

Молекулярная биология, 2019, T. 53, № 4, стр. 685-691
Подготовка образцов скелетной мышцы человека для протеомных исследований с использованием изобарической метки iTRAQ
Д. В. Попов a, b, *, О. Л. Виноградова a, b, В. Г. Згода c
a Институт медико-биологических проблем Российской академии наук
123007 Москва, Россия
b Факультет фундаментальной медицины Московского государственного университета им. М.В. Ломоносова
119192 Москва, Россия
c Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича
119121 Москва, Россия
* E-mail: danil-popov@yandex.ru
Поступила в редакцию 22.01.2019
После доработки 22.01.2019
Принята к публикации 04.02.2019
Аннотация
Использование масс-спектрометрических методов для изучения молекулярных механизмов, регулирующих метаболизм скелетных мышц, вызывает повышенный интерес. Большая представленность нескольких сократительных белков в скелетных мышцах значительно ограничивает глубину панорамного протеомного анализа. Использование изобарических меток при масс-спектрометрическом анализе позволяет улучшить оценку изменений содержания белков и анализировать одновременно до 10 проб за один запуск. В представленной работе сопоставлены различные методы фракционирования пептидов, полученных из мышечной ткани человека и меченных изобарической меткой iTRAQ. Пробы m. vastus lateralis брали с помощью игольчатой биопсии у восьми молодых мужчин. Подготовку проб проводили после гидролиза белков и мечения пептидов изобарической меткой в трех вариантах: 1) очистка пептидов–ВЭЖХ-МС/МС; 2) очистка пептидов–изоэлектрофокусировка–ВЭЖХ-МС/МС; 3) фракционирование по гидрофобности в оснóвных условиях– ВЭЖХ-МС/МС. Фракционирование меченых пептидов по гидрофобности в оснóвных условиях оказалось оптимальным для увеличения глубины протеомного анализа. Такой способ подготовки проб, помимо сократительных и митохондриальных, позволяет значительно увеличить количество детектируемых регуляторных белков, а именно белков, связанных с нуклеиновыми кислотами, шаперонов, рецепторов и транскрипционных факторов.
ВВЕДЕНИЕ
Скелетные мышцы составляют более трети массы тела человека и играют ключевую роль в регуляции углеводно-жирового обмена. Нарушение метаболизма скелетных мышц играет важную роль в развитии таких заболеваний, как метаболический синдром, сахарный диабет второго типа, ожирение, а также заболеваний сердечно-сосудистой системы, депрессии и саркопении [1–4]. В последнее десятилетние возрастает интерес к изучению молекулярно-генетических механизмов, регулирующих метаболизм скелетных мышц. Одним из перспективных современных методов изучения белков считается масс-спектрометрический анализ, поэтому не удивительно, что в последние годы наблюдается линейный рост количества работ, в которых скелетные мышцы изучали с использованием этого метода (рис. 1а).
Рис. 1.
Количество публикаций, посвященных изучению протеома скелетных мышц (а), и популярность разных подходов, используемых для количественной оценки различий в содержании белков в масс-спектрометрии (б). Поиск в PubMed проведен по запросу [skeletal muscle AND proteom* AND mass spectrometr*] (а) и по запросу [(TMT OR iTRAQ) AND proteom*], [(label-free OR label free OR labelfree) AND proteom*], [SILAC AND proteom*] и [(SRM OR MRM) AND proteom*] (б).
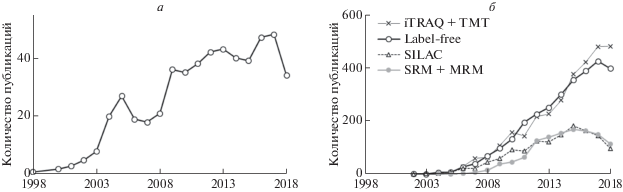
В масс-спектрометрии для количественной оценки различий в содержании белков в пробах используют различные подходы – без метки (Label-free) и с метками для белков и пептидов (SILAC, iTRAQ, TMT и др.). В настоящее время наиболее часто используют изобарические метки iTRAQ (isobaric Tags for Relative and Absolute Quantification; Sciex) и TMT (Tandem Mass Tag; Thermo Fisher Scientific) (рис. 1б). Преимущество изобарических меток заключается в улучшении оценки изменений в содержании белков и в возможности одновременного анализа нескольких проб (экспериментальных условий или повторов проб) за один запуск прибора (до восьми проб в случае iTRAQ и до 10 проб для TMT). Последнее особенно важно, поскольку позволяет значительно снизить вариативность, связанную с различиями в подготовке разных проб, с разными запусками прибора, а также кратно снизить время работы прибора. Поскольку метки изобарические, то при фрагментации родительского иона образуются (и детектируются) репортерные ионы, относящиеся ко всем меченым пробам. Это позволяет избегать пропуска данных по отдельным белкам, характерного для методов, в которых каждую пробу анализируют отдельно, и методов с ограниченным мультиплексированием, таких как SILAC.
Одним из факторов, лимитирующих количество детектируемых пептидов при панорамном протеомном анализе проб скелетных мышц, является то, что более 60% всех мышечных белков приходится на несколько высокопредставленных саркомерных и митохондриальных белков [5]. Снижение сложности анализируемой пробы за счет фракционирования смеси пептидов, например, с помощью изоэлектрофокусирования, позволяет значительно увеличить глубину панорамного протеомного анализа [5]. Мы предположили, что для дальнейшего повышения разрешения при фракционировании пептидов с изобарической меткой, полученных из пробы мышечной ткани, эффективным будет разделение смеси пептидов по гидрофобности в оснóвных условиях с помощью жидкостной хроматографии. Такой подход совместим с iTRAQ [6] и может быть эффективным не только для увеличения разрешения при фракционировании (и количества детектируемых пептидов и белков), но и для снижения эффекта занижения отношения репортерных ионов, характерного для iTRAQ, из-за коэлюции различных пептидов, совпадающих по времени удержания и m/z [7, 8].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пробы мышечной ткани брали из наружной головки четырехглавой мышцы бедра у восьми человек с помощью игольчатой микробиопсии под местной анестезией (2 мл 2% лидокаин), как описано нами ранее [9]. Пробу ткани немедленно помещали на фильтровальную бумагу для удаления избытка крови и замораживали в жидком азоте.
Кусочек замороженной ткани (15 мг) взвешивали и гомогенизировали в 10 объемах лизирующего буфера (4% додецилсульфата натрия в 0.1 M Трис-HCl pH 7.6, 0.1 M дитиотреитол) в 1.5 мл пробирке с помощью пластикового пестика, затем перемешивали на вортексе и инкубировали в течение 5 мин при 95°С. Пробы озвучивали (2 × 10 с при 100 Вт) с помощью УЗ-генератора и центрифугировали (5 мин, 16 000 g). Супернатант (8 мкл) наносили на фильтр YM-30 (“Millipore”, Ирландия) – FASP-метод [10], для алкилирования и трипсинолиза (12 ч, 2 мкг трипсина [Tripsin Gold, “Promega”, США] в 40 мкл 0.1 М бикарбоната тетраэтиламмония). Пептиды смывали с фильтра центрифугированием (10 мин, 14 000 g), затем на фильтр добавляли 40 мкл 0.1 М бикарбоната тетраэтиламмония и повторно центрифугировали.
Подготовку пептидов для масс-спектрометрии проводили в трех вариантах:
1) мечение пептидов iTRAQ–очистка пептидов–ВЭЖХ-МС/МС;
2) мечение пептидов iTRAQ–очистка пептидов–фракционирование (изоэлектрофокусировка)–ВЭЖХ-МС/МС;
3) мечение пептидов iTRAQ–фракционирование по гидрофобности в основных условиях– ВЭЖХ-МС/МС;
Мечение пептидов iTRAQ–очистка пептидов– ВЭЖХ-МС/МС. Концентрацию пептидов измеряли с помощью бицинхонинового метода, затем отбирали 30 мкг пептидов и концентрировали их на вакуумном концентраторе при 30°С до объема <4 мкл. В каждую метку из набора iTRAQ 8-plex добавляли 50 мкл изопропанола, 6 мкл 0.5 М бикарбоната тетраэтиламмония (все из набора iTRAQ 8-plex; окончательный объем около 65 мкл), затем 32 мкл раствора с меткой добавляли в пробирку с концентрированными пептидами. pH проверяли, нанося 0.3 мкл пробы на лакмусовую бумагу, при необходимости pH доводили до >7.5, добавляя 1–2 мкл 0.5 М бикарбоната тетраэтиламмония, а затем инкубировали в течение 2 ч. Затем объединяли восемь проб, концентрировали при 30°С до объема 70 мкл, добавляли трифторуксусную кислоту (ТФУК) до концентрации 0.1% и очищали на картридже C18-SD 3 ml SPE Cartridges (3 M Empore), как описано [10]. Пептиды последовательно смывали 0.45 мл 70 и 80% ацетонитрила, концентрировали при 30°С до 150 мкл и добавляли муравьиную кислоту до 0.1%. Очищенные пептиды трижды анализировали на ВЭЖХ-МС/МС.
Протеомный анализ пептидов осуществляли с использованием системы ВЭЖХ Ultimate 3000 RSLCnano (“Thermo Scientific”, США). Пептиды перед аналитическим разделением наносили на обогащающую колонку Accalaim μ-Precolumn (0.5 мм × 3 мм, размер частиц 5 мкм; “Thermo Scientific”) при скорости потока 10 мкл/мин в течение 5 мин в изократическом режиме подвижной фазы Б (2% ацетонитрила, 0.1% муравьиной кислоты). Затем пептиды разделяли на колонке Acclaim Pepmap C18 (75 мкм × 150 мм, размер частиц 2 мкм; “Thermo Scientific”) в градиентном режиме элюирования. Градиент формировали подвижными фазами А (0.1%-ная муравьиная кислота) и Б (80% ацетонитрила, 0.1% муравьиной кислоты) при скорости потока 0.3 мкл/мин. Колонку промывали 2%-ной подвижной фазой Б в течение 5 мин, после чего линейно увеличивали концентрацию подвижной фазы Б до 35% в течение 65 мин, затем за 5 мин линейно увеличивали концентрацию фазы Б до 99%. После промывки колонки (5 мин, 99% Б) концентрацию Б снижали до начальных условий – 2% фазы Б за 5 мин. Общая длительность анализа 90 мин.
Масс-спектрометрический анализ проводили на гибридном орбитальном масс-спектрометре Orbitrap Fusion (“Thermo Scientific”) в режиме положительной ионизации в источнике NESI (“Thermo Scientific”). Напряжение на эмиттере 2.1 кВ. Панорамное сканирование проводили в диапазоне масс от 400 до 1200 m/z, тандемное сканирование фрагментных ионов – от нижней границы 100 m/z до верхней границы, определяемой зарядным состоянием прекурсорного иона, но не более 2100 m/z. Для тандемного сканирования учитывали только ионы от z = 2+ до z = 6+ по зарядному состоянию. Максимальное число разрешенных для синхронной изоляции ионов в режиме MS2 не более 20. Максимальное время накопления прекурсорных ионов составило не более 50 мс, фрагментных ионов не более 110 мс.
Мечение пептидов iTRAQ–очистка пептидов– изоэлектрофокусировка–ВЭЖХ-МС/МС. Этапы мечения и очистки проводили, как описано выше. Затем пептиды концентрировали на вакуумном концентраторе при 30°С, растворяли в 200 мкл буфера (8 M мочевина, 0.1% [w/v] амфолины Bio-Lyte 3/10 (“Bio-Rad”), 0.001% бромфеноловый синий), наносили на дорожку в ванну для изоэлектрофокусировки и накрывали стрипом (11 cm 3–10 ReadyStrip IPG Strip, “Bio-Rad”) гелем вниз для пассивной регидратации в течение 12 ч. Изоэлектрофокусировку проводили с помощью Protean IEF Cell (“Bio-Rad”) при 20°C в режиме: 250 В в течение 30 мин, затем максимальное напряжение <8000 В до достижения 40 000 В ⋅ ч и удержание при 750 В.
Стрип нарезали стерильными лезвиями на 12 частей, каждую часть помещали в 1.5 мл пробирку. Пептиды экстрагировали из геля путем последовательной инкубации в трех буферах: 150 мкл 0.1% ТФУК в воде, 150 мкл 0.1% ТФУК в 35% ацетонитриле, 150 мкл 0.1% ТФУК в 70% ацетонитриле (20 мин каждый при медленном помешивании), после каждой отмывки раствор от каждой фракции объединяли в отдельной пробирке. Пептиды из каждой фракции концентрировали до <200 мкл, добавляли муравьиную кислоту до 0.1%, очищали на картридже С18 с помощью метода Stage Tips [11]. Затем пептиды концентрировали при 30°С до 20 мкл и анализировали на ВЭЖХ-МС/МС, как описано выше.
Мечение пептидов iTRAQ–фракционирование по гидрофобности в щелочных условиях–ВЭЖХ-МС/МС. Этап мечения проводили, как описано выше. Сразу после этого объединенную смесь меченых пептидов концентрировали до 30 мкл и фракционировали по гидрофобности в щелочных условиях. Фракционирование пептидов проводили на колонке XBridge C18 column (250 × 4.6 мм, размер частиц 5 мкм; “Waters”, Ирландия) с использованием ВЭЖХ Agilent 1200 Series, состоящей из бинарного насоса, термостата, колонок, автосемплера, УФ-детектора с диодной линейкой, коллектора фракций (“Agilent”, США). Образцы загружали на колонку в объеме 40 мкл, пептиды элюировали линейным градиентом подвижной фазы А (15 мМ ацетат аммония, pH 9.0) и мобильной фазы Б (ацетонитрил) при потоке 0.75 мл/мин. Градиент начинали с 2% фазы Б, концентрацию которой линейно увеличивали до 37% за 30 мин, затем резко повышали концентрацию Б до 92% за 3 мин и промывали колонку в течение 5 мин. На последнем этапе постепенно снижали концентрацию Б до начальных условий градиента (2% подвижной фазы Б) за 3 мин. Колонку уравновешивали при 2% мобильной фазы Б в течение 10 мин при потоке 0.75 мл/мин перед началом каждого разделения пептидов. В ходе анализа колонку термостатировали при 50°C. УФ-сигнал регистрировали при длине волны λ = 210 и 280 нм. Фракции собирали, начиная с 3 по 30 мин включительно с интервалом 1 мин. Собранные фракции высушивали с помощью вакуумного концентратора и растворяли в объеме 20 мкл, при необходимости объединяли и использовали для дальнейшего анализа. Пептиды из каждой фракции анализировали на ВЭЖХ-МС/МС, как описано выше.
Обработка результатов. Поиск, идентификацию пиков и оценку интенсивности пиков репортерных ионов проводили с использованием платформы MaxQuant со стандартными настройками при FDR 1%. В качестве фиксированных модификаций использовали модификации метками iTRAQ N-концевой аминогруппы и ε-аминогруппы лизина и карбамидометилирование цистеина, а для переменных модификаций – окисление метионина и ацилирование N-концевой аминогруппы белка. Эффективность мечения оценивали, проводя поиск и идентификацию пиков с такими же настройками, но в качестве фиксированных модификаций использовали: карбамидометилирование цистеина, а для переменных модификаций – окисление метионина и ацилирование N-концевой аминогруппы белка. Эффективность мечения оценивали как отношение количества пептидов, выявленных при поиске без метки, к количеству пептидов, детектированных при поиске с меткой.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Эффективность мечения во всех экспериментальных условиях превышала 95%.
При подготовке проб без фракционирования найдено 153 белка. Для снижения сложности проб (и увеличения количества обнаруженных белков) использованы различные методы фракционирования меченых пептидов. Максимальное количество белков – 981 – обнаружено при фракционировании по гидрофобности в оснóвных условиях. Количество детектированных уникальных пептидов и белков представлено в табл. 1.
Таблица 1.
Количество выявленных белков и уникальных пептидов
| Метод подготовки пробы | Уникальные пептиды | Белки |
|---|---|---|
| Мечение пептидов iTRAQ–ВЭЖХ-МС/МС | 936 | 153 |
| Мечение пептидов iTRAQ–изоэлектрофокусировка–ВЭЖХ-МС/МС | 1958 | 386 |
| Мечение пептидов iTRAQ–фракционирование по гидрофобности в оснóвных условиях–ВЭЖХ-МС/МС | 5969 | 981 |
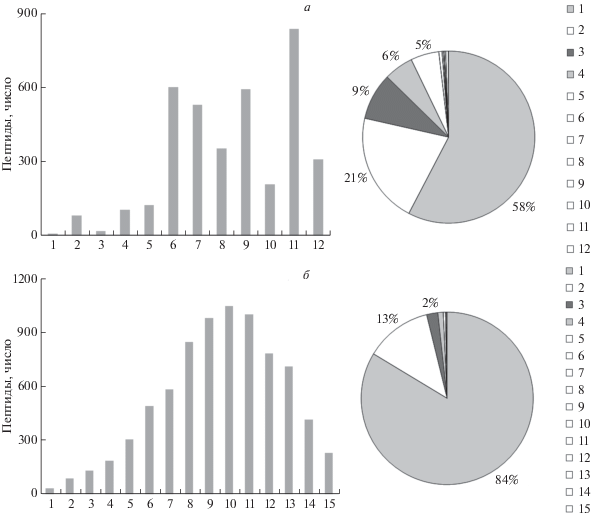
Наиболее эффективным для фракционирования меченых пептидов оказалось разделение по гидрофобности в оснóвных условиях: 84% всех пептидов обнаружено в одной из фракций, тогда как при изоэлектрофокусировке – 58% (рис. 2).
Рис. 2.
Эффективность фракционирования пептидов, меченных iTRAQ, с помощью изоэлектрофокусировки (а) и разделения по гидрофобности в оснóвных условиях (б). На гистограмме представлено распределение пептидов по фракциям. На круговой диаграмме показано содержание пептидов (%), обнаруженных только в одной, двух, трех и т.д. фракциях.
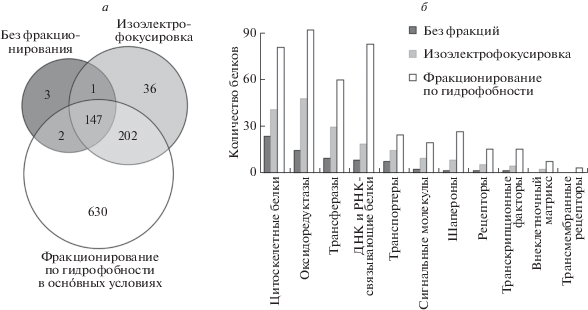
На рис. 3а представлено пересечение множеств белков, детектированных в различных экспериментальных условиях. Анализируемые множества сильно пересекались, т.е. увеличение количества обнаруженных белков при фракционировании связано с увеличением глубины прочтения, а не с обнаружением другого пула белков. С помощью инструмента PANTHER Protein Class показано, что фракционирование по гидрофобности в оснóвных условиях позволяет обнаружить не только высокопредставленные сократительные и митохондриальные белков, но и значительно увеличить количество выявляемых регуляторных белков, а именно, белков, связанных с нуклеиновыми кислотами, шаперонов, рецепторов и факторов транскрипции (рис. 3б).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Мы сравнивали количество белков скелетных мышц, детектируемых с использованием изобарической метки iTRAQ 8-plex, при различных способах подготовки проб. Как и ожидалось, наименьшее количество белков обнаружено при использовании проб, подготовленных без фракционирования. Это связано с тем, что большая часть всех детектированных в скелетной мышце белков относится к высокопредставленным белкам цитоскелета (в том числе к саркомерным) и к белкам окислительного фосфорилирования (рис. 3б), которые суммарно составляют более 60% от всех мышечных белков [5].
Фракционирование меченых пептидов по заряду и по гидрофобности в оснóвных условиях позволило значительно увеличить количество обнаруженных белков, причем последний подход оказался наиболее эффективным (табл. 1). Это согласуется с данными работ, в которых сопоставлены результаты, полученные двумя методами, при фракционировании немеченых пептидов [12, 13]. Эти данные показывают, что увеличение количества белков, обнаруженных при фракционировании по гидрофобности в оснóвных условиях, связано с большей эффективностью разделения пептидов, чем при использовании изоэлектрофокусировки (рис. 2). Следует отметить, что при отработке методики изоэлектрофокусировки меченых пептидов встречались проблемы, связанные с медленным выходом на целевое напряжение (8000 В) и перегревом стрипа. Возможно, дополнительная оптимизация условий изоэлектрофокусировки позволит преодолеть эти сложности и улучшить эффективность фракционирования.
Количество белков, детектированных нами при фракционировании меченых пептидов по гидрофобности в оснóвных условиях с использованием изобарической метки, было сопоставимым с результатами других исследований, в которых протеом скелетных мышц человека изучали без использования меток (см. обзоры [14, 15]). Следует отметить, что наименьшее количество пептидов выявлено нами в наиболее гидрофобных и гидрофильных фракциях (рис. 2б). По-видимому, дальнейшая стратегия оптимизации этой методики может быть связана с объединением нескольких наиболее гидрофобных (и наиболее гидрофильных) фракций перед масс-спектрометрией с одновременным увеличением количества промежуточных фракций.
Таким образом, масс-спектрометрический анализ пептидов, полученных из скелетной мышцы человека и меченных изобарической меткой iTRAQ, позволяет обнаружить менее двух сотен белков, что связано с большой долей высокопредставленных сократительных белков и белков окислительного фосфорилирования. Оптимальной стратегией для увеличения глубины протеомного анализа оказалось фракционирование меченых пептидов по гидрофобности в оснóвных условиях.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований в рамках научного проекта № 17-00-00308 (17-00-00242).
Проведение исследования одобрено комиссией по биомедицинской этике ГНЦ РФ-ИМБП РАН, все добровольцы дали информированное письменное согласие на участие в исследовании.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Pedersen B.K., Febbraio M.A. (2012) Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 8(8), 457–465.
Demontis F., Piccirillo R., Goldberg A.L., Perrimon N. (2013) The influence of skeletal muscle on systemic aging and lifespan. Aging Cell. 12(6), 943–949.
Agudelo L.Z., Femenia T., Orhan F., Porsmyr-Palmertz M., Goiny M., Martinez-Redondo V., Correia J.C., Izadi M., Bhat M., Schuppe-Koistinen I., Pettersson A.T., Ferreira D.M.S., Krook A., Barres R., Zierath J.R., Erhardt S., Lindskog M., Ruas J.L. (2014) Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell. 159(1), 33–45.
Cervenka I., Agudelo L.Z., Ruas J.L. (2017) Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science. 357(6349), pii: eaaf9794. https://doi.org/10.1126/science.aaf9794
Deshmukh A.S., Murgia M., Nagaraj N., Treebak J.T., Cox J., Mann M. (2015) Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol. Cell Proteomics. 14(4), 841–853.
Yang Y., Qiang X., Owsiany K., Zhang S., Thannhauser T.W., Li L. (2011) Evaluation of different multidimensional LC-MS/MS pipelines for isobaric tags for relative and absolute quantitation (iTRAQ)-based proteomic analysis of potato tubers in response to cold storage. J. Proteome. Res. 10(10), 4647–4660.
Evans C., Noirel J., Ow S.Y., Salim M., Pereira-Medrano A.G., Couto N., Pandhal J., Smith D., Pham T.K., Karunakaran E., Zou X., Biggs C.A., Wright P.C. (2012) An insight into iTRAQ: where do we stand now? Anal. Bioanal. Chem. 404(4), 1011–1027.
Rauniyar N., Yates J.R., 3rd. (2014) Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome. Res. 13(12), 5293–5309.
Popov D.V., Lysenko E.A., Bokov R.O., Volodina M.A., Kurochkina N.S., Makhnovskii P.A., Vyssokikh M.Y., Vinogradova O.L. (2018) Effect of aerobic training on baseline expression of signaling and respiratory proteins in human skeletal muscle. Physiological Rep. 6(17), e13868.
Wisniewski J.R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods. 6(5), 359–362.
Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2(8), 1896–1906.
Stein D.R., Hu X., McCorrister S.J., Westmacott G.R., Plummer F.A., Ball T.B., Carpenter M.S. (2013) High pH reversed-phase chromatography as a superior fractionation scheme compared to off-gel isoelectric focusing for complex proteome analysis. Proteomics. 13(20), 2956–2966.
Wang H., Sun S., Zhang Y., Chen S., Liu P., Liu B. (2015) An off-line high pH reversed-phase fractionation and nano-liquid chromatography-mass spectrometry method for global proteomic profiling of cell lines. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 974, 90–95.
Gonzalez-Freire M., Semba R.D., Ubaida-Mohien C., Fabbri E., Scalzo P., Hojlund K., Dufresne C., Lyashkov A., Ferrucci L. (2017) The Human Skeletal Muscle Proteome Project: a reappraisal of the current literature. J. Cachexia. Sarcopenia. Muscle. 8(1), 5–18.
Capitanio D., Moriggi M., Gelfi C. (2017) Mapping the human skeletal muscle proteome: progress and potential. Expert. Rev. Proteomics. 14(9), 825–839.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология