

Молекулярная биология, 2019, T. 53, № 3, стр. 402-410
Мутационный профиль подтипов острого миелоидного лейкоза у детей при отсутствии известных хромосомных перестроек
Л. Г. Гукасян a, Г. С. Краснов a, О. В. Муравенко a, Л. В. Байдун b, С. З. Ибрагимова c, Т. В. Наседкина a, d, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Российская детская клиническая больница Российского национального исследовательского медицинского университета им. Н.И. Пирогова Министерства здравоохранения Российской Федерации
119571 Москва, Россия
c Научно-исследовательский институт гематологии и переливания крови
Министерства здравоохранения Республики Узбекистан
100185 Ташкент, Узбекистан
d Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева
119117 Москва, Россия
* E-mail: nased@biochip.ru
Поступила в редакцию 21.11.2018
После доработки 07.12.2018
Принята к публикации 07.12.2018
Аннотация
Поиск значимых молекулярно-генетических маркеров имеет большое значение для постановки диагноза, определения прогноза и выбора терапии при злокачественных заболеваниях кроветворной системы. Характерные цитогенетические аберрации, приводящие к образованию слитых генов, встречаются более чем у 40% детей с острым миелоидным лейкозом (ОМЛ), однако у значительной части пациентов (около 20%) опухолевые клетки имеют нормальный кариотип. С целью выявления маркеров, характерных для ОМЛ, нами проанализирован мутационный профиль опухолевых клеток у детей с ОМЛ, не имеющих известных хромосомных перестроек. У 34 пациентов с различными подтипами ОМЛ методом массового параллельного секвенирования исследованы участки 26 генов, вовлеченных в патогенез этого заболевания. Выявлены соматические мутации в генах белков различных внутриклеточных сигнальных путей, в том числе в генах CEBPA, ETV, IDH1, JAK2, NRAS. Также обнаружены редкие генетические варианты генов CUX1, FLT3, TET2, PTPN11, NUP98. Полученные данные представляют интерес для понимания механизмов злокачественной трансформации клеток при лейкемогенезе.
Острый миелоидный лейкоз (ОМЛ) ‒ опухоль кроветворной системы миелоидного происхождения, которая возникает в результате генетических и эпигенетических изменений в гемопоэтических стволовых клетках-предшественниках. Эти изменения приводят к нарушению регуляции внутриклеточных сигнальных путей и появлению клонов недифференцированных клеток с неограниченным потенциалом размножения.
Видимые изменения кариотипа являются крае-угольным камнем в постановке диагноза ОМЛ и выборе стратегии лечения [1‒5]. Несмотря на значительное количество разнообразных цитогенетических аномалий при ОМЛ у детей, основную часть ОМЛ можно отнести к следующим подгруппам: 1) ОМЛ с транслокацией t(8;21) или инверсией inv(16) (18‒20%); 2) с транслокацией t(15;17) (9‒10%); 3) с перестройками с участием гена MLL (25%); 4) без перечисленных цитогенетических аномалий, включая ОМЛ (около 20%) с цитогенетически нормальным кариотипом (ОМЛ-НК).
Достаточно большая группа больных с ОМЛ-НК, формально относящаяся к категории промежуточного риска, крайне гетерогенна в отношении прогноза течения заболевания. К наиболее характерным мутациям при ОМЛ-НК относятся мутации в генах NPM1, FLT3 и CEBPA. Недавно выявлен новый класс мутаций, повреждающих гены, ответственные за эпигенетические процессы регуляции генома, в частности за метилирование ДНК или модификацию гистонов. Среди них наиболее изучены мутации в генах DNMT3A, IDH1/2, TET2 и ряде других [1, 5‒8]. Определенные мутации, наряду с известными хромосомными перестройками, могут служить маркерами различных подтипов опухолевых клеток.
Число исследований, посвященных генетическим мутациям при ОМЛ, постоянно растет. Накопление знаний о генетических изменениях при ОМЛ позволяет более полно понять механизм развития заболевания, сформировать новые диагностические и прогностические критерии, развить новые подходы к терапии.
Нами проведен анализ мутационного профиля у детей с ОМЛ, у которых не выявлены известные хромосомные перестройки, направленный на оценку генетической гетерогенности этой группы и обнаружение маркеров, характеризующих молекулярную природу заболевания.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Мутационный профиль анализировали у 34 пациентов с ОМЛ (18 мальчиков и 16 девочек), средний возраст 10.6 лет (от 1.2 до 15.4 лет). Для первичной диагностики лейкозов проводили две цитохимические реакции – на миелопероксидазу и на неспецифическую эстеразу. Иммунофенотипическое исследование проводили на клетках костного мозга в растворе антикоагулянта EDТА. Реакцию прямой иммунофлуоресценции и анализ на проточном цитофлуориметре выполняли по стандартному протоколу. Исследуемая панель включала следующие маркеры: СD34, СD13, СD33, СD19, СD41, СD61, СD7, СD14, CD15, СD4, CD56, СD11b, HLA-DR, внутриклеточная миелопероксидаза. Также исследованы лимфоидные маркеры CD7, CD2, CD3 (в цитоплазме), CD79a, CD22.
В соответствии с классификацией, предложенной Франко-Американо-Британской группой (FAB-группа), выделяли М0‒M7 варианты ОМЛ. Подтип М1 (острый миелоидный лейкоз без созревания) выявлен у четырех пациентов, М2/М4 (острый миелоидный лейкоз с созреванием/острый миеломоноцитарный лейкоз) ‒ у 10, М5 (острый монобластный и острый моноцитарный лейкоз) ‒ у четырех, М6 (острый эритроидный лейкоз) ‒ у четырех, М7 (острый мегакариоцитарный лейкоз) ‒ у пяти пациентов, ОМЛ с миелодисплазией (МДС) ‒ у трех пациентов, в четырех случаях подтип ОМЛ не был установлен (ОМЛ). Пациентов с М0 (ОМЛ с минимальной дифференцировкой) и М3 (острый промиелоцитарный лейкоз) в исследуемой выборке не было. От родителей пациентов получено письменное добровольное информированное согласие на использование результатов исследования в обезличенной форме в научных целях.
Определение хромосомных перестроек методом гибридизационного анализа на биочипе. Лейкоциты из образцов костного мозга подвергали гемолизу в 0.8%-ном растворе хлорида аммония, собирали центрифугированием и промывали фосфатно-солевым буфером. РНК из лейкоцитов выделяли при помощи набора RNeasy MiniKit (“Qiagen”, США) согласно инструкции производителя. РНК (2 мкг) инкубировали при 70°С в течение 5 мин со смесью праймеров для обратной транкрипции (ОТ-праймеров), специфичных для указанных транслокаций, и гена ABL, стабильно экспрессирующегося во всех клетках. В реакционную смесь вносили по 10 пмоль каждого праймера, проводили реакцию обратной транскрипции, используя обратную транскриптазу M-MLV (“Силекс”, Россия) в соотношении 40 ед. фермента на 40 мкл реакционной смеси; смесь содержит также 40 ед. ингибитора РНКазы (“Promega”, США), 1 мМ каждого dNTP (“Силекс”), 10 мМ дитиотрейтол, 50 мМ Трис-HCl pH 8.3, 75 мМ KCl и 3 мМ MgCl2 при 42°С в течение 90 мин. Реакцию останавливали, прогревая смесь при 70°С в течение 10 мин.
Полученную в ходе обратной транскрипции кДНК добавляли в качестве матрицы в мультиплексную ПЦР. Использовали вариант гнездовой ПЦР, включающий два этапа. Смесь первого этапа (25 мкл) содержит 67 мМ Трис-HCl pH 8.6, 166 мМ (NH4)2SO4, 0.01% Тритона Х-100, 1.5 мМ MgCl2, 0.2 мМ каждого из dNTP (“Силекс”, Россия), 1.5 ед. акт. Taq-полимеразы (“Силекс”), по 10 пмоль каждого праймера и 2 мкл кДНК. Состав ПЦР-смеси второго этапа аналогичен первому, но концентрация прямого праймера составляет 50 пмоль, обратного 10 пмоль (для преимущественной амплификации прямой цепи). В качестве матрицы брали 1 мкл ПЦР-продукта первого этапа. Второй этап ПЦР проводили в присутствии флуоресцентно меченного Сy5-dUTP.
Флуоресцентно меченный продукт второго этапа ПЦР гибридизовали на ЛК-биочипе для анализа 13 транслокаций (“БИОЧИП-ИМБ”, Россия), изготовленном методом фотоиндуцируемой совместной полимеризации олигонуклеотидов и компонентов полиакриламидного геля. Гибридизацию флуоресцентно меченного ПЦР-продукта на биочипе проводили в течение 18‒20 ч, как описано ранее [10]. Регистрацию флуоресцентных сигналов в ячейках биочипа и анализ изображения проводили с использованием программы ImageWare (“БИОЧИП-ИМБ”, Россия).
Массовое параллельное секвенирование (NGS, next-generation sequencing). Направленный отбор последовательностей проводили методом двойной гибридизации с библиотекой зондов NimbleGen SeqCap EZ согласно инструкции производителя (www.nimblegen.com). Библиотека зондов включала участки последовательностей 26 генов: ABL1 (экзоны 4–6), CBL (экзоны 8, 9), CEBPA (полностью), CUX1 (полностью), DNMT3A (полностью), ETV6 (полностью), EZH2 (полностью), FLT3 (экзоны 14, 15, 20), GATA1 (экзон 2), HRAS (экзоны 2, 3), IDH1 (экзон 4), IDH2 (экзон 4), IKZF1 (полностью), JAK2 (экзоны 12, 14), JAK3 (экзон 13), KIT (экзоны 2, 8–11, 13, 17), KMT2A (экзоны 5–8), NOTCH1 (экзоны 26–28, 34), NUP98 (экзоны 20‒25), NPM1 (экзон 12), NRAS (экзоны 2, 3), PTPN11 (экзоны 3, 13), RUNX1 (полностью), TET2 (экзоны 3, 4–11), TP53 (экзоны 2, 3–11), ZRSR2 (полностью), вовлеченных в патогенез ОМЛ. Приготовленные библиотеки ДНК секвенировали на приборе MiniSeq (“Illumina”, США) путем парноконцевых чтений 2 × 150. Мутации, выявленные методом NGS, верифицировали также секвенированием по Сэнгеру с использованием автоматического секвенатора Applied Biosystems 3730 DNA Analyzer (“Applied Biosystems”, США).
Биоинформатический анализ. Анализ данных секвенирования включал оценку качества прочтений, поиск избыточно представленных последовательностей (FastQC), удаление фрагментов адаптеров, фильтрацию прочтений, аннотацию вариантов. Первичный и вторичный анализ данных, аннотацию вариантов проводили с помощью программного обеспечения Illumina (https://www.basespace.illumina.com). Среднее покрытие на образец составило около ×500.
Работы по высокопроизводительному секвенированию выполнены с использованием оборудования ЦКП “Геном” ИМБ РАН (http://www.eimb.ru/ RUSSIAN_NEW/INSTITUTE/ccu_genome_c.php).
Анализ кривых плавления высокого разрешения (HRM, high resolution melting). Поиск мутаций в гене NPM1 проводили с помощью HRM-анализа. В реакции обратной транскрипции получали кДНК, далее фрагмент экзона 12 гена NPM1 амплифицировали с использованием праймеров NPM1_F (5'-GGTTGTTCTCTGGAGCAGCGT-TC-3') и NPM1_R (5'-CCTGGACAACATTTATCAAACACGGTA-3'). HRM-анализ выполняли на приборе LightCycler 96 (“Roche”, Швейцария) в присутствии флуоресцентного красителя EvaGreen.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Практически у всех 34 пациентов выявлена однa и более мутаций в анализируемых генах, описанных ранее как характерные для гемопоэтических клеток. У 14 пациентов мутации были представлены только синонимическими заменами, в то время как у 20 пациентов выявлена одна или несколько несинонимических мутаций, аннотированных в базах данных, включая каталог соматических мутаций при раке Cosmic Database (cancer.sanger.ac.uk). Результаты высокопроизводительного секвенирования c описанием несинонимических мутаций (частота в популяции 1% и менее) представлены в таблице.
Таблица.
Мутации, выявленные в образцах пациентов с острым миелоидным лейкозом
| Ген | Ref* | Alt** | ID*** | Мутация | Аминокислотная замена | MAF****, % | Образец (подтип ОМЛ) – частота аллельного варианта в образце, % |
|---|---|---|---|---|---|---|---|
| CEBPA | – | TCT | COSM120165 | c.928_929insAGA | p.E309_T310insK | – | 6 (M4) – 25 |
| CEBPA | GGGCGCGTGCG | – | COSM29240 | c.68_78del11 | p.P23Qfs*81 | – | 6 (M4) – 30 |
| CEBPA | G | A | rs535980233 | c.667G>A | p.G223S | 0.04 | 9 (M4) – 56 |
| CUX1 | C | G | rs138450169 | c.1525C>G | p.L509V | 0.26 | 20 (M7) – 67 |
| CUX1 | G | C | rs782489323 | c.1282G>C | p.A428P | 0 | 4 (ОМЛ) – 47, 6 (М4) – 48 |
| ETV6 | A | C | – | c.1031A>C | p.Y344S | – | 9 (М4) – 8 |
| ETV6 | G | T | – | c.1090G>T | stopgain | 8 (ОМЛ) – 43 | |
| ETV6 | TA | – | – | c.417_418delTA | p.I140Tfs*12 | – | 3 (ОМЛ) – 37 |
| EZH2 | G | A | rs397515548 | c.2233G>A | p.E745K | – | 3 (ОМЛ) – 86 |
| FLT3 | C | T | rs146030737 | c.580G>A | p.V194M | 0.2 | 13 (М2/М4) – 48 |
| IDH1 | C | A | rs121913499 | c.394C>A | p.R132S | 0 | 8 (ОМЛ) – 43 |
| IDH1 | G | A | rs121913500 | c.395G>A | p.R132H | 0 | 2 (М6) – 28 |
| JAK2 | G | T | rs77375493 | c.1849G>T | p.V617F | 0 | 5 (М7) – 40, 10 (М5а) – 14 |
| JAK2 | G | A | rs41316003 | c.3188G>A | p.R1063H | 1 | 20 (М7) – 46 |
| NOTCH1 | G | A | rs751275854 | c.3859C>T | p.R1287C | 0 | 17 (М2) – 53 |
| NUP98 | G | A | rs149360555 | c.2747C>T | p.T916M | 1 | 12 (М0) – 52, 14 (М6) – 44, 18 (М5а) – 56 |
| NRAS | T | A | rs11554290 | c.182A>G | p.Q61R | 0 | 7 (М7) – 8 |
| NRAS | C | T | rs121434596 | c.38G>A | p.G13D | 0 | 19 (М4) – 49 |
| NRAS | C | T | rs121913237 | c.35G>A | p.G12D | 0 | 15 (М0) – 51 |
| PTPN11 | G | A | rs397507534 | c.1052G>A | p.R351Q | 0.04 | 1 (МДС) – 52 |
| PTPN11 | A | G | rs121918459 | c.188A>G | p.Y63C | 0 | 16 (М6) – 28 |
| RUNX1 | C | T | – | c.592G>A | p.D198N | – | 13 (М2/М4) – 48 |
| TET2 | C | T | rs111948941 | c.100C>T | p.L34F | 1 | 14 (М6) – 62 |
| TET2 | C | A | rs146031219 | c.521C>A | p.P174H | 1 | 11 (М2/М4) – 56 |
| TET2 | C | T | rs373501577 | c.5977C>T | p.R1993W | 0 | 15 (М0) – 56 |
| ZRSR2 | G | A | rs199648317 | c.1319G>A | p.R440Q | 0.03 | 5 (М7) – 49 |
В образце 6 (подтип М4) выявлены две мутации в гене CEBPA, одна представлена инсерцией 3 п.н., приводящей к вставке аминокислоты, другая ‒ делецией 11 п.н., приводящей к сдвигу рамки считывания (рис. 1а). Мутации аннотированы в базе данных Cosmic и описаны ранее как соматические мутации при миелоидных лейкозах. Ген CEBPA кодирует белок C/EBPα ‒ важный фактор транскрипции, вовлеченный в нормальный гемопоэз [11]. Этот ген часто мутирует именно при ОМЛ с цитогенетически нормальным кариотипом, причем мутации поражают, как правило, оба аллеля этого гена [11, 12], что, скорее всего, имеет место и в нашем случае. Биаллельные мутации CEBPA у больных ОМЛ ассоциированы с благоприятным прогнозом при условии отсутствия мутации FLT3-ITD [13]. В классификации опухолей кроветворной и лимфоидной тканей ВОЗ 2008 г. вариант ОМЛ с мутантным CEBPA выделен в отдельную форму.
Рис. 1.
Результаты секвенирования по Сэнгеру. а ‒ CEBPA делеция со сдвигом рамки считывания (11 нуклеотидов) 68_78del P23Qfs*81; б ‒ ETV6 делеция со сдвигом рамки считывания (два нуклеотида) 417_418del, p. I140Tfs*12; в – ETV6 стоп-кодон 1090G>T, р. E364X.
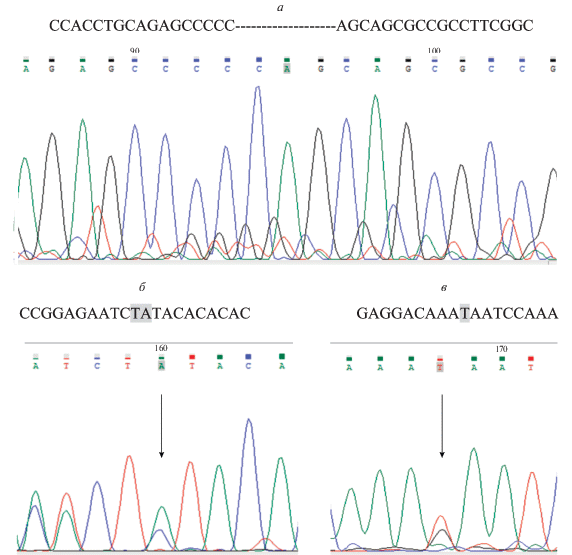
В образце 9 (М4) присутствует гетерозиготная мутация c.667G>A (rs535980233) в гене CEBPA. Согласно базам данных dbSNP и ClinVar это герминальный вариант с неизвестным клиническим значением, также обнаруженный ранее при ОМЛ.
Соматические мутации в гене ETV6 найдены в образцах 3 и 8 (ОМЛ), 9 (М4) (таблице, рис. 1б). Ген ETV6 кодирует белок-репрессор, негативный регулятор транскрипции. Более известны такие хромосомные перестройки с участием этого гена, как t(12;21) ETV6-RUNX1 при остром лимфобластном лейкозе у детей. Различные хромосомные транслокации с участием гена ETV6 и разнообразных генов-партнеров [14] и соматические мутации описаны также при ОМЛ.
К известным соматическим мутациям, обнаруженным в опухолевых клетках различного происхождения, относятся мутация p.V617F в гене JAK2, мутации в кодоне 132 гена IDH1, в кодонах 12, 13 и 61 гена NRAS [15].
Мутация p.V617F в гене JAK2 выявлена нами в 40% клеток образца 5 (М7) и в 14% клеток образца 10 (М5а). Белок JAK2 (Janus kinase, a protein tyrosine kinase) ‒ цитоплазматическая тирозинкиназа, участвующая в передаче сигнала по пути JAK-STAT (Signal transducers and activators of transcription) [16]. Связывание лигандов (эритропоэтина, тромбопоэтина, интерлейкинов, гранулоцитарного колониестимулирующего фактора) с рецепторами на поверхности клеток приводит к активации белка JAK2, который далее инициирует процесс димеризации белков STAT. Белки STAT проникают в ядро клетки и индуцируют транскрипцию генов-мишеней соответствующего цитокина. Мутация V617F повышает киназную активность белка JAK2, что приводит к конститутивной активации сигнального пути и клональной пролиферации клеток-предшественников миелоидного ростка кроветворения. Мутацию V617F гена JAK2 выявляют у 95% пациентов с истинной полицитемией и в 50% случаев эссенциальной тромбоцитемии и идиопатического миелофиброза, т.е. эта мутация является диагностическим маркером, в первую очередь, хронических миелопролиферативных заболеваний [15, 16].
В образцах 7 (М7), 15 (М0) и 19 (М4) нами выявлены мутации в гене NRAS. Мутации в онкогенах семейства RAS часто встречаются при ОМЛ, наиболее часто в генах NRAS (25%) и KRAS (15%) [17]. Мутации приводят к активации и повышенной экспрессии рецепторных тирозинкиназ без участия лиганда, что обеспечивает активацию внутриклеточных сигнальных путей, в первую очередь, RAS/RAF/MAPK и PI3K/AKT [17, 18]. Мутации в белке NRAS часто обнаруживают при миелодиспластическом синдроме (МДС), а также у пациентов со вторичными ОМЛ, которые развиваются после МДС. Прогностическое значение этих мутаций при ОМЛ не определено [17]. У детей с ОМЛ мутации в гене RAS часто сочетаются с благоприятными мутациями в гене NPM1, и их присутствие не ухудшает прогноз заболевания [19].
Соматические мутации в кодоне 132 гена IDH1 обнаружены в образцах 2 (М6) и 8 (ОМЛ). Гены IDH1/IDH2 кодируют изоцитратдегидрогеназы (IDH) 1 и 2 ‒ NADP-зависимые ферменты цикла Кребса, катализирующие реакцию превращения изоцитрата в α-кетоглутарат. IDH1 локализуется в ядре, а IDH2 в митохондриях. Мутации в генах IDH1 и IDH2, характерные для взрослых пациентов с ОМЛ, обнаруживают в 8–16% и 12–15% случаев ОМЛ соответственно [19]. Мутации в генах IDH1/2 приводят к образованию 2-гидроксиглутарата [20, 21], что нарушает процессы эпигенетической регуляции, опосредованной метилированием ДНК и модификацией гистонов. Данные о прогностической значимости мутаций в генах IDH1/2 противоречивы. Интересно, что мутации в генах IDH1/2 не характерны для ОМЛ у детей [1, 21].
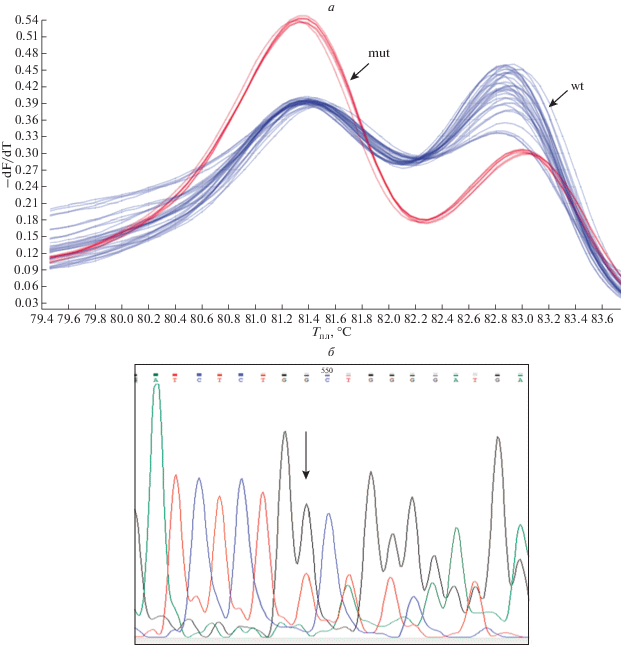
Анализ данных массового параллельного секвенирования, проведенный нами, не выявил мутации в гене NPM1, несмотря на то что этот ген наиболее часто мутирует при ОМЛ-НК [22]. Детальный анализ числа прочтений последовательности экзона 12 гена NPM1 в образцах пациентов, выполненный с помощью биоинформатического ресурса Integrative Genomics Viewer (IGV), показал, что во многих случаях этот участок практически не представлен в пуле последовательностей. Скорее всего, это обусловлено неудачным подбором зондов для гибридизации при приготовлении библиотек ДНК. Поэтому нами проведен поиск мутаций в гене NPM1 методом анализа кривых плавления высокого разрешения (HRM). Результаты HRM-анализа представлены на рис. 2а. Также секвенированы образцы ДНК двух групп пациентов с различными генотипами. Мутации в гене NPM1 обнаружены у трех пациентов из 34 (8.8%) (рис. 2б).
Рис. 2.
Определение мутаций в гене NPM1. а ‒ Анализ кривых плавления высокого разрешения (HRM, high resolution melting) экзона 12 гена NPM1 (стрелками указаны генотипы дикого типа и мутантные). б – Секвенирование по Сэнгеру (стрелка указывает на вставку четырех нуклеотидов TCTG). mut – мутация; wt – дикий тип.
Во многих образцах выявлены редкие мутации в генах CUX1, EZH2, FLT3, NUP98, PTPN11, TET2. В нашей выборке образцы костного мозга или крови представляли собой субстрат опухоли, поэтому отнесение мутации к герминальному или соматическому варианту носит характер предположения. Мутация в гене EZH2 (rs397515548) (представленность в образце 86%), скорее всего соматическая, так как биаллельные (или гомозиготные) мутации в этом гене характерны для ОМЛ [23]. Мутации в гене PTPN11 ассоциированы с синдромом Нунана, который характеризуется высоким риском развития лейкозов у носителей патогенных мутаций [24]. Мутацию p.Y63C (rs121918459) в гене PTPN11, выявленную в нашем исследовании, скорее всего следует отнести к соматическим, так как ее представленность в образце составляет 28% при отсутствии у пациента других фенотипических проявлений синдрома Нунана. Мутации в генах FLT3 (rs146030737), NUP98 (rs149360555), PTPN11 (rs397507534), TET2 (rs111948941, rs146031219, rs373501577) описаны ранее как герминальные варианты, которые встречаются как у пациентов с ОМЛ, так и в выборках здоровых доноров [25]. Тот факт, что в небольшой выборке из 34 человек обнаружены генетические варианты с частотой менее 1%, может свидетельствовать в пользу их участия в формировании генетической предрасположенности к миелопролиферативным заболеваниям. Дальнейшие исследования, проведенные на выборках большего размера, позволят ответить на эти вопросы.
Работа выполнена при финансовой поддержке Российского научного фонда (№ 18-15-00398).
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Schuback H.L., Arceci R.J., Meshinchi S. (2013) Somatic characterization of pediatric acute myeloid leukemia using next-generation sequencing. Semin. Hematol. 50(4), 325‒332.
Grimwade D. (2012) The changing paradigm of prognostic factors in acute myeloid leukaemia. Best Pract. Res. Clin. Haematol. 25(4), 419–425.
Renneville A., Roumier C., Biggio V., Nibourel O., Boissel N., Fenaux P., Preudhomme C. (2008) Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 22(5), 915–931.
Patel J.P., Gönen M., Figueroa M.E., Fernandez H., Sun Z., Racevskis J., Van Vlierberghe P., Dolgalev I., Thomas S., Aminova O., Huberman K., Cheng J., Viale A., Socci N.D., Heguy A., Cherry A., Vance G., Higgins R.R., Ketterling R.P., Gallagher R.E., Litzow M., van den Brink M.R., Lazarus H.M., Rowe J.M., Luger S., Ferrando A., Paietta E., Tallman M.S., Melnick A., Abdel-Wahab O., Levine R.L., Patel J.P., Gönen M., Figueroa M.E., Fernandez H., Sun Z., Racevskis J., Van Vlierberghe P., Dolgalev I., Thomas S., Aminova O., Huberman K., Cheng J., Viale A., Socci N.D., Heguy A., Cherry A., Vance G., Higgins R.R., Ketterling R.P., Gallagher R.E., Litzow M., van den Brink M.R., Laza-rus H.M., Rowe J.M., Luger S., Ferrando A., Paietta E., Tallman MS., Melnick A., Abdel-Wahab O., Levine R.L., Patel J.P., Gönen M., Figueroa M.E., Fernandez H., Sun Z., Racevskis J., Van Vlierberghe P., Dolgalev I., Thomas S., Aminova O., Huberman K., Cheng J., Viale A., Socci N.D., Heguy A., Cherry A., Vance G., Higgins R.R., Ketterling R.P., Gallagher R.E., Litzow M., van den Brink M.R., Lazarus H.M., Rowe J.M., Luger S., Ferrando A., Paietta E., Tallman M.S., Melnick A., Abdel-Wahab O., Levine R.L. (2012) Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 366(12), 1079–1089.
Döhner H., Estey E.H., Amadori S., Appelbaum F.R., Büchner T., Burnett A.K., Dombret H., Fenaux P., Grimwade D., Larson R.A., Lo-Coco F., Naoe T., Niederwieser D., Ossenkoppele G.J., Sanz M.A., Sierra J., Tallman M.S., Löwenberg B., Bloomfield C.D. (2010) Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 115(3), 453–474.
Schlenk R.F., Döhner K., Krauter J., Fröhling S., Corbacioglu A., Bullinger L., Habdank M., Späth D., Morgan M., Benner A., Schlegelberger B., Heil G., Ganser A., Döhner H. (2008) Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 358(18), 1909–1918.
Falini B., Mecucci C., Tiacci E., Alcalay M., Rosati R., Pasqualucci L., La Starza R., Diverio D., Colombo E., Santucci A., Bigerna B., Pacini R., Pucciarini A., Liso A., Vignetti M., Fazi P., Meani N., Pettirossi V., Saglio G., Mandelli F., Lo-Coco F., Pelicci P.G., Martelli M.F. (2005) Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 352(3), 254–266.
Ozeki K., Kiyoi H., Hirose Y., Iwai M., Ninomiya M., Kodera Y., Miyawaki S., Kuriyama K., Shimazaki C., Akiyama H., Nishimura M., Motoji T., Shinagawa K., Takeshita A., Ueda R., Ohno R., Emi N., Naoe T. (2004) Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood. 103(5), 1901–1908.
Nerlov C. (2004) C/EBPα mutations in acute myeloid leukaemias. Nat. Rev. Cancer. 4(5), 394–400.
Наседкина Т.В., Иконникова А.Ю., Цаур Г.А., Каратеева A.Ю., Амур Ю.И., Авдонина М.А., Карачунский А.И., Заседателев А.С. (2016) Биологический микрочип для определения структуры химерных транскриптов с участием гена MLL у детей, больных острыми лейкозами. Молекуляр. биология. 50(6), 968–977.
Taskesen E., Bullinger L., Corbacioglu A., Sanders M.A., Erpelinck C.A., Wouters B.J., van der Poel-van de Luytgaarde S.C., Damm F., Krauter J., Ganser A., Schlenk R.F., Löwenberg B., Delwel R., Döhner H., Valk P.J., Döhner K. (2011) Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 117(8), 2469–2475.
Cagnetta A., Adamia S., Acharya C., Patrone F., Miglino M., Nencioni A., Gobbi M., Cea M. (2014) Role of genotype-based approach in the clinical management of adult acute myeloid leukemia with normal cytogenetics. Leuk. Res. 38(6), 649–659.
Pastore F., Kling D., Hoster E., Dufour A., Konstandin N.P., Schneider S., Sauerland M.C., Berdel W.E., Buechner T., Woermann B., Braess J., Hiddemann W., Spiekermann K. (2014) Long-term follow-up of cytogenetically normal CEBPA-mutated AML. J. Hematol. Oncol. 7(1), 55.
Barjesteh van Waalwijk van Doorn-Khosrovani S., Spensberger D., de Knegt Y., Tang M., Löwenberg B., Delwel R. (2005) Somatic heterozygous mutations in ETV6 (TEL) and frequent absence of ETV6 protein in acute myeloid leukemia. Oncogene. 24, 4129‒4137.
de Noronha T.R., Mitne-Neto M., Chauffaille M.L. (2017) Mutational profiling of acute myeloid leukemia with normal cytogenetics in Brazilian patients: the value of next-generation sequencing for genomic classification. J. Investig. Med. 65(8), 1155‒1158.
Hidalgo-López J.E., Kanagal-Shamanna R., Medeiros L.J., Estrov Z., Yin C.C., Verstovsek S., Konoplev S., Jorgensen J.L., Mohammad M.M., Miranda R.N., Zhao C., Lee J., Zuo Z., Bueso-Ramos C.E. (2017) Morphologic and molecular characteristics of de novo AML with JAK2 V617F mutation. J. Natl. Compr. Canc. Netw. 15(6), 790‒796.
Johnson D.B., Smalley K.S., Sosman J.A. (2014) Molecular pathways: targeting NRAS in melanoma and acute myelogenous leukemia. Clin. Cancer Res. 20(16), 4186–4192.
Bacher U., Haferlach T., Schoch C., Kern W., Schnittger S. (2006) Implications of NRAS mutations in AML: a study of 2502 patients. Blood. 107(10), 3847–3853.
Berman J.N., Gerbing R.B., Alonzo T.A., Ho P.A., Miller K., Hurwitz C., Heerema N.A., Hirsch B., Raimondi S.C., Lange B., Franklin J.L., Gamis A., Meshinchi S. (2011) Prevalence and clinical implications of NRAS mutations in childhood AML: a report from the Children’s Oncology Group. Leukemia. 25(6), 1039–1042.
Marcucci G., Maharry K., Wu Y.Z., Radmacher M.D., Mrózek K., Margeson D., Holland K.B., Whitman S.P., Becker H., Schwind S., Metzeler K.H., Powell B.L., Carter T.H., Kolitz J.E., Wetzler M., Carroll A.J., Baer M.R., Caligiuri M.A., Larson R.A., Bloomfield C.D. (2010) IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J. Clin. Oncol. 28(14), 2348–2355.
Chotirat S., Thongnoppakhun W., Wanachiwanawin W., Auewarakul C.U. (2015) Acquired somatic mutations of isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2) in preleukemic disorders. Blood Cells Mol. Dis. 54(3), 286–291.
Paschka P., Schlenk R.F., Gaidzik V.I., Habdank M., Krönke J., Bullinger L., Späth D., Kayser S., Zucknick M., Götze K., Horst H.A., Germing U., Döhner H., Döhner K. (2010) IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 28(22), 3636–3643.
Makishima H., Jankowska A.M., Tiu R.V., Szpurka H., Sugimoto Y., Hu Z., Saunthararajah Y., Guinta K., Keddache M.A., Putnam P., Sekeres M.A., Moliterno A.R., List A.F., McDevitt M.A., Maciejewski J.P. (2010) Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. 24, 1799‒1804.
Maheshwari M., Belmont J., Fernbach S., Ho T., Molinari L., Yakub I., Yu F., Combes A., Towbin J., Craigen W.J., Gibbs R. (2002) PTPN11 mutations in Noonan syndrome type I: detection of recurrent mutations in exons 3 and 13. Hum. Mutat. 20(4), 298‒304.
Bodian D.L., McCutcheon J.N., Kothiyal P., Huddleston K.C., Iyer R.K., Vockley J.G., Niederhuber J.E. (2014) Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing. PLoS One. 9(4), e94554.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология