|---|---|---|---|---|---|---|
| 2 | 97.63/1.36 | 1 699 170 | 143 | 41.6 | 0.08 | p__Aquificota; g__Hydrogenobacter |
| 74 | 99.59/0.51 | 1 794 458 | 1** | 44 | 0.17 | p__Aquificota; s__Hydrogenobacter thermophilus |
| 58 | 99.59/0.41 | 1 722 082 | 1** | 43.1 | 2.13 | p__Aquificota; g__UBA11096 |
| 4 | 97.27/0.91 | 3 316 678 | 1** | 67.6 | 3.17 | p__Chloroflexota; s__Thermoflexus hugenholtzii |
| 7 | 85.55/1.87 | 2 784 472 | 202 | 65.1 | 0.03 | p__Chloroflexota; g__Thermomicrobium |
| 30 | 96.23/0 | 3 717 411 | 433 | 65.6 | 0.11 | p__Chloroflexota; f__UBA6265 |
| 92 | 100/0 | 2 424 424 | 1** | 64.9 | 48.83 | p__Deinococcota; s__Thermus antranikianii |
| 77 | 93.16/1.82 | 2 294 372 | 228 | 45 | 0.16 | p__Firmicutes; f__Amphibacillaceae |
| 39 | 98.09/0.68 | 2 502 348 | 201 | 37.5 | 0.12 | p__Firmicutes_A; s__Caldanaerobacter subterraneus |
| 35 | 94.06/0.2 | 2 3684 55 | 42 | 63.5 | 11.01 | p__Firmicutes_E; c__Thermaerobacteria |
| 62 | 92.57/13.9 | 4 585 313 | 204 | 68.9 | 2.34 | p__Firmicutes_E; g__Thermaerobacter |
| 82 | 92.25/2.97 | 3 307 331 | 619 | 58.5 | 0.09 | p__Firmicutes_G; o__DTU080 |
| 36 | 96.15/2.95 | 4 121 736 | 396 | 53.4 | 0.25 | p__Firmicutes_I; s__Bacillus_BB thermozeamaize |
| 40 | 84.97/4.92 | 2 109 662 | 127 | 64 | 0.37 | p__Firmicutes_I; s__Brockia lithotrophica |
| 8 | 91.28/1.72 | 2 288 228 | 67 | 61.8 | 0.48 | p__Firmicutes_I; g__Hydrogenibacillus |
| 49 | 92.44/8.08 | 2 964 420 | 72 | 66.1 | 10.75 | p__Firmicutes_I; s__Hydrogenibacillus schlegelii |
| 91 | 99.19/2.07 | 4 672 685 | 147 | 66.4 | 0.18 | p__Proteobacteria; g__Paracoccus |
| 48 | 99.38/1.91 | 3 146 152 | 24 | 70.2 | 0.29 | p__Proteobacteria; g__Lysobacter |
| 29 | 92.37/1.69 | 1 580 388 | 114 | 27.6 | 0.02 | p__WOR-3_A; o__LBFQ01 |
Таксономическая классификация MAG выявила те же самые основные бактериальные филумы, которые были обнаружены с помощью 16S рРНК. Относительные доли некоторых линий в пуле последовательностей генов 16S рРНК и в полном метагеноме отличались (рис. 1), что, вероятно, обусловлено разным числом копий гена 16S рРНК в геномах и разными размерами самих геномов. Около половины всего метагенома представлял один MAG, RBS10-92, отнесенный к филуму Deinococcus-Thermus. Около 26% метагенома составляли 10 MAG, представлявшие филум Firmicutes, три MAG были отнесены к Aquificae (2.4% метагенома), три – к Chloroflexi (3.3% метагенома), два – к Proteobacteria (0.5% метагенома) и 0.02% метагенома представлял MAG кандидатного филума WOR-3 (табл. 1).
С использованием длинных чтений, полученных с помощью нанопорового секвенирования, были собраны полные кольцевые последовательности четырех геномов, – Hydrogenobacter thermophiles (MAG RBS10-74), представителя кандидатного рода UBA11096 филума Aquificae (MAG RBS10-58), Thermoflexus hugenholtzii филума Chloroflexi (MAG RBS10-4) и Thermus antranikianii (MAG RBS10-92).
Чтобы получить представление о метаболических возможностях основных представителей микробного сообщества мы подробно проанализировали несколько MAG.
Геномы представителей Firmicutes. К наиболее многочисленным представителям микробного сообщества относились две бактерии филума Firmicutes, представленные MAG RBS10-35 и MAG RBS10-49. Геном RBS10-35 представлял 11.0% всего метагенома и был идентифицирован только до уровня класса Thermaerobacteria в геномной таксономической системе. По-видимому, этот геном соответствует ОТЕ, составлявшей 6.3% последовательностей 16S рРНК и отнесенной к неклассифицируемым фирмикутам. Поиск ближайших родственников RBS10-35 в GenBank выявил только одну последовательность 16S рРНК (FN687452) с идентичностью 91%, обнаруженную в термальном аэробном биореакторе для переработки отработанного активного ила (Hayes et al., 2011). Анализ генома RBS10-35 показал, что эта бактерия имеет полную аэробную дыхательную цепь и СО дегидрогеназу, что указывает на возможность окисления СО.
Вторым по относительной численности среди фирмикут (10.7% метагенома) был организм, представленный геномом RBS10-49. Этот генотип был идентифицирован как Hydrogenibacillus schlegelii на основании 97.68% AAI с геномом штамма H. schlegelii MA48 (Maker et al., 2017). Анализ генома RBS10-49 выявил наличие генов аэробной дыхательной цепи, мембранно-связанной поглощающей [NiFe] гидрогеназы группы 1d и респираторной СО дегидрогеназы, что указывает на способность этой бактерии получать энергию за счет окисления компонентов угольных газов – водорода и СО. В геноме также кодируется полный цикл Кальвина, функционирование которого может обеспечивать автотрофную фиксацию углерода.
Следует отметить, что среди собранных MAG отсутствовал геном, относящийся к роду Brevibacillus, хотя на его представителей приходилось около 15% всех последовательностей генов 16S рРНК. По-видимому, Brevibacillus были представлены несколькими близкими филотипами, что затрудняло сборку протяженных контигов и, соответственно, получение MAG.
Полный геном Thermus antranikiani RBS10-92. В результате секвенирования метагенома был собран полный геном бактерии RBS10-92. Это геном имеет 97.79% AAI с Thermus antranikianii DSM 12 462 (GCF_000423905), что позволяет отнести его к этому виду. Геном T. antranikianii RBS10-92 был просеквенирован с 5030-кратным средним покрытием, имеет длину 2 424 424 п.н. и является первым известным полным геномом T. antranikianii. Относительная численность RBS10-92 в метагеноме составляет 48.8%, что хорошо согласуется с 47.64% долей соответствующей ОТЕ в чтениях 16S рРНК. В результате аннотации генома RBS10-92 было идентифицировано 2636 потенциальных белок-кодирующих генов, функции только половины из которых были предсказаны. В геноме были идентифицированы две копии оперона рРНК и 50 генов транспортных РНК (тРНК).
Анализ генома T. antranikianii RBS10-92 показал, что эта бактерия, вероятно, является аэробным гетеротрофом, способным гидролизовать различные углеводы. Этот микроорганизм также имеет возможности для роста в анаэробных условиях, используя нитрат в качестве акцептора электронов. Эти предсказания согласуются с микробиологическими характеристиками изолятов T. antranikianii (Chung et al., 2000).
Полный геном Hydrogenobacter thermophilus RBS10-74. Собраны три генома представителей филума Aquificae с полнотой более 97% и загрязнением менее 2%. Для двух их этих организмов были получены полные кольцевые геномные последовательности. RBS10-74, на который приходится 0.17% всего метагенома, был классифицирован как Hydrogenobacter thermophilus на основании 95.7% ANI с H. thermophilus TK-6 (GCA_000010785), изолированным из горячего источника в Японии (Arai et al., 2010).
Анализ генома H. thermophiles RBS10-74 предсказал, что, как и культивируемые штаммы H. thermophiles, он является облигатным автотрофным организмом, способным окислять водород и фиксировать CO2 через восстановительный цикл трикарбоновых кислот. При росте в анаэробных условиях эта бактерия может использовать нитрат в качестве акцептора электронов. RBS10-74 также может окислять элементную серу или тиосульфат.
Полный геном MAG RBS10-58 – представителя кандидатного рода UBA11096. Второй полный геном представителя филума Aquificae был отнесен к роду UBA11096 семейства Aquificaceae в соответствии с геномной таксономической системой. До настоящего времени этот род не имеет культивируемых представителей и описан на основе нескольких собранных из метагеномов драфт-геномов, анализ которых ранее не проводился.
Геном RBS10-58 был просеквенирован с 309-кратным средним покрытием и собран в кольцевую хромосому длиной 1 722 082 п.н. Относительная для этого генотипа в метагеноме составляет 2.13%. В результате аннотации генома RBS10-58 было идентифицировано 1848 потенциальных белок-кодирующих генов, функции половины из которых были предсказаны, а также один оперон 16S–23S–5S рРНК и 42 гена тРНК.
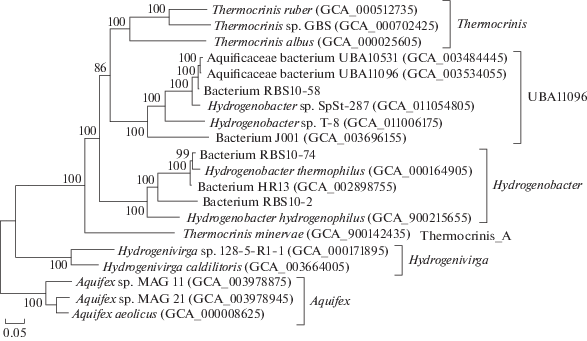
Для определения филогенетического положения бактерии RBS10-58 было построено филогенетическое дерево на основе конкатенированных аминокислотных последовательностей консервативных маркерных генов представителей всех родов семейства Aquificaceae, определяемых в геномной таксономической системе. Полученные результаты показывают, что RBS10-58 вместе с несколькими другими MAG представляет отдельную линию уровня рода, наряду с родами Aquifex, Hydrogenivirga, Hydrogenobacter, Thermocrinis и Thermocrinis minervae, представлявшим отдельный род Thermocrinis_А в геномной таксономии (рис. 2).
Рис. 2.
Филогенетическое положение RBS10-58 в семействе Aquificaceae. Филогенетическое дерево построено по методу maximum likelihood, основанной на основе конкатенированных последовательностей консервативных маркерных генов. Уровни поддержки ветвей определены с помощью Байесовского теста в PhyML. Таксономия показана на основе системы GTDB (g – род).
Анализ генома RBS10-58 выявил полный набор генов восстановительного цикла трикарбоновых кислот, который используется у Aquificae для автотрофной фиксации СО2 (Hügler et al., 2007), а также генов пути глюконеогенеза. Пентозофосфатный путь у RBS10-58 отсутствует. В геноме обнаружен полный набор генов пути окислительного фосфорилирования, в том числе NADH дегидрогеназы, сукцинатдегидрогеназы, цитохром bc1 комплекса и нескольких цитохром с оксидаз. Наличие поглощающих мембранно-связанных [NiFe] гидрогеназ групп 2a и 1d указывает на возможность использования молекулярного водорода в качестве донора электронов при дыхании, что характерно для представителей филума Aquificae. Другим субстратом может служить формиат, о чем свидетельствует наличие мембранно-связанной формиатдегидрогеназы.
В анаэробных условиях акцептором электронов может служить нитрат, на что указывает присутствие мембранно-связанной нитратредуктазы, которая восстанавливает нитрат до нитрита. Далее нитрит может восстанавливаться цитохром cd1 нитритредуктазой с образованием оксида азота(II). Последующие стадии денитрификации могут осуществляться редуктазой окиси азота (nitric-oxide reductase) и редуктазой закиси азота (nitrous oxide reductase) с образованием молекулярного азота.
В качестве донора электронов бактерия RBS10-58 может использовать и соединения серы. Окисление сероводорода может осуществляться сульфид:хинон оксидоредуктазой (sulfide:quinone oxidoreductase) и флавоцитохром с:сульфид дегидрогеназой (flavocytochrome c:sulfide dehydrogenase). В геноме также кодируется Sox-Hdr-Soe вариант пути окисления соединений серы до сульфата (Watanabe et al., 2019), включающий кластер sox-генов soxYZAXB, гены сульфитдегидрогеназы soeABC и кластер генов, кодирующих субъединицы гетеросуфльфидредуктазы hdrCBAhyphdrCB.
Таким образом, бактерия RBS10-58 является хемолитоавтотрофом, способным получать энергию за счет окисления молекулярного водорода и соединений серы в процессе аэробного дыхания, а в анаэробных условиях осуществлять все стадии денитрификации.
ОБСУЖДЕНИЕ
Несмотря на то, что один из первых описанных термофилов, Thermoplasma acidophilum, был выделен из проб отходов горящего угля группой Томаса Брока в конце 60-х годов прошлого столетия (Darling et al., 1970), состав микробного сообщества этих экосистем остается малоизученным. Исследования состава микробных сообществ почв в районах подземных угольных пожаров были выполнены в Пенсильвании, США (Tobin-Janzen et al., 2005; Lee et al., 2017). Анализ микробных сообществ по 16S рРНК выявил присутствие архей филума Crenarchaeota, а также бактерий филумов Chloroflexi, Acidobacteria, Proteobacteria, Bacteroidetes, Elusimicrobia и Gemmatimonadetes. В последующих метагеномных исследованиях было показано, что микроорганизмы с меньшими размерами клеток и геномов преобладают на участках почвы с более высокой температурой (Sorensen et al., 2019).
Микробные сообщества нагретой почвы в районе выходов на поверхность горячих угольных газов в Синьцзяне, Китай, были исследованы с использованием T-RFLP анализа и клональных библиотек генов 16S рРНК (Zhang et al., 2013). Среди доминирующих групп микроорганизмов обнаружены представители филумов Firmicutes, Proteobacteria, Acidobacteria, Bacteroidetes, Planctomycetes и Actinobacteria. Самой многочисленной группой были Firmicutes, в основном представители родов Bacillus и Paenibacillus.
В обоих этих случаях объектами исследования были нагретые за счет выхода горячих газов “обычные”, богатые органикой почвы, что обусловило преобладание в сообществах типичных почвенных групп микроорганизмов. В настоящей же работе объектом исследования были не почвы, а отвалы карьера по добыче угля, представляющие собой углесодержащие горные породы. Предполагается, что в такой экосистеме будут формироваться специфические сообщества термофилов, развитие которых поддерживается за счет угольных газов. Наиболее близким аналогом исследованного нами объекта было микробное сообщество нагретых горных пород в районе подземного горения угля на территории Горного Алтая (Kadnikov et al., 2018). Оно было простым по составу и включало всего три доминирующих филотипа, все они представляли филум Firmicutes. Это аэробный гетеротроф Ca. Carbobacillus altaicus, анаэробный хемолитоавтотроф Brockia lithotrophica и аэробная бактерия Hydrogenibacillus schlegelii, способная как использовать органические соединения, так и расти автотрофно. Все эти микроорганизмы могут получать энергию за счет окисления молекулярного водорода (а некоторые также и СО; Kadnikov et al., 2018). Также в этом сообществе были обнаружены некультивируемые линии фирмикут, относящиеся к Thermaerobacteria.
Еще одним близким аналогом являются горящие отвалы угольного разреза у города Киселевск Кемеровской области. В микробном сообществе поверхностного слоя нагретого до 58°С грунта доминировали представители Ktedonobacteria (филум Chloroflexi), способные окислять водород и СО, а термофильные гидрогенотрофные фирмикуты составляли небольшую часть сообщества (Kadnikov et al., 2021). Возможно, отличия от исследованного в этой работе объекта обусловлены различной температурой и влажностью.
В исследованном нами сообществе RBS10 фирмикуты составляли около трети и были представлены термофильными группами. H. schlegelii и некультивируемые представители Thermaerobacteria входили в число доминирующих групп, в небольших количествах была обнаружена и Brockia lithotrophica. Однако, помимо фирмикутов, доминирующими группами в сообществе RBS10 были представители рода Thermus и семейства Aquificaceae, характерные для гидротермальных экосистем (Counts et al., 2017; Bonch-Osmolovskaya, 2020). Ранее об их присутствии в почвах в районах подземного горения угля не сообщалось. Будучи способными к автотрофной фиксации углерода и получению энергии за счет окисления водорода и соединений веры, Aquificaceae, как и фирмикуты, представляют автотрофную часть сообщества, поддерживаемую угольными газами. Составляющий около половины микробного сообщества T. antranikianii, в свою очередь, использует в качестве субстратов органические вещества, образуемые автотрофными фирмикутами и Aquificaceae.
Исследованная нами термальная экосистема, ассоциированная с горящими угольными отвалами, и другие подобные объекты в Кемеровской области и на Алтае, возникли не более нескольких десятков лет назад и являются “молодыми” по сравнению с геотермальными объектами. Ранее было высказано предположение, что термофильные Firmicutes, споры которых могут распространяться на большие расстояния (Bonjour et al., 1988; Aullo et al., 2013), могут быть первыми “колонизаторами” таких новых термальных экологических ниш (Kadnikov et al., 2018). Обнаружение нами представителей Thermus и Aquificaceae свидетельствует о том, что распространение неспорообразующих термофилов, источником которых могут быть встречающиеся на юге Сибири горячие источники и другие геотермальные объекты, также может быть достаточно быстрым.
Список литературы
Arai H., Kanbe H., Ishii M., Igarashi Y. Complete genome sequence of the thermophilic, obligately chemolithoautotrophic hydrogen-oxidizing bacterium Hydrogenobacter thermophilus TK-6 // J. Bacteriol. 2010. V. 192. P. 2651–2652.
Aüllo T., Ranchou-Peyruse A., Ollivier B., Magot M. Desulfotomaculum spp. and related gram-positive sulfate-reducing bacteria in deep subsurface environments // Front. Microbiol. 2013. V. 4. P. 362.
Bonch-Osmolovskaya E. Aquificales // eLS. 2020. V. 1. P. 433–441.
Bonjour F., Graber A., Aragno M. Isolation of Bacillus schlegelii, a thermophilic, hydrogen oxidizing, aerobic a-utotroph, from geothermal and nongeothermal environments // Microb. Ecol. 1988. V. 16. P. 331–337.
Brettin T., Davis J.J., Disz T., Edwards R.A., Gerdes S., Olsen G.J., Olson R., Overbeek R., Parrello B., Pusch G.D., Shukla M., Thomason J.A. III, Stevens R., Vonstein V., Wattam A.R., Xia F. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes // Sci. Rep. 2015. V. 5. Art. 8365. https://doi.org/10.1038/srep08365
Cao M.D., Nguyen S.H., Ganesamoorthy D., Elliott A.G., Cooper M.A., Coin L.J. Scaffolding and completing genome assemblies in real-time with nanopore sequencing // Nat. Commun. 2017. V. 8. P. 1–10.
Chaumeil P.A., Mussig A.J., Hugenholtz P., Parks D.H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database // Bioinformatics. 2020. V. 36. P. 1925–1927.
Chung A.P., Rainey F.A., Valente M., Nobre M.F., da Costa M.S. Thermus igniterrae sp. nov. and Thermus antranikianii sp. nov., two new species from Iceland // Int. J. Syst. Evol. Microbiol. 2000. V. 50. P. 209–217.
Counts J.A., Zeldes B.M., Lee L.L., Straub C.T., Adams M.W.W., Kelly R.M. Physiological, metabolic and biotechnological features of extremely thermophilic microorganisms // Wiley Interdiscip. Rev. Syst. Biol. Med. 2017. V. 9. e1377.
Darling G., Brock T.D., Samsonoff W., Conti S.F. A thermophilic, acidophilic mycoplasma isolated from a coal refuse pile // Science. 1970. V. 170. P. 1416–1418.
Edgar R.C. Search and clustering orders of magnitude faster than BLAST // Bioinformatics. 2010. V. 26. P. 2460–2461.
Engle M.A., Radke L.F., Heffern E.L., O’Keefe J.M., Hower J.C., Smeltzer C.D., Hower J.M., Olea R.A., Eatwell R.J., Blake D.R., Emsbo-Mattingly S.D., Stout S.A., Queen G., Aggen K.L., Kolker A., Prakash A., Henke K.R., Stracher G.B., Schroeder P.A., Román-Colón Y., Schure A. Gas emissions, minerals, and tars associated with three coal fires, Powder River Basin, USA // Sci. Total Environ. 2012. V. 420. P. 146–159.
Frey B., Rime T., Phillips M., Stierli B., Hajdas I., Widmer F., Hartmann M. Microbial diversity in European alpine permafrost and active layers // FEMS Microbiol. Ecol. 2016. V. 92. fiw018.
Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0 // Syst. Biol. 2010. V. 59. P. 307–321.
Hayes D., Izzard L., Seviour R. Microbial ecology of autothermal thermophilic aerobic digester (ATAD) systems for treating waste activated sludge // Syst. Appl. Microbiol. 2011. V. 34. P. 127–138.
Hügler M., Huber H., Molyneaux S.J., Vetriani C., Sievert S.M. Autotrophic CO2 fixation via the reductive tricarboxylic acid cycle in different lineages within the phylum Aquificae: evidence for two ways of citrate cleavage // Environ. Microbiol. 2007. V. 9. P. 81–92.
Kadnikov V.V., Mardanov A.V., Beletsky A.V., Grigoriev M.A., Karnachuk O.V., Ravin N.V. Thermophilic Chloroflexi dominate in the microbial community associated with coal-fire gas vents in the Kuznetsk Coal Basin, Russia // Microorganisms. 2021. V. 9. P. 948.
Kadnikov V.V., Mardanov A.V., Ivasenko D.A., Antsiferov D.V., Beletsky A.V., Karnachuk O.V., Ravin N.V. Lignite coal burning seam in the remote Altai Mountains harbors a hydrogen-driven thermophilic microbial community // Sci. Rep. 2018. V. 8. P. 1–12.
Kämpfer P., Glaeser S.P., Busse H.J. Transfer of Bacillus schlegelii to a novel genus and proposal of Hydrogenibacillus schlegelii gen. nov., comb. nov. // Int. J. Syst. Bacteriol. 2013. V. 63. P. 1723–1727.
Kang D.D., Froula J., Egan R., Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities // PeerJ. 2015. V. 3. e1165.
Khalil A.B., Sivakumar N., Arslan M., Saleem H., Qarawi S. Insights into Brevibacillus borstelensis AK1 through whole genome sequencing: a thermophilic bacterium isolated from a hot spring in Saudi Arabia // Biomed. Res. Int. 2018. Art. 5862437.
Kolmogorov M., Yuan J., Lin Y., Pevzner P.A. Assembly of long, error-prone reads using repeat graphs // Nat. Biotechnol. 2019. V. 37. P. 540–546.
Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2 // Nat. Methods. 2012. V. 9. P. 357–359. https://doi.org/10.1038/nmeth.1923
Lee S.H., Sorensen J.W., Grady K.L., Tobin T.C., Shade A. Divergent extremes but convergent recovery of bacterial and archaeal soil communities to an ongoing subterranean coal mine fire // ISME J. 2017. V. 11. P. 1447–1459.
Li H., Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform // Bioinformatics. 2010. V. 26. P. 589–595.
Magoč T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies // Bioinformatics. 2011. V. 27. P. 2957–2963.
Maker A., Hemp J., Pace L.A., Ward L.M., Fischer W.W. Draft genome sequence of Hydrogenibacillus schlegelii MA48, a deep-branching member of the Bacilli class of Firmicutes // Genome Announc. 2017. V. 5. e00380-16.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads // EMBnet. J. 2011. V. 17. P. 10–12.
Nurk S., Meleshko D., Korobeynikov A., Pevzner P.A. metaSPAdes: a new versatile metagenomic assembler // Genome Res. 2017. V. 27. P. 824–834.
Parks D.H., Chuvochina M., Waite D.W., Rinke C., Skarshewski A., Chaumeil P.A., Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life // Nat. Biotechnol. 2018. V. 36. P. 996–1004.
Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes // Genome Res. 2015. V. 25. P. 1043–1055.
Pone J.D.N., Hein K.A., Stracher G.B., Annegarn H.J., Finkleman R.B., Blake D.R., McCormack J.K., Schroeder P. The spontaneous combustion of coal and its by-products in the Witbank and Sasolburg coalfields of South Africa // Int. J. Coal Geol. 2007. V. 72. P. 124–140.
Rattigan J.H. Phenomena about Burning Mountain, Wingen, NSW // Aust. J. Sci. 1967. V. 30. P. 183–184.
Reysenbach A.L., Ehringer M., Hershberger K. Microbial diversity at 83°C in Calcite Springs, Yellowstone National Park: another environment where the Aquificales and “Korarchaeota” coexist // Extremophiles. 2000. V. 4. P. 61–67.
Rodriguez-R L.M., Konstantinidis K.T. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes // PeerJ Preprints. 2016. e1900v1.
Rognes T., Flouri T., Nichols B., Quince C., Mahé F. VSEARCH: A versatile open source tool for metagenomics // PeerJ. 2016. V. 4. e2584.
Schenk A., Aragno M. Bacillus schlegelii, a new species of thermophilic, facultatively chemolithoautotrophic bacterium oxidizing molecular hydrogen // Microbiology (SGM). 1979. V. 115. P. 333–341.
Shafirovich E., Varma A. Underground coal gasification: A brief review of current status // Ind. Eng. Res. 2009. V. 48. P. 7865–7875.
Shida O., Takagi H., Kadowaki K., Udaka S., Nakamura L.K., Komagata K. Proposal of Bacillus reuszeri sp. nov., Bacillus formosus sp. nov., nom. rev., and Bacillus borstelensis sp. nov., nom. rev. // Int. J. Syst. Bacteriol. 1995. V. 45. P. 93–100.
Sorensen J.W., Dunivin T.K., Tobin T.C., Shade A. Ecological selection for small microbial genomes along a temperate-to-thermal soil gradient // Nat. Microbial. 2019. V. 4. P. 55–61.
Stracher G.B., Taylor T.P. Coal fires burning out of control around the world: Thermodynamic recipe for environmental catastrophe // Int. J. Coal Geol. 2004. V. 59. P. 7–17.
Takacs-Verbach C., Inskeep W.P., Jay Z.J., Herrgard M.J., Rusch D.B., Tringe S.G., Kozubal M.A., Hamamura N., Macur R.E., Fouke B.W., Reysenbach A.L., McDermott T.R., Jennings R.deM., Hengartner N.W., Xie G. Metagenome sequence analysis of filamentous microbial communities obtained from geochemically distinct geothermal channels reveals specialization of three Aquificales lineages // Front. Microbiol. 2013. V. 4. P. 84.
Tatusova T., DiCuccio M., Badretdin A., Chetvernin V., Nawrocki E.P., Zaslavsky L., Lomsadze A., Pruitt K.D., Borodovsky M., Ostell J. NCBI prokaryotic genome annotation pipeline // Nucleic Acids Res. 2016. V. 44. P. 6614–6624.
Tobin-Janzen T., Shade A., Marshall L., Torres K., Beblo C., Janzen C., Lenig J., Martinez A., Ressler D. Nitrogen changes and domain bacteria ribotype diversity in soils overlying the Centralia, Pennsylvania underground coal mine fire // Soil Sci. 2005. V. 170. P. 191–201.
Urbieta M.S., Donati E.R., Chan K.G., Shahar S., Sin L.L., Goh K.M. Thermophiles in the genomic era: Biodiversity, science, and applications // Biotechnol. Adv. 2015. V. 33. P. 633–647.
Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement // PLoS One. 2014. V. 9. e112963.
Watanabe T., Kojima H., Umezawa K., Hori C., Takasuka T.E., Kato Y., Fukui M. Genomes of neutrophilic sulfur-oxidizing chemolithoautotrophs representing 9 proteobacterial species from 8 genera // Front. Microbiol. 2019. V. 10. Art. 316.
Zhang T., Xu J., Zeng J., Lou K. Diversity of prokaryotes associated with soils around coal-fire gas vents in MaNasi county of Xinjiang, China // Antonie van Leeuwenhoek. 2013. V. 103. P. 23–36.
Дополнительные материалы отсутствуют.