

Молекулярная биология, 2019, T. 53, № 4, стр. 574-599
Генетические маркеры персонализированной терапии полигенных заболеваний: фармакогенетика рассеянного склероза
Е. Ю. Царева a, О. О. Фаворова a, А. Н. Бойко a, О. Г. Кулакова a, *
a Российский национальный исследовательский медицинский университет им. Н.И. Пирогова
Министерства здравоохранения Российской Федерации
117997 Москва, Россия
* E-mail: olga.koulakova@gmail.com
Поступила в редакцию 11.03.2019
После доработки 18.03.2019
Принята к публикации 18.03.2019
Аннотация
Фармакогенетика – наука, изучающая наследственные варианты генома человека, которые обусловливают индивидуальные различия в процессах метаболического превращения лекарственного средства, его доставки и механизма действия. В случае полигенных заболеваний, к которым относится большинство болезней человека, необходимо учитывать не только вклад отдельных вариантов генов, но и их кумулятивный эффект. Рассеянный склероз (РС) – тяжелейшее аутоиммунное нейродегенеративное заболевание центральной нервной системы полигенной природы. Понимание роли иммунной системы в патогенезе РС привело к разработке препаратов для его патогенетической терапии, получивших общее название “препараты, изменяющие течение РС”. Эффективность двух таких препаратов – интерферона-β и глатирамера ацетата – и их безопасность в долгосрочной перспективе была доказана в многочисленных клинических испытаниях, однако их влияние на течение РС варьирует у разных пациентов в диапазоне от высоко эффективного до отсутствия ответа. Выявление предиктивных генетических биомаркеров эффективности их лечения дает возможность определять группы больных РС, которым предпочтительно (или даже строго показано) назначать тот или другой препарат. В этом обзоре представлены данные фармакогенетических исследований, посвященных изучению эффективности лечения больных РС препаратами интерферона-β и глатирамера ацетата, включая собственные результаты авторов.
ВВЕДЕНИЕ
Одним из важнейших достижений молекулярной медицины XXI века, в постгеномную эру, стало развитие концепции предиктивной, превентивной и персонализированной медицины, которая представляет собой совокупность методов прогноза и профилактики заболеваний, их диагностики и подбора соответствующей терапии, основанных на индивидуальных особенностях пациента. Методы оценки индивидуальных особенностей человека основаны на всей совокупности геномных (“омиксных”) технологий, среди которых ключевое значение имеет выявление генетических маркеров.
Согласно современному определению, фармакогенетика (ФГ) – это наука, изучающая варианты последовательностей генома человека, которые обусловливают индивидуальные различия в процессах метаболического превращения лекарственного средства, его доставки и механизма действия, что, в конечном счете, приводит к различиям в ответе организма на действие лекарств. Становление ФГ как науки связано с именами Werner Kalow, Friedrich Vogel и Arno Motulsky, которые с конца 50-х годов XX века начали развивать представления о том, что в основе формирования индивидуальных различий в способности метаболизировать синтетические лекарственные препараты (ксенобиотики) и/или развивать неблагоприятные побочные реакции может лежать генетическая вариабельность (см. [1]). Бурное развитие ФГ, начиная с 60-х годов XX века, было связано с исследованием побочных явлений в ответ на введение ксенобиотиков, а также с изучением индивидуальных особенностей фармакокинетических процессов (всасывание препарата, его распределение, метаболизм и выведение) и фармакодинамики (механизм действия, эффективность и токсичность препарата). Обусловленные характером функционирования различных ферментов, структурных и транспортных белков, рецепторов, ионных каналов, систем вторичных мессенджеров и т.д., эти процессы находятся под строгим генетическим контролем [2]. Важная составляющая ФГ – популяционные исследования, которые выявили расовые, этнические и географические различия в реакциях на лекарства [3].
Если на начальных этапах ФГ-исследования касались в основном моногенных заболеваний, то большинство современных исследований направлено на анализ эффективности лечения полигенных заболеваний, обусловленных наследственной предрасположенностью с неменделевским типом наследования. К числу полигенных относится большинство заболеваний человека: аутоиммунные, сердечно-сосудистые, неврологические, онкологические и многие другие. Важно отметить, что, как правило, эти заболевания требуют длительного, а иногда и пожизненного лечения, часто сопряженного с развитием тяжелых побочных эффектов. На этом фоне возможность предсказания характера действия препарата на основании анализа вариантов генома приобретает исключительную важность.
Вклад генетических факторов в вариабельность ответа на лекарства может составлять от 20 до 95% [4]. Генетическая вариабельность во многом основана на явлении генетического полиморфизма. К полиморфным вариантам генома относят однонуклеотидные замены (single nucleotide polymorphism, SNP), которые определяют 85–90% вариабельности генома, а также инсерционно-делеционные мутации, вариации числа копий отдельных участков генома (copy number variations) размером от 1000 п.н. и более, тандемные повторы различной длины (мини- и микросателлиты). Полиморфизмы могут затрагивать различные области генома. Мутации в кодирующей области гена могут изменять аминокислотную последовательность белка и приводить к изменению структуры и функции белкового продукта, а в случае синонимических замен кодирующих триплетов (так называемые “молчащие” полиморфизмы) влиять на сплайсинг, пространственную структуру и стабильность мРНК или же на скорость трансляции [5]. К такому же результату, что и синонимические замены кодонов, может приводить генетическая вариабельность транскрибируемой некодирующей области гена (интроны, 5'- и 3'-концевые области). В последние годы из результатов проекта ENCODE (Encyclopedia of DNA Elements) стало ясно, что более 80% генома обладает определенной биологической функцией, во многом связанной с регуляцией экспрессии генов, кодирующих белки. Среди выявленных функциональных элементов наиболее представлены гены, кодирующие регуляторные РНК различного типа [6]. Как следствие, анализ генетической вариабельности сместился в сторону исследования полиморфизмов, находящихся в регуляторных областях генов (сайты связывания с транскрипционными факторами, энхансеры, сайленсеры) и в генах регуляторных РНК, то есть вариантов, влияющих на экспрессию гена.
При ФГ-исследованиях полигенных заболеваний для выявления генетических маркеров, обладающих предиктивным эффектом, используют различные методологические подходы, в первую очередь, подход “ген-кандидат”, когда выбор анализируемых генов осуществляют исходя из функциональной роли их белковых продуктов. Традиционно основными генами-кандидатами при ФГ-исследованиях служили гены, продукты которых играют роль в процессах метаболического превращения лекарственного средства и его доставки либо участвуют в механизмах реализации эффекта препарата. В последние годы в ФГ-исследованиях полигенных заболеваний активно используется подход, основанный на полногеномном поиске ассоциаций (GWAS), что позволяет исследовать генетическую архитектуру этих заболеваний, не прибегая к гипотезам об их этиопатогенезе (hypothesis-free approach) [7]. Благодаря развитию методов высокопроизводительного секвенирования, GWAS позволяет одновременно проводить анализ тысяч полиморфных маркеров. Однако из-за высоких требований к уровню значимости при анализе методом GWAS (величины р < 5 × 10–8), обусловленных множественностью сравнений, воспроизводимость результатов невелика.
На сегодняшний день ФГ-анализ прочно вошел в медицинскую практику. Для некоторых введенных ранее лекарственных препаратов проведены ретроспективные ФГ-исследования, основанные на предшествующих клинических испытаниях [8, 9], а в протоколы многих текущих клинических испытаний включен последующий ФГ-анализ. В таких странах как США, Канада и Великобритания одобрен соответствующими комиссиями по контролю за пищевыми продуктами и медикаментами ряд ФГ-алгоритмов, которые учитывают генетический статус пациента при назначении конкретных лекарственных препаратов. Это распространяется на лечение миелодиспластического синдрома, опухолей молочной железы, гастроинтестинальных стромальных и некоторых других опухолей, СПИДа, ревматоидного артрита, эпилепсии, шизофрении, депрессии, артериальной гипертензии и др. [10]. По результатам анализа ассоциации полиморфизма генов CYP2C9 и VCORC1 с риском развития геморрагических осложнений при антикоагулянтной терапии варфарином рекомендуется осуществлять индивидуальный подбор дозы препарата [11–13]; при этом учитывается этническая принадлежность пациентов, поскольку индивидуальные особенности процессов обмена веществ в организме представителей разных рас сильно отличаются.
Таким образом, ФГ-анализ, учитывающий уникальные генетические особенности пациента и индивидуальные клинико-патогенетические особенности течения заболевания, создает возможность с большой долей вероятности предсказывать чувствительность конкретного индивида к исследуемому препарату и назначать ему оптимальный препарат или дозу препарата на самых ранних этапах лечения.
В последние 15 лет ФГ-исследования стали активно проводиться в отношении одного из тяжелых инвалидизирующих аутоиммунных неврологических заболеваний – рассеянного склероза (РС). РС, как правило, развивается в молодом возрасте, а в последнее время все чаще диагностируется у детей и подростков. Распространенность РС в мире составляет порядка 2.5 млн человек [14 ] , и его лечение остается одной из наиболее серьезных проблем неврологии. Благодаря углублению знаний о патогенетических механизмах РС разработаны эффективные препараты, изменяющие течение РС (ПИТРС). В настоящее время существует более 10 разрешенных ПИТРС с разной степенью эффективности лечения [15]. Среди них интерферон-β (ИФН-β) и глатирамера ацетат (ГА) в долгосрочной перспективе проявили себя как наиболее безопасные и остаются препаратами первой линии для лечения РС во всем мире. Предлагаемый обзор посвящен ФГ-исследованиям оценки эффективности применения этих препаратов у больных РС.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ РАССЕЯННОГО СКЛЕРОЗА
РС – хроническое аутоиммунное демиелинизирующее заболевание центральной нервной системы (ЦНС), которым в России страдает около 200 тыс. человек. В ряде регионов России заболеваемость РС довольно высока и находится в пределах 35–70 случаев на 100 тыс. населения [16]. Помимо наиболее распространенной ремиттирующей формы течения РС (РРС), которая характеризуется обострениями и ремиссиями и в большинстве случаев со временем переходит во вторичное прогрессирование, у 10–15% пациентов с самого начала наблюдается первично-прогрессирующий тип течения РС с быстрым нарастанием инвалидности [17].
В настоящее время РС рассматривают как многофакторное полигенное заболевание, развивающееся при воздействии внешних факторов у генетически предрасположенных индивидов [18]. Внешние воздействия запускают каскад патологических процессов, приводящих к необратимой потере неврологических функций [19]. В качестве внешних факторов, связанных с повышенным риском развития РС, чаще всего называют различные инфекционные агенты, и в первую очередь, некоторые вирусы (вирус простого герпеса типа VI, вирус Эпштейна–Барр, некоторые ретро- и полиомавирусы и др.), а также климатические и географические условия, пониженное содержание витамина D, неблагоприятную экологическую обстановку и изменение содержания микроэлементов в воде [20, 21].
Этиология РС остается до конца не выясненной, хотя многочисленные исследования, а также успехи иммуномодулирующей и иммуносупрессивной терапии позволяют считать, что в основе развития РС большую роль играет аутоиммунный процесс, опосредованный, в первую очередь, Т-клетками. Это способствует развитию в ЦНС воспалительного ответа, приводящего к разрушению миелиновой оболочки нервного волокна в ЦНС (демиелинизации), гибели олигодендроцитов, формирующих миелиновую оболочку, разрушению аксонов, глиозу и последующей нейродегенерации [22, 23] (рис. 1).
Рис. 1.
Основные этапы иммунопатогенеза рассеянного склероза. Описание см. в тексте. АПК – антигенпрезентирующая клетка; ФНО – фактор некроза опухолей; ИФН-γ – интерферон-гамма; Th – Т-хелперы.
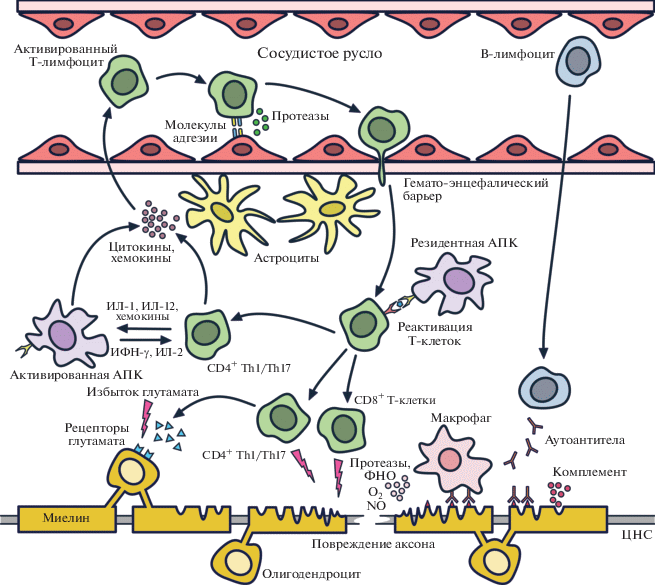
Необходимое условие развития аутоиммунного повреждения – активация анергичных аутореактивных Т-лимфоцитов (в первую очередь CD4+) на периферии. Такую активацию могут запускать микробные суперантигены или собственные антигены, иммуногенность которых повышена вследствие различных причин, в частности, при хроническом воспалении [24]. Одновременно с активацией Т-клеток происходит и нарушение В-клеточной толерантности, сопровождающееся нарастанием титров антител к различным структурам миелина и олигодендроглии [25]. Активированные Тh1- и Th17-клетки, специфичные к компонентам миелина, проникают через гемато-энцефалический барьер (ГЭБ) в ЦНС и там реактивируются резидентными антигенпрезентирующими клетками (АПК), презентирующими антигены ЦНС. Это запускает новую волну воспаления, при которой образуются многочисленные цитокины, хемокины и другие молекулы, такие как NO, глутамат и свободные радикалы. В дальнейшие процессы демиелинизации активно вовлекаются CD8+ Т-лимфоциты и В-лимфоциты [26, 27], что в конечном итоге приводит к повреждению миелиновой оболочки, олигодендроцитов и нейронов.
Нейродегенеративные процессы в патогенезе РС, отчетливо проявляющиеся по мере прогрессирования заболевания, связаны с повреждением аксонов нейронов, которое наблюдается не только в острых и хронических активных очагах, но и в неизменном белом веществе. По-видимому, демиелинизированные аксоны становятся уязвимы для таких продуцируемых иммунными и глиальными клетками факторов, как различные протеолитические ферменты, цитокины, активные формы кислорода [19].
В целом, на основании данных последних лет можно говорить о гетерогенности механизмов патогенеза РС, включающих триаду “воспаление, демиелинизация и дегенерация”, которая реализуется с прямым участием иммунной системы [22].
ИММУНОМОДУЛИРУЮЩИЕ ПРЕПАРАТЫ ИНТЕРФЕРОН-β И ГЛАТИРАМЕРА АЦЕТАТ В ТЕРАПИИ БОЛЬНЫХ РС
Иммуномодулирующие препараты ИФН-β и ГА стали первыми зарегистрированными ПИТРС, введенными в практику лечения РС (в 1993 г. и в 1998 г. соответственно) [28]. Терапевтический эффект ИФН-β и ГА на активность РС проявляется в уменьшении частоты и тяжести обострений, снижении скорости прогрессирования РС по шкале инвалидизации (EDSS) и задержке возникновения новых очагов демиелинизации в головном мозге (так называемых бляшек) по данным магнитно-резонансной томографии (МРТ). Эффективность этих препаратов при лечении РРС, а ИФН-β также при лечении вторично-прогрессирующего РС с обострениями и клинически изолированного синдрома (так называется первый эпизод клинического проявления симптомов РС) продемонстрирована в международных мультицентровых рандомизированных клинических испытаниях с использованием двойного слепого метода [29]. Длительное наблюдение за пациентами, получающими эти препараты, подтвердило, что появилась возможность вмешиваться в механизмы развития патологического процесса, снижая активность воспалительного и аутоиммунного процессов, приводящих к демиелинизации и аксональной дегенерации. Остановимся на основных механизмах действия ИФН-β и ГА.
Механизмы действия интерферона-β при лечении РС
Интерфероны играют ключевую роль в регуляторных механизмах врожденного иммуннитета и обладают широким спектром биологического действия. ИФН-β – плейотропный цитокин, относящийся к ИФН типа I. Его биологическое действие, как и действие всех ИФН этого типа, опосредовано взаимодействием с гетеродимерным клеточным рецептором IFNAR, который состоит из двух субъединиц: IFNAR1 и IFNAR2 [30]. Это взаимодействие активирует сигнальный путь JAK-STAT, что в итоге приводит к активации транскрипции множества ИФН-индуцибельных генов.
Хотя точные механизмы терапевтического эффекта ИФН-β при РС до конца не выяснены, описанный спектр его действия весьма широк. Показано, что ИФН-β может ингибировать пролиферацию и активацию аутореактивных Т-клеток за счет подавления экспрессии HLA-антигенов класса II и изменения экспрессии некоторых костимулирующих молекул на поверхности АПК [31]. Также ИФН-β повышает чувствительность Т-лимфоцитов к апоптотическим сигналам, например, через усиление экспрессии поверхностной молекулы Fas; в результате аутореактивные клетки больных РС подвергаются апоптозу [32]. В ряде исследований in vitro и in vivo показано, что при действии ИФН-β наблюдается изменение баланса цитокинов в сторону уменьшения продукции различными клетками провоспалительных цитокинов, таких как фактор некроза опухолей (ФНО), ИФН-γ, интерлейкин (ИЛ)-12, ИЛ-17, и увеличения продукции противовоспалительных цитокинов (ИЛ-4, ИЛ-10, трансформирующего фактора роста β1 и некоторых других) [33]. Кроме того, в исследованиях in vivo выявлено значимое увеличение количества и функциональной активности CD4+/СD8+CD25+Foxp3 регуляторных Т-клеток в периферической крови больных РС, получавших препараты ИФН-β, по сравнению с показателями на момент начала лечения, что может быть частично связано с изменением цитокинового баланса [34–36]. ИФН-β также уменьшает миграцию активированных аутореактивных иммунных клеток через ГЭБ – in vitro ИФН-β ингибирует экспрессию различных матриксных металлопротеиназ, способствующих увеличению проницаемости ГЭБ [37], и уменьшает продукцию различных молекул адгезии, некоторых хемокинов и их рецепторов [38]. Еще один важный механизм действия ИФН-β – стимуляция в ЦНС экспрессии некоторых нейтротрофических факторов [39].
Механизмы действия глатирамера ацетата при лечении РС
Глатирамера ацетат – синтетический аналог оснóвного белка миелина (ОБМ). ОБМ – самый представленный белок в миелиновой оболочке. Молекулярная масса ГА варьирует в диапазоне от 4.7 до 11 кДа – это уксуснокислая соль смеси синтетических полипептидов, состоящих из L-изомеров глутаминовой кислоты, лизина, аланина и тирозина (отсюда и название – глатирамера ацетат) в том же молярном соотношении: 4.2 : 3.4 : 1.4 : 1.0, – в каком они представлены в ОБМ [40].
Хотя точный механизм действия ГА при РС до конца не известен, его благоприятный эффект связывают, в первую очередь, с индукцией ГА-специфичных клонов Т-хелперов фенотипа Th2, которые продуцируют противовоспалительные цитокины и подавляют аутоиммунное воспаление в ЦНС [41–44]. Кроме того, ГА конкурирует с аутоантигенами ОБМ за связывание с молекулами HLA класса II на поверхности АПК, что приводит к ингибированию презентации антигенов и, как следствие, к подавлению активации миелинреактивных Тh1-клеток [45, 46]. Длительный прием ГА способствует увеличению экспрессии Foxp3 под влиянием трансформирующего ростового фактора β1 (TGF- β1), что приводит к увеличению пула регуляторных CD4+CD25+Foxp3 Т-клеток [47, 48]. Показано также, что в ответ на введение ГА у пациентов, помимо CD4+ T-клеток, активируются CD8+ T-клетки с регуляторными/супрессорными свойствами, причем их уровень гораздо выше тех, которые регистрируют как у пациентов, не получавших ГА, так и у здоровых индивидов [49, 50].
ГА также обладает свойством модуляции В-клеточного звена иммунитета. При введении ГА модельным животным в В-клетках активируется синтез противовоспалительных цитокинов и уменьшается экспрессия костимулирующих молекул [51]. Более того, антитела против ГА индуцируют опосредованную олигодендроцитами ремиелинизацию в хронических бляшках у животных, тем самым обеспечивая благоприятный эффект [52].
Одним из важнейших механизмов действия ГА считается обеспечение нейропротективного эффекта в ЦНС за счет синтеза нейротрофических факторов, таких как нейротрофический фактор мозга (BDNF; brain-derevied neurotrophic factor,) и фактор роста нервов (NGF; nerve growth factor,), влияющих на рост и дифференцировку нейронов и олигодендроцитов [53, 54]. В целом, проникновение в ЦНС ГА-специфичных клеток с противовоспалительным и нейропротективным фенотипом – существенный фактор, определяющий эффективность лечения больных РС препаратом ГА [55].
Таким образом, хотя механизмы действия ИФН-β и ГА и нуждаются в дальнейшем изучении, уже сейчас ясно, что область приложения каждого из них не ограничивается воздействием на конкретное патогенетическое звено аутоиммунного процесса. Терапевтическое действие этих ПИТРС, вероятнее всего, реализуется как через сходные механизмы, например, регуляцию цитокинового баланса и регуляцию активации и пролиферации Т-клеток, так и посредством совершенно различных механизмов, таких как контроль миграции аутореактивных клеток через ГЭБ для ИФН-β или конкуренция с антигенными пептидами за молекулы HLA класса II на АПК для ГА.
ФАРМАКОГЕНЕТИЧЕСКИЕ ИССЛЕДОВАНИЯ ЭФФЕКТИВНОСТИ ПРЕПАРАТОВ, ИЗМЕНЯЮЩИХ ТЕЧЕНИЕ РС
Хотя в многочисленных клинических испытаниях в целом выявлена высокая эффективность лечения РС препаратами ИФН-β и ГА, у значительной части пациентов (30–50%) она невелика, вплоть до полного отсутствия влияния препарата на течение РС [56, 57]. Важно заметить, что сформированные перед началом лечения группы пациентов, по-разному ответивших на лечение, клинически и демографически не отличались [58]. Учитывая, что вывод о действенности терапии ПИТРС для каждого больного РС делают на основании клинических критериев (снижение числа обострений, снижение скорости прогрессирования заболевания и др.) только после достаточно длительного периода приема препарата (не менее 9–12 мес.), до момента его отмены у неответивших пациентов, как правило, происходит ухудшение неврологического состояния. Именно поэтому принципиальную важность приобретает поиск ранних прогностических маркеров эффективности лечения.
Одной из основных причин индивидуальных различий в чувствительности к отдельным препаратам может быть генетический статус пациентов. Это предположение послужило пусковым механизмом для поиска генетических маркеров, ассоциированных с эффективностью лечения ПИТРС. Такие ФГ-исследования до настоящего времени проводили, в основном, для иммуномодулирующих препаратов первой линии: ИФН-β и ГА. Первое ФГ-исследование эффективности лечения РС иммуномодулирующими препаратами ИФН-β и ГА опубликовано в 2001 г. [59]. К настоящему времени проведено около 40 исследований эффективности лечения больных РС препаратами ИФН-β и немногим более 10 – препаратом ГА.
Следует отметить определенную ограниченность проведенных ФГ-исследований терапии РС. Во-первых, в подавляющем большинстве пациенты, включенные в эти исследования, были европеоидами (иногда смешанных этносов), причем численность выборок колебалась от нескольких десятков пациентов [59–61] до, максимум, 1385 [62]. Во-вторых, серьезным ограничением в ФГ-исследованиях была длительность наблюдения за ответом больных РС на лечение. Если в ранних работах срок наблюдения за пациентами составлял 6–12 мес., то в последние годы эффект препаратов оценивается как положительный при наблюдении за пациентами в течение двух лет и более. В-третьих, отсутствуют единые критерии оценки эффективности лечения иммуномодулирующими препаратами, и в частности ИФН-β и ГА. Действительно, эффективность терапии оценивали на основании различных клинических характеристик: отсутствие или снижение числа обострений, отсутствие или снижение прогрессирования РС по шкале инвалидизации EDSS, отсутствие новых бляшек по данным МРТ и др. В большинстве исследований в качестве положительного (оптимального) ответа принимали критерий “отсутствие неблагоприятных клинических событий” (“event-free”), который включает “отсутствие обострений” и “отсутствие прогрессирования по шкале EDSS” за весь срок наблюдения (сейчас – за период не менее двух лет). В некоторых работах проводили сравнение “крайних групп”, когда больных с оптимальным ответом на терапию сравнивали с больными, у которых неблагоприятные события наступали существенно раньше, например, менее чем через 1 год; при этом часть пациентов с промежуточным ответом исключали из рассмотрения. Кроме того, хотя важным этапом генетических исследований считается валидация полученных результатов с привлечением независимых групп пациентов [63], валидация полученных результатов на независимых выборках была проведена всего в нескольких ФГ-исследованиях эффективности терапии препаратами ИФН-β [9, 62, 64–66]. И, наконец, всего в нескольких работах уровень значимости полученных результатов оценивали по значению величины p с поправкой на множественные сравнения [66–71], а в исследовании Gross и соавт. [9] введена поправка на влияние демографических и клинических факторов, искажающих результаты на исходном уровне (baseline confounders).
Фармакогенетические исследования эффективности лечения рассеянного склероза препаратами интерферона-β
Результаты ФГ-исследований, проведенных до 1 января 2019 г. для ИФН-β с использованием подхода “ген-кандидат”, в которых были получены значимые результаты (с величиной p < 0.05), представлены в табл. 1. В основном гены выбирали, исходя из участия их продуктов в иммунопатогенезе РС, в механизмах действия ИФН-β или в механизмах передачи сигнала после взаимодействия эндогенного или экзогенного ИФН-β со своим рецептором.
Таблица 1.
Значимые результаты фармакогенетических исследований эффективности лечения больных РС препаратами ИФН-β, проведенные методом “ген-кандидат”
| Ген | Хромосомная локализация | Этническая принадлежность больных РС, численность выборки (длительность наблюдения) |
Аллель/генотип/гаплотип, ассоциированный с позитивным (+) или негативным (−) ответом на лечение ИФН-β | Значение Р (Ркорр)a |
Ссылки |
|---|---|---|---|---|---|
| CTSS | 1q21 | Ирландцы, n = 162 (6–9 мес.) | rs1136774*С (+) | 0.020 | [72] |
| IL10 | 1q31-q32 | Норвежцы, n = 25 (6 мес.) | Отсутствие гаплотипа rs1800896*G/ rs1800871*C/rs1800872*C (+) |
0.040 | [60] |
| CD46 | 1q32 | Испанцы, n = 163 (1 год) | rs2724385*T/T (+) | 0.006 (<0.01) | [71] |
| СD58 | 1p13 | Иранцы, n = 120 (2 года) | rs12044852*С/C (–) | <0.05 | [73] |
| IFIH1 | 2q24 | Европейцы, I группа, n = 830 (2 года) Европейцы, II группа, n= 555 (2 года) I + II группы, n = 1385 |
rs3747517*A (+) | 0.010 Н.з.b Н.з. |
[62] |
| GABRR3 | 3q11.2 | Европейцы, I группа, n= 830 (2 года) Европейцы, II группа, n = 555 (2 года) I + II группы, n= 1385 |
rs832032*A (+) | 0.0006 Н.з. 0.006 |
[62] |
| CCR5 | 3p21.31 | Русские, n = 253 (2 года) | rs333*del (+) | 0.036 | [67] |
| Египтяне, n = 80 (2 года) | rs333*del (+) | 0.01 | [69] | ||
| CXCL1 | 4q21 | Европейцы, I группа, n = 830 (2 года) Европейцы, II группа, n = 555 (2 года) I + II группы, n = 1385 |
rs4422395*A (+) | 0.017 Н.з. Н.з. |
[62] |
| LMP7 | 6p21.3 | Ирландцы, n = 162 (6–9 мес.) | rs2071543*C (+) | 0.002 | [72] |
| HLA-DRB1 HLA-B |
6p21.32 6p21.33 |
Иранцы, n = 231 (2 года) | HLA-DRB1*04 (+) HLA-B*15 (–) |
0.008 0.03 |
[68] |
| IRF5 | 7q32 | Европейцы, n = 73; (1–2 года) Американцы, n = 261 (1–2 года) | rs2004640*T/T (–) | 0.01 0.037 |
[64] |
| IKBKB | 8p11.2 | Европейцы, I группа, n = 830 (2 года) Европейцы, II группа, n = 555 (2 года) I + II группы, n = 1385 |
rs10958713*G (+) | 0.0030 Н.з. 0.016 |
[62] |
| TRAILR1 | 8p21 | Испанцы, I группа, n = 509 (2 года) Испанцы, II группа , n = 226 (2 года) I + II группы n = 735 |
rs20576*C/C (+)a | Н.з. 0.005 0.0009 (0.048) |
[66] |
| PELI3 | 11q13.2 | Европейцы, I группа, n = 830 (2 года) Европейцы, II группа, n = 555 (2 года) I + II группы n = 1385 |
rs2277302*A (+) | 0.017 Н.з. 0.008 |
[62] |
| IFNG | 12q14 | Испанцы, n = 110 (2 года) | Варианты числа повторов микросателлита (CA)n в интроне 1: | [74] | |
| (CA)12 (–) | 0.013 | ||||
| (CA)13 (–) | 0.040 | ||||
| (CA)14 (–) | 0.009 | ||||
| (CA)15 (+) | 0.005 | ||||
| GPC5 | 13q32 | Испанцы, n = 199 (2 года) | rs10492503*A/A (+) rs1411751*G/G (+) |
0.003 (0.018) 0.002 (0.012) |
[70] |
| IRF8 | 16q24.1 | Американцы, n = 424, (до 600 недель) Немцы, n = 211 (до 600 недель) |
rs17445836*A/A (–) | 0.017 Н.з. |
[9] |
| STUB1 | 16p13.3 | Европейцы, I группа n = 830 (2 года) Европейцы, II группа, n = 555 (2 года) I + II группы, n = 1385 |
rs6597*A (+) | 0.019 Н.з. 0.04 |
[62] |
| ACE | 17q23 | Хорваты, словенцы, n = 275 (2 года) Мужчины, n = 64 |
rs1799752*del (-–) | Н.з. 0.022 |
[75] |
| TGFB1 | 19q13.1 | Русские, n = 253(2 года) | rs1800469*C (+) | 0.0062 (0.042) | [67] |
| MХA | 21q22.3 | Ирландцы, n = 162 (6–9 мес.) | rs2071430*G (+) rs17000900*G/G (+) |
0.015 0.018 |
[72] |
| Иранцы, n = 231 (2 года) | rs464138*A/A (+) гаплотипы rs17000900/ rs2071430/rs464138: | <0.0001 | [76] | ||
| CGC (+) | 0.003 | ||||
| ATA (+) | 0.007 | ||||
| AGA (+) | 0.036 | ||||
| IFNAR1 | 21q22.11 | Испанцы, n =147 (2 года) | rs1012334*A (+) | 0.03 | [77] |
| Ирландцы, n =162 (6–9 мес.) | rs55884088*GTn (+) | 0.036 | [72] | ||
| Европейцы, I группа, n = 830 (2 года) Европейцы, II группа, n = 555 (2 года) I + II группы, n = 1385 |
rs2834202*G (+) | 0.03 Н.з. 0.048 |
[62] | ||
| USP18 | 22q11.21 | Испанцы, n = 225 (2 года) | rs2542109*A/A (+) | 0.041 | [78] |
Первыми в качестве генов-кандидатов для ФГ-анализа были выбраны гены локуса главного комплекса гистосовместимости (HLA класса I и II), некоторые из которых были описаны ранее как основные гены предрасположенности к РС [18]. Во всех исследованиях на европейских выборках не наблюдали значимых ассоциаций носительства отдельных аллелей и/или гаплотипов генов HLA класса I и II с эффективностью лечения больных РС препаратами ИФН-β [59, 67, 68, 79–81]. Только в одной работе на иранской популяции выявлена ассоциация аллеля HLA-DRB1*04 с оптимальным ответом на ИФН-β и аллеля HLA-B*15 с отсутствием ответа на лечение [68].
Вполне логично, что при анализе эффективности лечения РС препаратами ИФН-β исследовали полиморфизм генов IFNAR1 и IFNAR2, кодирующих две субъединицы белка IFNAR – общего рецептора ИФН типа I. Значимые ассоциации различных полиморфных вариантов гена IFNAR1, но не IFNAR2, с эффективностью препарата ИФН-β наблюдали в нескольких исследованиях [62, 72, 77]. В одной из этих работ проведена валидация ассоциации полиморфных вариантов гена IFNAR1 и последующий анализ в объединенной группе пациентов, однако значимых результатов получено не было [62]. Для гена IFNAR2 значимые ассоциации обнаружены при полногеномном анализе [82] и мультилокусном анализе [83] (см. ниже).
Механизм действия ИФН-β, как и других ИФН типа I, опосредован, по крайней мере частично, взаимодействием специфических транскрипционных факторов с элементами ИФН-индуцированного ответа (interferon-stimulated response elements, ISREs), которые в основном расположены в промоторных областях многих ИФН-индуцибельных генов [84]. Проведено исследование ассоциации полиморфных вариантов в последовательностях ISRE (или вблизи от них), расположенных в промоторных областях 100 ИФН-индуцибельных генов, с эффективностью терапии ИФН-β у ирландцев [72].
Помимо упомянутой выше ассоциации полиморфного варианта гена IFNAR1 (число (GT)n повторов), наблюдали также ассоциацию с эффективностью лечения ИФН-β полиморфных вариантов генов LMP7, CTSS и MХA (у последнего – два полиморфных участка, находящихся в неравновесном сцеплении). Продукты гена LMP7 − β-субъединица протеасомы – и гена CTSS − протеолитический фермент катепсин S – участвуют в процессировании и презентации антигенов молекулами HLA I и II класса, что, по мнению авторов исследования [72], может указывать на особое значение этих процессов в формировании гетерогенного ответа на лечение ИФН-β. Особый интерес представляет ген MХA, кодирующий белок MxA (myxovirus resistance protein A), который влияет на ряд клеточных функций и обладает сильной антивирусной активностью. Полученные в одном их исследований [72] результаты по ассоциации его вариантов rs2071430*G и rs17000900*G/G с эффективной терапией ИФН-β позднее не получили подтверждения в работе [85], проведенной на американских пациентах (без указания этнического происхождения). В то же время в работе [76], проведенной на иранцах, была показана ассоциация оптимального ответа на лечение ИФН-β с другим SNP в области этого гена, rs464138, а также с двумя гаплотипами, включающими rs2071430, rs17000900 и rs464138.
Гены-кандидаты, кодирующие молекулы из семейства транскрипционных факторов, которые регулируют систему интерферонов (interferon re-gulatory factor, IRF), также стали объектами ФГ-исследований. Продукты этих генов относятся к важнейшим регуляторам ISRE, участвуют в сигнальном каскаде цитокинов, дифференцировке гематопоэтических клеток, регуляции врожденного антивирусного ответа и т.д. [86]. В исследовании [64], проведенном на европейских больных РС с использованием данных МРТ в качестве критерия оценки эффективности лечения ИФН-β, наблюдали ассоциацию генотипа rs2004640*T/T гена IRF5 с неоптимальным ответом; эта ассоциация была валидирована авторами на независимой выборке американских пациентов, где критерием эффективности лечения была длительность периода до первого обострения. В то же время для больных испанского происхождения значимых различий в распределении полиморфных вариантов гена IRF5 в группах пациентов с различной эффективностью лечения препаратами ИФН-β не выявлено [65]. В исследовании [9], проведенном на американских больных РС, получающих терапию ИФН-β, выявлена ассоциация полиморфизма rs17445836 гена IRF8 с длительностью периода без неблагоприятных клинических событий (“event-free”), хотя этот результат в дальнейшем не был валидирован. Продукт гена USP18 кодирует убиквитинспецифическую пептидазу – негативный регулятор ИНФ-индуцированного сигнального пути. Показано, что носительство генотипа USP18 rs2542109*A/A значимо ассоциировано с эффективной терапией ИФН-β [78]. Авторы исследования считают, что USP18 может играть важную роль и в патогенезе РС, и в формировании ответа на лечение ИФН-β.
Особо следует остановиться на недавнем ФГ-исследовании [62], где на большой этнически гетерогенной выборке больных РС, получающих терапию ИФН-β, анализировали 384 полиморфизма в генах белков, вовлеченных в сигнальные пути ИФН типа I, Toll-подобных рецепторов, нейротрансмиттеров и глутаматных рецепторов. На этапе скрининга c участием 830 больных РС получены значимые ассоциации полиморфных вариантов генов GABRR3, STUB1, IFIH1, PELI3, IKBKB, CXCL1 и упомянутого ранее гена IFNAR1 с различной эффективностью лечения ИФН-β, но на репликативной выборке найденные ассоциации не подтвердились. Однако при объединенном анализе с участием 1385 пациентов значимые ассоциации с эффективностью терапии ИФН-β сохранились для полиморфных вариантов генов GABRR3 и PELI3 (p < 0.01) и, с меньшей значимостью, для генов STUB1, IKBKB, IFNAR1 (p < 0.05). Белковые продукты этих генов вовлечены в сигнальный путь ИФН типа I и Toll-подобных рецепторов, а также в работу глутаматэргической системы. Безусловно, для уточнения роли полиморфных вариантов этих генов в эффективности лечения больных РС препаратами ИФН-β необходима дальнейшая валидация результатов.
В нескольких работах исследована ассоциация эффективности лечения ИФН-β с полиморфизмом генов, кодирующих белки, вовлеченные во взаимодействие иммунных клеток и поддержание механизмов нормального клеточного развития. Выявлена ассоциация полиморфных вариантов генов CD58 и CD46 с различной эффективностью лечения ИФН-β [71, 73]. Более того, показано, что белок CD46 вовлечен в механизмы нарушения целостности ГЭБ [87], и у больных РС этот сигнальный путь нарушен [88]. При ФГ-исследовании на двух выборках испанских пациентов 54 полиморфизмов в генах TRAIL/TRAILR1-4 [66], кодирующих членов супресемейства ФНО: TRAIL и их рецепторы, – ассоциация генотипа TRAILR1 rs20576*C/C с ответом на лечение ИФН-β на этапе скрининга проявилась как тренд, но была значима на стадии репликации, а при объединении выборок сохранила значимость после поправки на множественные сравнения. Учитывая, что при аутоиммунных заболеваниях и воспалительных заболеваниях ЦНС эти белки вовлечены в индукцию апоптоза [89, 90], можно предположить, что аберрантная передача сигналов программируемой клеточной смерти важна для клеточного гомеостаза при аутоиммунном воспалении, характерном для РС.
В последние годы выявлено участие ренин-ангиотензин-альдостероновой системы в патогенезе РС [91]. Ключевой активатор этой системы – ангиотензинпревращающий фермент, кодируемый геном ACE, – помимо других функций играет роль провоспалительного фактора, вовлеченного в демиелинизацию, связанную с аутоиммунным воспалением. Наблюдали увеличение уровня этого фермента в бляшках больных РС [92], а также в сыворотке крови [93] и спинно-мозговой жидкости [94]. На небольшой выборке словаков и хорватов показана ассоциация функционального инсерционно-делеционного полиморфизма в гене ACE c эффективностью лечения ИФН-β только у мужчин [75]. Безусловно, эти результаты еще необходимо подтвердить.
В отдельных исследованиях была показана ассоциация с эффективностью лечения РС препаратом ИФН-β полиморфных генов некоторых цитокинов и их рецепторов: IFNG [74], IL10 [60], TGFB1 и CCR5 [67]. Для ряда генов иммунного ответа, кодирующих цитокины или их рецепторы, не обнаружено ассоциации с эффективностью ответа [60, 66, 67, 69, 72, 77, 83, 95, 96]. Отрицательные результаты получены и при поиске ассоциаций эффективности лечения с полиморфными вариантами генов инфламмасом [97, 98].
В целом, учитывая ограниченные размеры многих выборок больных РС и отсутствие валидации, следует признать, что проведенные ФГ-исследования эффективности терапии ИФН-β в большинстве случаев не позволяют сделать окончательных выводов о наличии/отсутствии ассоциации с генами рассмотренных выше систем. Однако в этих исследованиях подтверждена генетическая детерминированность ответа на лечение препаратами ИФН-β, которая, вероятнее всего, определяется участием не одного, а нескольких генов, продукты которых так или иначе связаны с патогенетическими механизмами развития РС и механизмами передачи сигнала от ИФН типа I.
В последние годы стало ясно, что анализ независимого вклада аллелей/генотипов генов-кандидатов поодиночке, каждый из которых может оказывать малый и трудно выявляемый эффект на общую эффективность лечения заболевания тем или иным препаратом, может оказаться недостаточно информативным. Благодаря развитию биоинформатических подходов перспективным направлением ФГ-исследований стало выявление совместного вклада генов-кандидатов с помощью мультилокусного анализа, при котором анализируют ассоциации носительства сочетаний аллелей/генотипов нескольких генов с исследуемым фенотипом. Такой анализ проводили в нескольких исследованиях с помощью программного обеспечения APSampler, использующего метод Монте-Карло марковскими цепями и байесовскую непараметрическую статистику [99]. Это программное обеспечение поддерживает также различные способы проверки выявленных ассоциаций с помощью широкого спектра традиционных статистических методов, в том числе использует пермутационный тест для учета множественности сравнений [99]. Среди ряда программ, позволяющих проводить мультилокусный анализ для полигенных заболеваний, APsampler характеризуется наиболее высокой информативностью результатов благодаря высокой статистической мощности анализа [100]. Далее приведены результаты наших ФГ-исследований с использованием этого программного обеспечения.
Проведен поиск совместного вклада аллельных комбинаций в формирование ответа на лечение ИФН-β у ирландских больных РС [83]. В мультилокусный анализ был включен 61 полиморфный участок в 34 генах, продукты которых участвуют в передаче сигнала от ИФН типа I или в поддержании цитокинового статуса. С эффективностью лечения ИФН-β не выявлено ни одной значимой ассоциации аллелей/генотипов исследованных полиморфных участков поодиночке, в то же время показано, что носительство некоторых комбинаций из 3, 4 и 5 аллелей генов IFNAR2, IL10, JAK2, JAK1, TYK2, IL10RB, GBP1, PIAS1, CASP7, CASP3, PNPT1, DUSP1, PRKCA, PRKR высокозначимо ассоциировано с эффективностью лечения ИФН-β. Верификацию значимости полученных ассоциаций провели с помощью пермутационного анализа (значения pperm от 0.0023 до 0.0008). Таким образом, действительно, вклад в эффективность лечения РС вносят несколько генов, носительство сочетаний которых определяет гетерогенность клинического ответа на лечение препаратами ИФН-β.
При мультилокусном ФГ-анализе, проведенном на 253 больных РС русской этнической принадлежности [67], исследованы полиморфизмы генов иммунного ответа, включая гены цитокинов, рецепторов цитокинов и хемокинов, костимуляторующих молекул, а также гена HLA-DRB1. Как указывалось выше, значимую ассоциацию поодиночке с эффективной терапией ИФН-β наблюдали для аллелей гена TGFB1 и CCR5 (табл. 1). В то время как полиморфный вариант гена TGFB1 не входит ни в одно из аллельных сочетаний, значимо ассоциированных с эффективностью лечения, аллель CCR5 rs333*d обнаружен в составе двух триаллельных сочетаний с аллелями генов IFNAR1, IFNB1 и IFNG, индивидуальную ассоциацию которых с эффективностью лечения не наблюдали поодиночке. Найденные сочетания характеризуются более высоким уровнем значимости, чем для одиночного аллеля CCR5*d. Носительство этих сочетаний, представляющих собой композитные (составные) маркеры: (CCR5 rs333*d + IFNAR1 rs1012335*G + IFNG rs2430561*T) и (CCR5 rs333*d + IFNAR1 rs1012335*G + IFNB1 rs1051922*T/T), – увеличивает шансы эффективной терапии соответственно в 2.8 и 14.3 раза (pperm = 0.035 и 0.017 соответственно).
Наблюдаемый кумулятивный эффект различных генов в составе аллельных сочетаний может возникать вследствие аддитивного или же эпистатического (нелинейного) механизмов взаимодействия между исследуемыми аллелями [100]. Впервые в рамках ФГ-исследования был проведен анализ природы кумулятивного эффекта аллелей, вошедших в состав композитных маркеров. Показано, что между аллелями CCR5*d, IFNAR1*G и IFNG*T в первом сочетании имеют место аддитивные взаимодействия, а между аллелем CCR5*d и генотипом IFNB1*T/T во втором сочетании – эпистатические [67]. В последнем случае входящие в это эпистатическое сочетание полиморфные варианты CCR5*d и IFNB1*T/T следует рассматривать как один композитный биаллельный маркер (CCR5*d + IFNB1*T/T), ассоциированный с эффективностью лечения. Таким образом, полученные с помощью программного обеспечения APSampler результаты позволили выявить ассоциацию совместного носительства алеллей/генотипов отдельных генов с эффективностью терапии препаратами ИФН-β больных РС. Благодаря этому удалось перейти порог значимости и обнаружить слабый (независимый или усиленный взаимодействием) вклад в эффективность терапии иммуномодулирующими препаратами отдельных генов, индивидуальное влияние которых не выявлено поодиночке.
Полногеномный поиск ассоциаций с эффективностью лечения интерфероном-β
К настоящему времени проведено пять исследований эффективности терапии ИФН-β методом GWAS [82, 101–104]. Длительность наблюдения пациентов для оценки эффективности терапии ИФН-β составляла от 2 до 4 лет. Клинические критерии при стратификации больных по эффективности лечения ИФН-β в разных исследованиях отличались, однако в большинстве случаев в фокусе исследований было сравнение “крайних групп”. В двух работах в анализ включали также группу пациентов с промежуточным ответом на лечение [102, 103]. Параллельно, для проверки специфичности найденных ассоциаций с эффективностью лечения ИФН-β, в нескольких исследованиях проводили анализ в группах больных РС, получавших другой иммуномодулирующий препарат – ГА [102–104]. В табл. 2 представлены результаты ФГ-исследований эффективности ПИТРС при лечении РС, полученные методом GWAS при сравнении “крайних групп”. В эту таблицу включены только генетические маркеры, расположенные в пределах конкретных генов.
Таблица 2.
Результаты фармакогенетических исследований эффективности лечения больных РС препаратами интерферона-β и глатирамера ацетата, проведенных методом полногеномного поиска ассоциаций (GWAS)
| Исходная группа, размер выборки (платформа) | Валидационная группа, размер выборки (платформа/метод) | Ген | Полиморфный вариантa |
Значение pb | Ссылка |
|---|---|---|---|---|---|
| Ассоциации полиморфных участков генома с эффективностью лечения препаратами ИФН-β | |||||
| Испанцы/французы, n = 206 (Affymetrix 100K GeneChips) | Расширенная исходная группа испанцев/французов, n = 287 | HAPLN1 | rs4466137 | 0.004 | [101] |
| GPC5 | rs10492503 | 0.007 | |||
| rs9301789 | 0.01 | ||||
| TAFA1 | rs4855469 | 0.01 | |||
| LOC442331 | rs6944054 | 0.014 | |||
| SYN2 | rs60834 | 0.022 | |||
| NPAS3 | rs4128599 | 0.024 | |||
| TRAF6 | rs10501154 | 0.035 | |||
| CASP1 | rs580253 | 0.037 | |||
| COL25A1 | rs794143 | 0.037 | |||
| CAST | rs10510779 | 0.042 | |||
| Испанцы, n = 106 (Affymetrix 500K GeneChips) |
Испанцы, n = 94 | GRIA3 CIT ADAR ZFAT STARD13 ZFHX4 IFNAR2 |
rs12557782 rs7308076 rs2229857 rs733254 rs9527281 rs11787532 rs2248202 |
0.002 0.006 0.02 0.02 0.02 0.04 0.04 |
[82] |
| Итальянцы, n = 116 (Illumina Human 660-Quad chip) |
Американцы, n = 281 (Affymetrix Human SNP array 6.0); Итальянцы, n = 275 (TaqMan); Французы, n = 325 (TaqMan); |
SLC9A9 | rs9828519 | 4.43 × 10–8 (исходная выборка); 7.78 × 10–4 (мета-анализ трех валидационных выборок); 8.39 × 10–7 (суммарный мета-анализ) |
[103] |
| Когорта 1: Итальянцы, n = 251 (Illumina OmniExpress BeadChip) Когорта 2: Итальянцы, n = 86 (Illumina Human 660-Quad) |
– | NINJ2 | rs7298096 | 9.82 × 10–5 | [104] |
| TBXAS1 | rs4726460 | 7.41 × 10–5 | |||
| GRIK2 | rs1475919 | 1.89 × 10–4 | |||
| GRM3 | rs2237562 | 2.21 × 10–4 | |||
| Австралийцы,
n = 151 (Illumina 370K, Illumina Human 660-Quad chip) |
Австралийцы, испанцы, итальянцы, n = 479 (Sequenom iPLEX assay) |
FHIT | rs760316 | 6.74 × 10–6 | [102] |
| GAPVD1 | rs10819043 | 5.83 × 10–5 | |||
| rs2291858 | 1.67 × 10–4 | ||||
| rs10760397 | 6.51 × 10–5 | ||||
| Вблизи ZNF697 | rs10494227 | 8.15 × 10–5 | |||
| Ассоциации полиморфных участков генома с эффективностью лечения глатирамера ацетатом | |||||
| Когорта 1 (GALA): Многонациональная группа, n = 318; Когорта 2 (FORTE): Многонациональная группа, n = 262. Illumina OMNI-5M |
Многонациональная группа, n = 311 (TaqMan) Американцы, n = 112 (TaqMan) Европейцы/Канадцы, n = 124 (TaqMan) Многонациональная группа, n = 372 (TaqMan) |
HLA-DRB1*15 | rs3135391 | 0.04/0.0499 | [105] |
| HLA-DQB2/DOB | rs28724893 | 0.0006/0.00037 | |||
| HLA-DOB/TAP2 | rs1894408 | 0.003/0.0093 | |||
| MBP | rs1789084 | 0.036/0.01 | |||
| PTPRT | rs117602254 | 0.0037/0.016 | |||
| ALOX5AP | rs10162089 | 0.0078/0.032 | |||
| MAGI2 | rs16886004 | 0.0023/3.3 × 10–5 | |||
| ZAK (CDCA7) | rs139890339 | 3.4 × 10–5/0.011 | |||
| SLC5A4 (RFPL3) | rs73166319 | 0.006/0.015 | |||
| UVRAG | rs80191572 | 0.0024/3.4 × 10–5 | |||
| SLC1A5 | rs759458 | 4.4 × 10–5/0.049 | |||
В двух ранних работах по GWAS, 2008 и 2009 гг. [82, 101], с помощью платформы Affymetrix генотипировали пулированные образцы ДНК пациентов, получавших ИФН-β, а затем на этапе валидации провели индивидуальное генотипирование отобранных полиморфных участков, для которых наблюдали номинальную значимость (p < 0.05). В качестве валидационной группы в первом исследовании увеличили на 81 человека исходную выборку пациентов (206 человек), а во втором валидацию проводили на независимой выборке из 94 пациентов. В результате получили данные о номинальных ассоциациях ряда полиморфных вариантов в различных участках генома. Так, в работе [101] среди 18 выявленных SNP только 8 были локализованы непосредственно в генах HAPLN1, GPC5 (2 SNP), TAFA1, LOC442331, NPAS3, COL25A1 и CAST, преимущественно кодирующих белки внеклеточного матрикса, а также глутаматные рецепторы и нейротрансмиттеры. В этой работе для сопоставления с результатами предыдущих ФГ-исследований провели также индивидуальное генотипирование полиморфных вариантов, расположенных в областях локализации сотни ИФН-индуцибельных генов, представленных на микрочипе Affymetrix 100K GeneChips. Выявлены значимые ассоциации эффективности лечения РС с тремя SNP, локализованными непосредственно в генах SYN2, TRAF6 и CASP1 (см. табл. 2), и еще c 12 SNP в областях рядом с другими 8 генами. Не найдено ассоциаций с полиморфизмами вблизи/в генах IFNAR1 и IFNAR2, а также в генах LMP7, CTSS и MXA, ассоциация с которыми была показана ранее [72]. Однако необходимо отметить, что проанализированные на этом этапе SNP, за исключением одного, ранее не исследовались.
В GWAS, опубликованном в 2009 г. [82], ассоциации обнаружены с полиморфными вариантами генов GRIA3, CIT, ADAR, ZFAT, STARD13, ZFHX4 и IFNAR2, продукты которых являются компонентами глутаматэргической системы (ионные каналы, нейтротрасмиттеры) и участвуют в передаче сигналов от ИФН типа I и связанных с противовирусным ответом ИФН. В этой работе с помощью программы MDR также проанализирована возможность взаимодействия аллелей выбранных генов в сочетаниях из двух или трех аллелей у 94 пациентов с различной эффективностью лечения, но значимых результатов не получено. Проведенные исследования, хотя в них и не было достигнуто полногеномного уровня значимости, обратили внимание исследователей на гены и белки глутаматэргической системы и внеклеточного матрикса как на возможные биомаркеры эффективности терапии ИФН-β.
В исследовании [70] была предпринята попытка валидировать две наиболее значимые ассоциации из первого GWAS [101] – в генах GPC5 и HAPLN1. Для двух из трех исследованных полиморфизмов, расположенных в интроне гена GPC5, включая GWAS-идентифицированный rs10492503 [101], выявили ассоциации с эффективным лечением ИФН-β, значимые после поправки на множественные сравнения (см. табл. 1). Для локусов гена HAPLN1 значимых результатов не выявлено [70].
В GWAS, опубликованном в 2015 г. [103], провели анализ более 500 тыс. аутосомных полиморфных маркеров с использованием платформы Illumina Human 660-Quad chip. На исходной группе пациентов выявлена значимая ассоциация аллеля rs9828519*G в интроне гена SLC9A9 с неэффективным лечением (p = 4.43 × 10–8), т.е. впервые значение p достигло полногеномного уровня значимости [106]. В рамках этого же исследования полученная ранее ассоциация была валидирована с помощью мета-анализа в трех независимых группах пациентов итальянского, французского и американского происхождения с уровнем значимости p = 7.78 × 10–4. В итоге уровень значимости суммарного мета-анализа в четырех группах составил p = 8.39 × 10–7. Дополнительный анализ в группах больных РС, получавших ГА, показал отсутствие ассоциации полиморфизма rs9828519 с эффективностью лечения. Белковый продукт гена SLC9A9 (Solute Carrier Family 9 Member A9) связан с работой Na+/H+ ионных каналов преимущественно в эндосомах и широко представлен в ЦНС и в иммунных клетках [107, 108]. Для исследования возможной роли rs9828519 гена SLC9A9 оценили потенциальное влияние этого SNP и ряда SNP, находящихся с ним в неравновесном сцеплении (его прокси-SNP) на экспрессию этого гена в разных отделах мозга человека [109]. Действительно, в разных областях мозга обнаружена дифференциальная экспрессия SLC9A9, при этом идентифицировано 15 сцепленных с rs9828519 SNPs, которые существенно регулировали экспрессию SLC9A9 в нескольких областях мозга: в затылочной коре, интралобулярном белом веществе и черной субстанции. Эти результаты могут свидетельствовать о вовлечении rs9828519 SLC9A9 в формирование ответа на лечение ИФН-β.
Полногеномное исследование 2017 года [104] интересно тем, что в нем была предпринята попытка выявить генетические факторы, связанные с длительным (более 4 лет) оптимальным ответом на лечение ИФН-β у итальянских больных РС, при этом анализ результатов проводили при сравнении “крайних групп”, исключив пациентов с промежуточным ответом. Это двухстадийное ФГ-исследование проведено с привлечением платформ OmniExpress BeadChip (когорта 1) и Illumina Human 660-Quad (когорта 2).
При мета-анализе результатов, полученных на двух когортах, выявлено 43 ассоциированных с эффективностью лечения ИФН-β полиморфизма с уровнем значимости p < 10–4. Для оценки специфичности выявленных ассоциаций провели также исследование связи этих полиморфизмов с эффективностью другого ПИТРС – ГА – и не нашли значимых ассоциаций. Результаты ассоциативных исследований были сопоставлены 1) с данными по экспрессии генов в мононуклеарных клетках здоровых индивидов, стимулированных ИФН-β, 2) с выявленными локусами, определяющими количественные характеристики экспрессии (eQTL, expression quantitative trait loci), и 3) с результатами анализа генных сетей. Из обнаруженных маркеров два SNP (rs7298096 и rs4726460) локализованы в генах NINJ2 и TBXAS1, экспрессия которых была значимо снижена при действии ИФН-β, и еще два SNP (rs2237562 и rs1475919) – в генах GRIK2 и GRM3, выявленных при анализе генных сетей. Белковые продукты этих генов вовлечены в процессы, связанные с нейрональным ростом, регуляцией воспаления и работой глутаматэргической системы.
В 2017 г. опубликованы результаты еще одного двухстадийного GWAS с привлечением разных платформ, проведенного на австралийских больных РС, получавших лечение ИФН-β [102]. На первом этапе использовали данные двух ранее проведенных полногеномных исследований по анализу предрасположенности к РС [110, 111 ] . После унификации данных и контроля качества проанализировали 286 592 полиморфных участка. На этом этапе не было выявлено ассоциаций с заданным порогом значимости (≤1.74 × 10−7). Валидацию наиболее значимых ассоциаций проводили на независимой группе больных РС. Для объединенных групп пациентов из исходной и валидационной выборок при сравнении “крайних групп” выявлены полиморфные варианты, ассоциированные с эффективностью лечения ИФН-β (p <10–4). Они включали один SNP в гене FHIT, три SNP в области гена GAPVD1, один SNP вблизи гена ZNF697 и два SNP в межгенных участках. При дополнительном анализе, проведенном в группах больных РС, получавших ГА, ни один из полиморфных вариантов, значимо ассоциированных с эффективностью лечения ИФН-β, не был ассоциирован с эффективностью лечения ГА.
Таким образом, ФГ-исследования эффективности лечения РС препаратами ИФН-β, проведенные к настоящему времени методом GWAS, не привели к желаемым результатам. Только в одном GWAS значение величины p – для SNP в гене SLC9A9 – достигло полногеномного уровня значимости [103], в других случаях уровень значимости был существенно ниже, а обнаруженные разными авторами значимо ассоциированные полиморфные участки, за редким исключением, не совпадали. Это может быть связано как с недостаточным размером выборок и отсутствием унифицированных протоколов проведения ФГ-исследований РС, так и с отсутствием “главного гена”, вклад которого в эффективность терапии больных РС препаратами ИФН-β можно выявить на малых выборках. В то же время выявление в этих исследованиях высокозначимой ассоциации SNP в гене SLC9A9, участвующего в работе Na+/H+ ионного канала, и менее значимых ассоциаций с несколькими генами глутаматэргической системы имеет принципиальное значение, поскольку указывают на возможное влияние терапии ИФН-β не только на иммунные, но и на нейродегенеративные процессы.
В целом, с использованием различных подходов (“ген-кандидат”, мультилокусный анализ и GWAS) получены данные по ассоциации ряда генов с эффективностью лечения РС препаратами ИФН-β. Это представленные в табл. 3 гены: IFNAR1, IFNAR2, IRF5, IFNG, IL10, GPC5, и SLC9A9, – для которых эта ассоциация показана, по крайней мере, в двух исследованиях. На основании полученных результатов можно говорить о кумулятивности эффекта генов, вовлеченных в формирование положительного ответа на лечение, и о плейотропности терапевтического воздействия ИФН-β. Тот факт, что действие ИФН-β при терапии РС варьирует у пациентов с различным генетическим статусом, может отражать комплексный характер природы заболевания и проявления клинических фенотипов в ответ на введение препарата. Для успешного поиска генетических вариантов и их сочетаний, ассоциированных с ответом на лечение РС препаратами ИФН-β, необходимо проводить анализ на более репрезентативных группах пациентов, а также включать в анализ валидацию полученных результатов на этнически независимых группах больных РС.
Таблица 3.
Гены, для полиморфных локусов которых показана ассоциация с эффективностью лечения РС препаратами ИФН-β по меньшей мере в двух исследованиях
| Ген (белковый продукт) |
Полиморфный участок |
Аллель/генотип, связанный с оптимальным ответом на лечение ИФН-β | Подход [ссылка] |
|---|---|---|---|
| IFNAR1 (субъединица 1 рецептора ИФН типа I) |
rs1012334 | A | Ген-кандидат [77] |
| rs55884088 | (GT)n | Ген-кандидат [72] | |
| rs2834202 | G | Ген-кандидат [62] | |
| rs1012335 | G | Мультилокусный анализ [67] | |
| IFNAR2 (субъединица 2 рецептора ИФН типа I) |
rs2248202 | C/C | GWAS [82] |
| rs8127890 | A | Мультилокусный анализ [82] | |
| IRF5 (регуляторный фактор 5 интерферона) |
rs2004640 | G* | Ген-кандидат [64] |
| IFNG (ИФН-γ) |
– | микросателлит (CA)15 | Ген-кандидат [74] |
| rs2430561 | T | Мультилокусный анализ [67] | |
| IL10 (ИЛ-10) |
rs1800896/rs1800871/rs1800872 | Не-GCC-гаплотип | Ген-кандидат [60] |
| rs1800872 | A | Мультилокусный анализ [83] | |
| GPC5 (глипикан-5) |
rs10492503 rs1411751 |
A/A G/G |
Ген-кандидат [70] |
| rs10492503 rs9301789 |
A/A T/T |
GWAS [101] | |
| SLC9A9 | rs9828519 | G* | GWAS [103] |
Фармакогенетические исследования эффективности лечения рассеянного склероза глатирамера ацетатом
Хотя ФГ-исследования, направленные на предсказание эффективности лечения РС препаратом ГА [59], начались в то же время, что и ФГ-исследования для ИФН-β, они развивались существенно медленнее. Численность выборок, состоящих исключительно из европеоидов, варьировала от нескольких десятков [8, 59, 112, 113] до нескольких сотен пациентов – максимум 332 пациента [9]. В табл. 4 представлены данные по ассоциации полиморфных участков генома с эффективностью лечения РС препаратом ГА, полученные методом “ген-кандидат”. Исходя из возможного механизма действия ГА при РС, опосредованного формированием тримолекулярного комплекса молекул генов HLA класса II с пептидами ГА и Т-клеточным рецептором, эти исследования в первую очередь были сфокусированы на поиске ассоциации эффективности лечения с полиморфизмом генов HLA класса II и, в первую очередь, высоко полиморфного гена HLA-DRB1.
Таблица 4.
Значимые результаты фармакогенетических исследований эффективности лечения РС глатирамера ацетатом (проведены методом “ген-кандидат”)
| Ген | Хромосомная локализация | Этническая принадлежность больных РС, численность выборки (длительность наблюдения) | Аллель/генотип/гап-лотип, ассоцииро-ванный с позитив- ным (+) или нега-тивным (−) ответом на лечение ГА | Значение Р (Ркорр) |
Ссылка |
|---|---|---|---|---|---|
| IL12RB2 | 1p31.3-p31.2 | Американцы, n = 73 (2 года) | rs946685*G (+) | 0.045 | [8] |
| CTSS | 1q21 | Европейцы/канадцы, n = 101 (9 мес.) | rs2275235*G (+) rs1415148*A (+) |
0.0009 (0.049) 0.0018 |
[8] |
| IL1R1 | 2q12 | Европейцы/канадцы, n = 101 (9 мес.) | rs956730*A (+) | 0.049 | [8] |
| CCR5 | 3p21.31 | Русские, n = 285 (2 года) | rs333*Wild type/Wild type (+) | 0.046 | [114] |
| EOMES | 3p24.1 | Русские, n = 296 (2 года) | rs2371108*T (+) | 0.00092 (0.018) | [115] |
| CD86 | 3q21 | Европейцы/канадцы, n = 101 (9 мес.) | rs1129055*C (–) | 0.04 | [8] |
| Локус генов HLA класса II | 6p21.3 | Итальянцы, n = 44 (2 года) | HLA-DRB1*1501 (+) | 0.016 | [59] |
| Американцы, n = 332 (до 600 недель) | rs3135388*AA (+)# | 0.015 (0.048) | [9] | ||
| Американцы, n = 64 (2 года) | DR15 (+) DQ6 (+) DR17 (–) DQ2 (–) |
0.02 0.014 0.012 0.02 |
[112] | ||
| Гаплотипы: | |||||
| DR15-DQ6 (+) | 0.0062 (0.044) | ||||
| DR17-DQ2 (–) | 0.0077 (0.046) | ||||
| Русские, n = 285 (2 года) | HLA-DRB1*04 (+) | 0.027 | [114] | ||
| Русские, n = 296 (2 года) | HLA-DRB1*04 (+) | 0.015 | [115] | ||
| IL22RA2 | 6q23.3 | Русские, n = 296 (2 года) | rs202573*G/G (+) | 0.021 | [115] |
| TCRB | 7q34 | Европейцы/канадцы, n = 101 (9 мес.) | rs71878*C (+) | 0.006 | [8] |
| Американцы, n = 73 (2 года) | 0.039 | ||||
| PVT1 | 8q24.21 | Русские, n = 296 (2 года) | rs2114358*A (+) | 0.032 | [115] |
| FAS | 10q24.1 | Европейцы/канадцы, n = 101 (9 мес.) | rs982764*C (+) | 0.03 | [8] |
| CLEC16A | 16p13 | Русские, n = 296 (2 года) | rs6498169*A (+) | 0.0017 (0.024) | [115] |
| MBP | 18q23 | Европейцы/канадцы, n = 101 (9 мес.) | rs470929*T (+) | 0.0038 | [8] |
В исследовании, проведенном в 2001 году [59] на группе из 44 итальянских больных РС, выявили значимую ассоциацию эффективного лечения ГА с носительством аллеля DRB1*1501, или DR15. Позже этот вывод подтвержден на группе из 332 американских пациентов [9]. Показано, что у носителей генотипа rs3135388*A/A, аллель A которого находится в неравновесном сцеплении с аллелелем DRB1*1501, период без неблагоприятных неврологических событий при лечении ГА был более длительным по сравнению с носителями гетерозиготного и гомозиготного по альтернативному аллелю генотипов. В другом исследовании [112], проведенном также на американских пациентах, наблюдали, что носительство аллелей DR15 и DQ6 по отдельности или в составе гаплотипа, но при отсутствии аллелей DR17 и DQ2, также по отдельности или в составе гаплотипа, связано с оптимальным ответом на лечение ГА. В нашем ФГ-исследовании эффективности лечения ГА 285 русских больных РС [114], при сравнении пациентов с оптимальным ответом на лечение ГА (без неблагоприятных клинических событий) с остальными пациентами, найдена ассоциация с аллелем HLA-DRB1*4. В то же время другие группы исследователей [8, 113] не нашли ассоциации одиночных аллеей/генотипов гена HLA-DRB1 с ответом на лечение ГА. Анализируя вклад вариабельности 9 генов иммунного ответа в эффективность терапии ГА, мы обнаружили также ассоциацию генотипа CCR5*w/w с оптимальным ответом [114].
Отдельного внимания заслуживает работа Grossman и соавт. [8]. Это исследование проведено с привлечением двух групп больных РС европеоидного происхождения: американской (исходная, 73 человека) и европейской/канадской (валидационная, 101 человек), участвовавших в мультицентровых двойных слепых плацебоконтролируемых клинических испытаниях эффективности лечения ГА. Кроме гена HLA-DRB1, также анализировали 61 полиморфизм в 27 генах, кодирующих Т‑клеточный рецептор, некоторые цитокины и хемокины, их рецепторы, костимулирующие молекулы, аутоантигены, протеазы и белки, участвующие в апоптозе. Только один полиморфизм TCRB, rs71878*С, был значимо ассоциирован с эффективностью ГА в обеих выборках. Кроме того, в валидационной группе обнаружена ассоциация двух SNP гена CTSS с эффективной терапией ГА, причем ассоциация rs2275235*G выдержала поправку на множественные сравнения. Также найдены полиморфные варианты ряда генов, номинально ассоциированные с эффективностью терапии ГА только в одной их групп: гена IL12RB2 в исходной группе и генов MBP, CD86, FAS и IL1R1 в валидационной группе. Важно отметить, что в этом исследовании также проводили анализ для группы больных РС, получавших плацебо вместо ПИТРС, и не выявили ни одной значимой ассоциации с эффектом приема плацебо. Таким образом, в результате проведенного исследования выявлены статистически значимые ассоциации эффективности лечения ГА больных РС с генетическими вариантами генов, продукты которых непосредственно участвуют в иммунопатогенезе РС, причем значимость ассоциаций выявлена только в группах пациентов, принимавших ГА, но не плацебо. В работе не удалось валидировать результаты по ассоциации DRB1*1501 с эффективностью лечения ГА, полученные Fusco и др. [59]. По мнению авторов, причиной несоответствия результатов двух исследований может быть различный этнический состав исследуемых групп, небольшой размер выборок и возможные ошибки первого рода.
Недавно на группе из 296 русских больных РС нами проанализирован [115] вклад в формирование ответа на лечение ГА 17 полиморфных вариантов генов иммунного ответа, ассоциированных с риском развития РС по результатам GWAS. Найдены ассоциации полиморфных вариантов генов EOMES, CLEC16A, IL22RA2 и PVT1 с оптимальным ответом на лечение при двух типах сравнения: группу пациентов с оптимальным ответом на лечение ГА сравнивали или с группой пациентов, у которых развились неблагоприятные события уже в течение 1 года (не ответившие на лечение ГА), или с объединенной группой, в которую вошли не ответившие пациенты и пациенты с промежуточным ответом на лечение ГА. На основе полученных результатов можно предположить, что GWAS-идентифицированные гены предрасположенности к РС вовлечены в механизм действия ГА.
При ФГ-анализе ассоциации полиморфных вариантов генов-кандидатов TRAILR1 [66] и IL12В [113] не выявлено ассоциации с эффективностью лечения ГА, что не удивительно для исследований, проведенных на малых выборках.
Нами впервые проведен мультилокусный ФГ-анализ, позволяющий выявлять совместной вклад генов-кандидатов в эффективность лечения ГА [116]. На выборке из 285 русских больных РС, получающих терапию ГА, убедительно показано участие генов CCR5, HLA-DRB1, IFNG, TGFB1, IFNAR1 и IL7RA в формировании ответа полигенной природы на лечение [116]. Действительно, найдено два высокоспецифичных композитных (составных) маркера, носительство которых ассоциировано с эффективностью ответа больных РС на лечение ГА, причем ассоциации выдерживали поправку на множественные сравнения. Это, во-первых, биаллельное сочетание (DRB1*4 + IL7RA rs6897932*T) (pperm = 0.036), ассоциированное с оптимальным ответом на лечение ГА, при носительстве которого в 3.7 раза повышены шансы эффективной терапии; и, во-вторых, сочетание из четырех аллелей (CCR5 rs333*d + DRB1*15 + TGFB1 rs1800469*T + + IFNAR1 rs1012335*G), ассоциированное с отсутствием оптимального ответа на лечение (pperm = = 0.0056), при носительстве которого в 26 раз увеличены шансы неэффективной терапии. При анализе природы кумулятивного эффекта аллелей, входящих в состав сочетаний, выявлено наличие как аддитивных, так и эпистатических взаимодействий между аллелями в последнем сочетании. Очевидно, что больным РС, носителям этого сочетания, не показано назначать ГА в качестве препарата первой линии.
Мультилокусный анализ проведен нами еще в одной работе [115], описанной выше. В результате выявлены би- и триаллельные композитные маркеры эффективности терапии ГА, которые помимо аллелей/генотипов генов EOMES, CLEC16A, IL22RA2, PVT1 и DRB1*4, выявленных поодиночке, включают также полиморфные варианты генов TYK2, CD6, IL7RA и IRF8. При исследовании характера взаимодействия компонентов в составе сочетаний с помощью анализа двух метрик – фактора синергии (SF) и Pflint [117] – обнаружено эпистатическое взаимодействие между компонентами двух биаллельных сочетаний (EOMES rs2371108*G/G + TYK2 rs2304256*C) и (PVT1 rs2114358*A + IRF8 rs17445836*G), которые были значимо ассоциированы с оптимальным ответом на ГА. Для всех остальных сочетаний обнаружен только аддитивный эффект. Таким образом, эти два сочетания можно рассматривать как композитные маркеры эффективности ГА.
Для анализа предсказательной силы маркеров эффективности терапии ГА при РС (как одиночных вариантов, так и эпистатических сочетаний) использовали метод множественной логистической регрессии. Значимые коэффициенты регрессии композитной модели, включающей одиночные полиморфные варианты, были получены для аллелей генов EOMES rs2371108, CLEC16A rs6498169 и IL22RA2 rs202573. В результате анализа построенных ROC-кривых (receiver operating characteristic) и оценки площади под кривой (area under curve, AUC) для одиночных маркеров показано, что композитная регрессионная модель дает средний уровень предсказательной способности (AUC = 0.657). Включение в анализ эпистатических композитных маркеров (EOMES rs2371108*G/G + TYK2 rs2304256*C) и (PVT1 rs2114358*A + IRF8 rs17445836*G) позволило повысить предсказательную силу (AUC = 0.701). В обоих случаях композитные модели обладают предсказательной силой, которая значительно выше по сравнению с таковой для отдельных предикторов (диапазон AUC от 0.560 до 0.607). Таким образом, продемонстрированы преимущества композитных генетических моделей, особенно если они учитывают существование эпистатических взаимодействий между отдельными вариантами генов. Подобный анализ может быть полезен в клинической практике для первоначального назначения ГА в качестве препарата первой линии при лечении РС.
Полногеномный поиск ассоциаций с эффективностью лечения глатирамера ацетатом
К настоящему времени проведено всего одно исследование методом GWAS эффективности лечения больных РС препаратом ГА [105]. Его результаты представлены в табл. 2. Это самое репрезентативное полногеномное ФГ-исследование при РС: в него включено 1171 человек из разных этнических групп. В качестве критерия эффективности терапии использован показатель снижения ежегодной частоты обострений; сравнение проведено на “крайних группах” c исключением из анализа пациентов с промежуточным ответом на лечение. Для подтверждения оптимального ответа пациентов на лечение ГА дополнительно использовали радиологический критерий – отсутствие прогрессии по результатам МРТ. К недостаткам проведенного анализа относится длительность наблюдения – всего 1 год.
На первом этапе Ross и соавт. [105] анализировали полногеномные данные двух когорт пациентов, участвовавших в разных клинических испытаниях (GALA и FORTE), и одной когорты больных РС, принимавших плацебо (GALA). С помощью платформы Illumina OMNI-5M проанализировали более 4 млн полиморфизмов. Ни одна из ассоциаций не достигла порога значимости, принятого для GWAS. При значительном снижении порога значимости до 0.05 обнаружено 11 полиморфизмов, ассоциированных с верифицированным оптимальным ответом на лечение ГА и локализованных в области генов HLA-DRB1*15, HLA-DQB2/DOB, HLA-DOB/TAP2, MBP, PTPRT, ALOX5AP, MAGI2, ZAK (CDCA7), SLC5A4 (RFPL3), UVRAG и SLC1A5; при этом в группе, получавшей плацебо, эти ассоциации не выявлены. На следующем этапе проводили генотипирование выбранных 11 SNP на полной выборке из 1171 пациента. Методом логистической регрессии проведен поиск вариантов, обеспечивающих наибольшую предсказательную силу при определении эффективности терапии ГА (по критерию отсутствия неблагоприятных клинических событий). Наиболее эффективным оказался композитный маркер, включающий 4 полиморфных варианта: rs80191572 в гене UVRAG, rs28724893 в гене HLA-DQB2, rs1789084 в гене MBP и rs139890339 в гене ZAK (CDCA7). И в исходных, и в валидационных группах у носителей этого маркера наблюдали существенно бóльшее снижение ежегодной частоты обострений, чем у пациентов без него (“неносителей”). Важно отметить отсутствие этого эффекта в группах, получавших плацебо или ИФН-β. Авторы полагают, что продукты генов UVRAG, ZAK (CDCA7), HLA-DQB2 и MBP вовлечены в патогенетические механизмы при РС, а также в реализацию механизма действия ГА.
Безусловно, ФГ-исследования эффективности ГА при терапии больных РС еще очень немногочисленны. Однако уже сейчас можно сказать, что для аллелей генов HLA класса II даже на небольших выборках получены более или менее воспроизводимые результаты. Более того, для некоторых не-HLA-генов получены впечатляющие результаты, которые позволяют рассматривать их как потенциальные маркеры. Проведенные исследования, включая и единственный GWAS, с убедительностью показали, что анализ кумулятивного вклада полиморфных генетических вариантов в формирование ответа на лечение ГА обладает более высокой предсказательной силой, чем анализ вклада одиночных аллелей/генотипов. В целом показано, что гены, ассоциированные с эффективностью лечения ГА, могут быть вовлечены в различные этапы механизма его действия. Однако эти результаты нуждаются в воспроизведении на репрезентативных выборках из разных этносов.
ЗАКЛЮЧЕНИЕ
Несомненный успех длительной иммуномодулирующей терапии больных РС препаратами ИФН-β и ГA подтвердил, что иммунный ответ – важнейшая стадия патогенеза РС. В то же время различия в эффективности лечения этими ПИТРС в группах клинически идентичных пациентов приводят к заключению о необходимости поиска критериев отбора оптимальных препаратов для каждого пациента. ФГ-исследования терапии ПИТРС при РС направлены на выявление генетических биомаркеров, которые в совокупности с клиническими характеристиками помогут клиницистам выбирать оптимальную терапию для каждого больного РС.
ФГ-исследования полигенных заболеваний, как и изучение генетической предрасположенности к этим заболеваниям, не всегда приводят к воспроизводимым результатам, как видно на примере ФГ-исследований эффективности препаратов ИФН-β и ГА при лечении больных РС. Это, безусловно, может быть связано как с неоднородностью исследуемых выборок по размерам, этническому составу и клиническим характеристикам, так и с использованием разных критериев для оценки эффективности терапии ПИТРС. Кроме того, необходимо учитывать, что отдельные гены-кандидаты могут оказывать слабый эффект на результативность лечения ИФН-β и ГА вследствие плеойтропности действия этих иммуномодулирующих препаратов и полигенной природы формирования ответа на лечение; поэтому ФГ-анализ значимости их полиморфных вариантов поодиночке может оказаться неэффективным. Действительно, уже первые мультилокусные исследования выявили кумулятивное влияние полиморфных вариантов ряда генов на формирование ответа при терапии ПИТРС и показали значимость этого подхода для персонализированной медицины. Более того, продемонстрирована эффективность сравнительного ФГ-исследования при выборе наиболее предпочтительного препарата для конкретного пациента из имеющихся альтернативных вариантов (например, ИФН-β и ГА) – такой подход расширяет дискриминирующие возможности анализа [118]. Принимая во внимание появление новых препаратов для лечения РС, можно предполагать, что значение этого подхода будет возрастать. Сравнительный ФГ-анализ может быть использован в персонализированной терапии не только РС, но и других полигенных заболеваний, если для их лечения имеются альтернативные препараты.
По мере развития геномных технологий наряду с ФГ-исследованиями, вероятно, будет возрастать число работ, направленных на анализ транскриптома и эпигенома для выбора тактики ведения конкретного пациента. Однако, учитывая лабильность транскриптомных и эпигенетических профилей, можно полагать, что анализ генетического статуса пациента, неизменного в течение жизни, останется самым надежным подходом при поиске предиктивных ФГ-маркеров.
Авторы благодарят И.С. Киселева за помощь в оформлении рукописи.
Работа поддержана грантом РФФИ № 17-00-00206.
Все процедуры, выполненные в данной работе, соответствуют этическим стандартам институционального комитета по исследовательской этике и Хельсинкской декларации 1964 года и ее последующим изменениям или сопоставимым нормам этики.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Kalow W. (2002) Pharmacogenetics and personalised medicine. Fundam. Clin. Pharmacol. 16, 337–342.
Середенин С.Б. (2004) Лекции по фармакогенетике. М., Медицинское информационное агентство (МИА), 303 с.
Степанов В.А. (2010) Геномы, популяции, болезни: этническая геномика и персонифицированная медицина. Acta Nature. 4, 18–34.
Laing R.E., Hess P., Shen Y., Wang J., Hu S.X. (2011) The role and impact of SNPs in pharmacogenomics and personalized medicine. Curr. Drug. Metab. 12, 460–486.
Sauna Z.E., Kimchi-Sarfaty C., Ambudkar S.V., Gottesman M.M. (2007) Silent polymorphisms speak: how they affect pharmacogenomics and the treatment of cancer. Cancer Res. 67, 9609–9612.
Consortium E.P., Birney E., Stamatoyannopoulos J.A., Dutta A., Guigo R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Kuehn M.S., Taylor C.M., Neph S., Koch C.M., Asthana S., Malhotra A., Adzhubei I., Greenbaum J.A., Andrews R.M., Flicek P., Boyle P.J., Cao H., Carter N.P., Clelland G.K., Davis S., Day N., Dhami P., Dillon S.C., Dorschner M.O., Fiegler H., Giresi P.G., Goldy J., Hawrylycz M., Haydock A., Humbert R., James K.D., Johnson B.E., Johnson E.M., Frum T.T., Rosenzweig E.R., Karnani N., Lee K., Lefebvre G.C., Navas P.A., Neri F., Parker S.C., Sabo P.J., Sandstrom R., Shafer A., Vetrie D., Weaver M., Wilcox S., Yu M., Collins F.S., Dekker J., Lieb J.D., Tullius T.D., Crawford G.E., Sunyaev S., Noble W.S., Dunham I., Denoeud F., Reymond A., Kapranov P., Rozowsky J., Zheng D., Castelo R., Frankish A., Harrow J., Ghosh S., Sandelin A., Hofacker I.L., Baertsch R., Keefe D., Dike S., Cheng J., Hirsch H.A., Sekinger E.A., Lagarde J., Abril J.F., Shahab A., Flamm C., Fried C., Hackermuller J., Hertel J., Lindemeyer M., Missal K., Tanzer A., Washietl S., Korbel J., Emanuelsson O., Pedersen J.S., Holroyd N., Taylor R., Swarbreck D., Matthews N., Dickson M.C., Thomas D.J., Weira-uch M.T., Gilbert J., Drenkow J., Bell I., Zhao X., Srinivasan K.G., Sung W.K., Ooi H.S., Chiu K.P., Foissac S., Alioto T., Brent M., Pachter L., Tress M.L., Valencia A., Choo S.W., Choo C.Y., Ucla C., Manzano C., Wyss C., Cheung E., Clark T.G., Brown J.B., Ganesh M., Patel S., Tammana H., Chrast J., Henrichsen C.N., Kai C., Kawai J., Nagalakshmi U., Wu J., Lian Z., Lian J., Newburger P., Zhang X., Bickel P., Mattick J.S., Carninci P., Hayashizaki Y., Weissman S., Hubbard T., Myers R.M., Rogers J., Stadler P.F., Lowe T.M., Wei C.L., Ruan Y., Struhl K., Gerstein M., Antonarakis S.E., Fu Y., Green E.D., Karaoz U., Siepel A., Taylor J., Liefer L.A., Wetterstrand K.A., Good P.J., Feingold E.A., Guyer M.S., Cooper G.M., Asimenos G., Dewey C.N., Hou M., Nikolaev S., Montoya-Burgos J.I., Loytynoja A., Whelan S., Pardi F., Massingham T., Huang H., Zhang N.R., Holmes I., Mullikin J.C., Ureta-Vidal A., Paten B., Seringhaus M., Church D., Rosenbloom K., Kent W.J., Stone E.A., Program N.C.S., Baylor College of Medicine Human Genome Sequencing C., Washington University Genome Sequencing C., Broad I., Children’s Hospital Oakland Research I., Batzoglou S., Goldman N., Hardison R.C., Haussler D., Miller W., Sidow A., Trinklein N.D., Zhang Z.D., Barrera L., Stuart R., King D.C., Ameur A., Enroth S., Bieda M.C., Kim J., Bhinge A.A., Jiang N., Liu J., Yao F., Vega V.B., Lee C.W., Ng P., Shahab A., Yang A., Moqtaderi Z., Zhu Z., Xu X., Squazzo S., Oberley M.J., Inman D., Singer M.A., Richmond T.A., Munn K.J., Rada-Iglesias A., Wallerman O., Komorowski J., Fowler J.C., Couttet P., Bruce A.W., Dovey O.M., Ellis P.D., Langford C.F., Nix D.A., Euskirchen G., Hartman S., Urban A.E., Kraus P., Van Calcar S., Heintzman N., Kim T.H., Wang K., Qu C., Hon G., Luna R., Glass C.K., Rosenfeld M.G., Aldred S.F., Cooper S.J., Halees A., Lin J.M., Shulha H.P., Zhang X., Xu M., Haidar J.N., Yu Y., Ruan Y., Iyer V.R., Green R.D., Wadelius C., Farnham P.J., Ren B., Harte R.A., Hinrichs A.S., Trumbower H., Clawson H., Hillman-Jackson J., Zweig A.S., Smith K., Thakkapallayil A., Barber G., Kuhn R.M., Karolchik D., Armengol L., Bird C.P., De Bakker P.I., Kern A.D., Lopez-Bigas N., Martin J.D., Stranger B.E., Woodroffe A., Davydov E., Dimas A., Eyras E., Hallgrimsdottir I.B., Huppert J., Zody M.C., Abecasis G.R., Estivill X., Bouffard G.G., Guan X., Hansen N.F., Idol J.R., Maduro V.V., Maskeri B., Mcdowell J.C., Park M., Thomas P.J., Young A.C., Blakesley R.W., Muzny D.M., Sodergren E., Wheeler D.A., Worley K.C., Jiang H., Weinstock G.M., Gibbs R.A., Graves T., Fulton R., Mardis E.R., Wilson R.K., Clamp M., Cuff J., Gnerre S., Jaffe D.B., Chang J.L., Lindblad-Toh K., Lander E.S., Koriabine M., Nefedov M., Osoegawa K., Yoshinaga Y., Zhu B., De Jong P.J. (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 447, 799–816.
Bashinskaya V.V., Kulakova O.G., Boyko A.N., Favorov A.V., Favorova O.O. (2015) A review of genome-wide association studies for multiple sclerosis: classical and hypothesis-driven approaches. Hum. Genet. 134, 1143–1162.
Grossman I., Avidan N., Singer C., Goldstaub D., Hayardeny L., Eyal E., Ben-Asher E., Paperna T., Pe’er I., Lancet D., Beckmann J.S., Miller A. (2007) Pharmacogenetics of glatiramer acetate therapy for multiple sclerosis reveals drug-response markers. Pharmacogenet. Genomics. 17, 657–667.
Gross R., Healy B.C., Cepok S., Chitnis T., Khoury S.J., Hemmer B., Weiner H.L., Hafler D.A., De Jager P.L. (2011) Population structure and HLA DRB1 1501 in the response of subjects with multiple sclerosis to first-line treatments. J. Neuroimmunol. 233, 168–174.
Beaulieu M., De Denus S., Lachaine J. (2010) Systematic review of pharmacoeconomic studies of pharmacogenomic tests. Pharmacogenomics. 11, 1573–1590.
King C.R., Porche-Sorbet R.M., Gage B.F., Ridker P.M., Renaud Y., Phillips M.S., Eby C. (2008) Performance of commercial platforms for rapid genotyping of polymorphisms affecting warfarin dose. Am. J. Clin. Pathol. 129, 876–883.
Marin-Leblanc M., Perreault S., Bahroun I., Lapointe M., Mongrain I., Provost S., Turgeon J., Talajic M., Brugada R., Phillips M., Tardif J.C., Dube M.P. (2012) Validation of warfarin pharmacogenetic algorithms in clinical practice. Pharmacogenomics. 13, 21–29.
Carlquist J.F., Anderson J.L. (2011) Using pharmacogenetics in real time to guide warfarin initiation: a clinician update. Circulation. 124, 2554–2259.
Wellcome Trust Case Control C., Australo-Anglo-American Spondylitis C., Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., Mccarthy M.I., Ouwehand W.H., Samani N.J., Todd J.A., Donnelly P., Barrett J.C., Davison D., Easton D., Evans D.M., Leung H.T., Marchini J.L., Morris A.P., Spencer C.C., Tobin M.D., Attwood A.P., Boorman J.P., Cant B., Everson U., Hussey J.M., Jolley J.D., Knight A.S., Koch K., Meech E., Nutland S., Prowse C.V., Stevens H.E., Taylor N.C., Walters G.R., Walker N.M., Watkins N.A., Winzer T., Jones R.W., Mcardle W.L., Ring S.M., Strachan D.P., Pembrey M., Breen G., St Clair D., Caesar S., Gordon-Smith K., Jones L., Fraser C., Green E.K., Grozeva D., Hamshere M.L., Holmans P.A., Jones I.R., Kirov G., Moskivina V., Nikolov I., O’donovan M.C., Owen M.J., Collier D.A., Elkin A., Farmer A., Williamson R., Mcguffin P., Young A.H., Ferrier I.N., Ball S.G., Balmforth A.J., Barrett J.H., Bishop T.D., Iles M.M., Maqbool A., Yuldasheva N., Hall A.S., Braund P.S., Dixon R.J., Mangino M., Stevens S., Thompson J.R., Bredin F., Tremelling M., Parkes M., Drummond H., Lees C.W., Nimmo E.R., Satsangi J., Fisher S.A., Forbes A., Lewis C.M., Onnie C.M., Prescott N.J., Sanderson J., Matthew C.G., Barbour J., Mohiuddin M.K., Todhunter C.E., Mansfield J.C., Ahmad T., Cummings F.R., Jewell D.P., Webster J., Brown M.J., Lathrop M.G., Connell J., Dominiczak A., Marcano C.A., Burke B., Dobson R., Gungadoo J., Lee K.L., Munroe P.B., Newhouse S.J., Onipinla A., Wallace C., Xue M., Caulfield M., Farrall M., Barton A., Biologics In R.a.G., Genomics Study Syndicate Steering C., Bruce I.N., Donovan H., Eyre S., Gilbert P.D., Hilder S.L., Hinks A.M., John S.L., Potter C., Silman A.J., Symmons D.P., Thomson W., Worthington J., Dunger D.B., Widmer B., Frayling T.M., Freathy R.M., Lango H., Perry J.R., Shields B.M., Weedon M.N., Hattersley A.T., Hitman G.A., Walker M., Elliott K.S., Groves C.J., Lindgren C.M., Rayner N.W., Timpson N.J., Zeggini E., Newport M., Sirugo G., Lyons E., Vannberg F., Hill A.V., Bradbury L.A., Farrar C., Pointon J.J., Wordsworth P., Brown M.A., Franklyn J.A., Heward J.M., Simmonds M.J., Gough S.C., Seal S., Breast Cancer Susceptibility C., Stratton M.R., Rahman N., Ban M., Goris A., Sawcer S.J., Compston A., Conway D., Jallow M., Newport M., Sirugo G., Rockett K.A., Bumpstead S.J., Chaney A., Downes K., Ghori M.J., Gwilliam R., Hunt S.E., Inouye M., Keniry A., King E., Mcginnis R., Potter S., Ravindrarajah R., Whittaker P., Widden C., Withers D., Cardin N.J., Davison D., Ferreira T., Pereira-Gale J., Hallgrimsdo’ttir I.B., Ho-wie B.N., Su Z., Teo Y.Y., Vukcevic D., Bentley D., Brown M.A., Compston A., Farrall M., Hall A.S., Hattersley A.T., Hill A.V., Parkes M., Pembrey M., Stratton M.R., Mitchell S.L., Newby P.R., Brand O.J., Carr-Smith J., Pearce S.H., Mcginnis R., Keniry A., Deloukas P., Reveille J.D., Zhou X., Sims A.M., Dowling A., Taylor J., Doan T., Davis J.C., Savage L., Ward M.M., Learch T.L., Weisman M.H., Brown M. (2007) Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39(11), 1329–1337.
Baecher-Allan C., Kaskow B.J., Weiner H.L. (2018) Multiple sclerosis: mechanisms and immunotherapy. Neuron. 97, 742–768.
Гусев Е.И., Бойко А.Н., Завалишин И.А., Быкова О.В. (2004) Современная эпидемиология рассеянного склероза. Рассеянный склероз и другие демиелинизирующие заболевания. Ред. Гусев Е.И., Завалишин И.А., Бойко А.Н. Москва: Миклош, с. 8–29.
Габибов А.Г., Фаворова О.О., Кулакова О.Г., Бойко А.Н., Белогуров А.А., Пономаренко Н.А., Гусев Е.И. (2010) Рассеянный склероз. Нейродегенеративные заболевания: фундаментальные и прикладные аспекты. Ред. Угрюмов М.В. Москва: Наука, с. 382–442.
Фаворова O.O., Кулакова О.Г., Бойко А.Н. (2010) Рассеянный склероз как полигенное заболевание: современное состояние проблемы. Генетика. 46, 302–313.
Bjartmar C., Wujek J.R., Trapp B.D. (2003) Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J. Neurol. Sci. 206, 165–171.
Ascherio A., Munger K.L. (2007) Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann. Neurol. 61, 288–299.
Ascherio A., Munger K.L. (2007) Environmental risk factors for multiple sclerosis. Part II: noninfectious factors. Ann. Neurol. 61, 504–513.
Stadelmann C., Wegner C., Bruck W. (2011) Inflammation, demyelination, and degeneration – recent insights from MS pathology. Biochim. Biophys. Acta. 1812, 275–282.
Dendrou C.A., Fugger L., Friese M.A. (2015) Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558.
Tauber S.C., Nau R., Gerber J. (2007) Systemic infections in multiple sclerosis and experimental autoimmune encephalomyelitis. Arch. Physiol. Biochem. 113, 124–130.
Dobson R., Meier U.C., Giovannoni G. (2011) More to come: humoral immune responses in MS. J. Neuroimmunol. 240–241, 13–21.
Lassmann H., Bradl M. (2017) Multiple sclerosis: experimental models and reality. Acta Neuropathol. 133(2), 223–244.
Von Budingen H.C., Bar-Or A., Zamvil S.S. (2011) B cells in multiple sclerosis: connecting the dots. Curr. Opin. Immunol. 23, 713–720.
Soelberg Sorensen P. (2017) Safety concerns and risk management of multiple sclerosis therapies. Acta Neurol. Scand. 136, 168–186.
Hanson K.A., Agashivala N., Wyrwich K.W., Raimundo K., Kim E., Brandes D.W. (2014) Treatment selection and experience in multiple sclerosis: survey of neurologists. Patient Prefer Adherence. 8, 415–422.
Uze G., Schreiber G., Piehler J., Pellegrini S. (2007) The receptor of the type I interferon family. Curr. Top. Microbiol. Immunol. 316, 71–95.
Jiang H., Milo R., Swoveland P., Johnson K.P., Panitch H., Dhib-Jalbut S. (1995) Interferon beta-1b reduces interferon gamma-induced antigen-presenting capacity of human glial and B cells. J. Neuroimmunol. 61, 17–25.
Hallal-Longo D.E., Mirandola S.R., Oliveira E.C., Farias A.S., Pereira F.G., Metze I.L., Brandao C.O., Ruocco H.H., Damasceno B.P., Santos L.M. (2007) Diminished myelin-specific T cell activation associated with increase in CTLA4 and Fas molecules in multiple sclerosis patients treated with IFN-beta. J. Interferon Cytokine Res. 27, 865–873.
Kieseier B.C. (2011) The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs. 25, 491–502.
Namdar A., Nikbin B., Ghabaee M., Bayati A., Izad M. (2010) Effect of IFN-beta therapy on the frequency and function of CD4(+)CD25(+) regulatory T cells and Foxp3 gene expression in relapsing-remitting multiple sclerosis (RRMS): a preliminary study. J. Neuroimmunol. 218, 120–124.
Korporal M., Haas J., Balint B., Fritzsching B., Schwarz A., Moeller S., Fritz B., Suri-Payer E., Wildemann B. (2008) Interferon beta-induced restoration of regulatory T-cell function in multiple sclerosis is prompted by an increase in newly generated naive regulatory T cells. Arch. Neurol. 65, 1434–1439.
Aristimuno C., De Andres C., Bartolome M., De Las Heras V., Martinez-Gines M.L., Arroyo R., Fernandez-Cruz E., Sanchez-Ramon S. (2010) IFNbeta-1a therapy for multiple sclerosis expands regulatory CD8+ T cells and decreases memory CD8+ subset: a longitudinal 1-year study. Clin. Immunol. 134, 148–157.
Waubant E., Goodkin D., Bostrom A., Bacchetti P., Hietpas J., Lindberg R., Leppert D. (2003) IFNbeta lowers MMP-9/TIMP-1 ratio, which predicts new enhancing lesions in patients with SPMS. Neurology. 60, 52–57.
Teleshova N., Pashenkov M., Huang Y.M., Soderstrom M., Kivisakk P., Kostulas V., Haglund M., Link H. (2002) Multiple sclerosis and optic neuritis: CCR5 and CXCR3 expressing T cells are augmented in blood and cerebrospinal fluid. J. Neurol. 249, 723–729.
Biernacki K., Antel J.P., Blain M., Narayanan S., Arnold D.L., Prat A. (2005) Interferon beta promotes nerve growth factor secretion early in the course of multiple sclerosis. Arch. Neurol. 62, 563–568.
Arnon R. (1996) The development of Cop 1 (Copaxone), an innovative drug for the treatment of multiple sclerosis: personal reflections. Immunol. Lett. 50, 1–15.
Duda P.W., Schmied M.C., Cook S.L., Krieger J.I., Hafler D.A. (2000) Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J. Clin. Invest. 105, 967–976.
Vieira P.L., Heystek H.C., Wormmeester J., Wierenga E.A., Kapsenberg M.L. (2003) Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J. Immunol. 170, 4483–4488.
Chen M., Gran B., Costello K., Johnson K., Martin R., Dhib-Jalbut S. (2001) Glatiramer acetate induces a Th2-biased response and crossreactivity with myelin basic protein in patients with MS. Mult. Scler. 7, 209–219.
Weder C., Baltariu G.M., Wyler K.A., Gober H.J., Lienert C., Schluep M., Radu E.W., De Libero G., Kappos L., Duda P.W. (2005) Clinical and immune responses correlate in glatiramer acetate therapy of multiple sclerosis. Eur. J. Neurol. 12, 869–878.
Teitelbaum D., Milo R., Arnon R., Sela M. (1992) Synthetic copolymer 1 inhibits human T-cell lines specific for myelin basic protein. Proc. Natl. Acad. Sci. USA. 89, 137–141.
Fridkis-Hareli M., Teitelbaum D., Gurevich E., Pecht I., Brautbar C., Kwon O.J., Brenner T., Arnon R., Sela M. (1994) Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells – specificity and promiscuity. Proc. Natl. Acad. Sci. USA. 91, 4872–4876.
Haas J., Korporal M., Balint B., Fritzsching B., Schwarz A., Wildemann B. (2009) Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4(+)CD25(+)FOXP3(+)CD31(+) T-cells in patients with multiple sclerosis. J. Neuroimmunol. 216, 113–117.
Hong J., Li N., Zhang X., Zheng B., Zhang J.Z. (2005) Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc. Natl. Acad. Sci. USA. 102, 6449–6454.
Tennakoon D.K., Mehta R.S., Ortega S.B., Bhoj V., Racke M.K., Karandikar N.J. (2006) Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J. Immunol. 176, 7119–7129.
Karandikar N.J., Crawford M.P., Yan X., Ratts R.B., Brenchley J.M., Ambrozak D.R., Lovett-Racke A.E., Frohman E.M., Stastny P., Douek D.C., Koup R.A., Racke M.K. (2002) Glatiramer acetate (Copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J. Clin. Invest. 109, 641–649.
Kala M., Rhodes S.N., Piao W.H., Shi F.D., Campagnolo D.I., Vollmer T.L. (2010) B cells from glatiramer acetate-treated mice suppress experimental autoimmune encephalomyelitis. Exp. Neurol. 221, 136–145.
Ure D.R., Rodriguez M. (2002) Polyreactive antibodies to glatiramer acetate promote myelin repair in murine model of demyelinating disease. FASEB J. 16, 1260–1262.
Aharoni R., Kayhan B., Eilam R., Sela M., Arnon R. (2003) Glatiramer acetate-specific T cells in the brain express T helper 2/3 cytokines and brain-derived neurotrophic factor in situ. Proc. Natl. Acad. Sci. USA. 100, 14157–14162.
Ziemssen T., Kumpfel T., Schneider H., Klinkert W.E., Neuhaus O., Hohlfeld R. (2005) Secretion of brain-derived neurotrophic factor by glatiramer acetate-reactive T-helper cell lines: implications for multiple sclerosis therapy. J. Neurol. Sci. 233, 109–112.
Racke M.K., Lovett-Racke A.E., Karandikar N.J. (2010) The mechanism of action of glatiramer acetate treatment in multiple sclerosis. Neurology. 74(Suppl. 1), S25–S30.
Kappos L., Weinshenker B., Pozzilli C., Thompson A.J., Dahlke F., Beckmann K., Polman C., Mcfarland H., European Interferon Beta-1b in Secondary Progressive Multiple Sclerosis Trial Steering C., Independent Advisory B. North American Interferon Beta-1b in Secondary Progressive Multiple Sclerosis Trial Steering C. Independent Advisory B. (2004) Interferon beta-1b in secondary progressive MS: a combined ana-lysis of the two trials. Neurology. 63, 1779–1787.
Kappos L., Traboulsee A., Constantinescu C., Eralinna J.P., Forrestal F., Jongen P., Pollard J., Sandberg-Wollheim M., Sindic C., Stubinski B., Uitdehaag B., Li D. (2006) Long-term subcutaneous interferon beta-1a therapy in patients with relapsing-remitting MS. Neurology. 67, 944–953.
Бойко А.Н. Хачанова Н.В., Буглак А.В., Демина Т.Л., Беляева И.А., Лащ Н.В., Серков С.В., Гусев Е.И. (2000) Возможность использования клинических, генетических показателей и данных магнитно-резонансной томографии для прогнозирования эффектов лечения бета-интерфероном-1b рассеянного склероза. Журнал неврологии и психиатрии им. С.С. Корсакова. 100, 53–59.
Fusco C., Andreone V., Coppola G., Luongo V., Guerini F., Pace E., Florio C., Pirozzi G., Lanzillo R., Ferrante P., Vivo P., Mini M., Macri M., Orefice G., Lombardi M.L. (2001) HLA-DRB1*1501 and response to copolymer-1 therapy in relapsing-remitting multiple sclerosis. Neurology. 57, 1976–1979.
Wergeland S., Beiske A., Nyland H., Hovdal H., Jensen D., Larsen J.P., Maroy T.H., Smievoll A.I., Vedeler C.A., Myhr K.M. (2005) IL-10 promoter haplotype influence on interferon treatment response in multiple sclerosis. Eur. J. Neurol. 12, 171–175.
Guerrero A.L., Tejero M.A., Gutierrez F., Martin-Polo J., Iglesias F., Laherran E., Martin-Serradilla J.I., Merino S. (2011) Influence of APOE gene polymorphisms on interferon-beta treatment response in multiple sclerosis. Neurologia. 26, 137–142.
Bustamante M.F., Morcillo-Suarez C., Malhotra S., Rio J., Leyva L., Fernandez O., Zettl U.K., Killestein J., Brassat D., Garcia-Merino J.A., Sanchez A.J., Urcelay E., Alvarez-Lafuente R., Villar L.M., Alvarez-Cermeno J.C., Farre X., Lechner-Scott J., Vandenbroeck K., Rodriguez-Antiguedad A., Drulovic J.S., Martinelli Boneschi F., Chan A., Oksenberg J., Navarro A., Montalban X., Comabella M. (2015) Pharmaco-genomic study in patients with multiple sclerosis: responders and nonresponders to IFN-beta. Neurol. Neuroimmunol. Neuroinflamm. 2, e154.
Konig I.R. (2011) Validation in genetic association studies. Brief. Bioinform. 12, 253–258.
Vosslamber S., Van Der Voort L.F., Van Den Elskamp I.J., Heijmans R., Aubin C., Uitdehaag B.M., Crusius J.B., Van Der Pouw Kraan T.C., Comabella M., Montalban X., Hafler D.A., De Jager P.L., Killestein J., Polman C.H., Verweij C.L. (2011) Interferon regulatory factor 5 gene variants and pharmacological and clinical outcome of Interferonβ therapy in multiple sclerosis. Genes Immun. 12, 466–472.
Vandenbroeck K., Alloza I., Swaminathan B., Antiguedad A., Otaegui D., Olascoaga J., Barcina M.G., De Las Heras V., Bartolome M., Fernandez-Arquero M., Arroyo R., Alvarez-Lafuente R., Cenit M.C., Urcelay E. (2011) Validation of IRF5 as multiple sclerosis risk gene: putative role in interferon beta therapy and human herpes virus-6 infection. Genes Immun. 12, 40–45.
Lopez-Gomez C., Pino-Angeles A., Orpez-Zafra T., Pinto-Medel M.J., Oliver-Martos B., Ortega-Pinazo J., Arnaiz C., Guijarro-Castro C., Varade J., Alvarez-Lafuente R., Urcelay E., Sanchez-Jimenez F., Fernandez O., Leyva L. (2013) Candidate gene study of TRAIL and TRAIL receptors: association with response to interferon beta therapy in multiple sclerosis patients. PLoS One. 8, e62540.
Kulakova O.G., Tsareva E.Y., Boyko A.N., Shchur S.G., Gusev E.I., Lvovs D., Favorov A.V., Vandenbroeck K., Favorova O.O. (2012) Allelic combinations of immune-response genes as possible composite markers of IFN-β efficacy in multiple sclerosis patients. Pharmacogenomics. 13, 1689–1700.
Mazdeh M., Taheri M., Sayad A., Bahram S., Omrani M.D., Movafagh A., Inoko H., Akbari M.T., Noroozi R., Hajilooi M., Solgi G. (2016) HLA genes as modifiers of response to IFN-β-1a therapy in relapsing-remitting multiple sclerosis. Pharmacogenomics. 17, 489–498.
Karam R.A., Rezk N.A., Amer M.M., Fathy H.A. (2016) Immune response genes receptors expression and polymorphisms in relation to multiple sclerosis susceptibility and response to INF-β therapy. IUBMB Life. 68, 727–734.
Cenit M.D., Blanco-Kelly F., De Las Heras V., Bartolome M., De La Concha E.G., Urcelay E., Arroyo R., Martinez A. (2009) Glypican 5 is an interferon-beta response gene: a replication study. Mult. Scler. 15, 913–917.
Alvarez-Lafuente R., Blanco-Kelly F., Garcia-Montojo M., Martinez A., De Las Heras V., Dominguez-Mozo M.I., Bartolome M., Garcia-Martinez A., De La Concha E.G., Urcelay E., Arroyo R. (2011) CD46 in a Spanish cohort of multiple sclerosis patients: genetics, mRNA expression and response to interferon-beta treatment. Mult. Scler. 17, 513–520.
Cunningham S., Graham C., Hutchinson M., Droogan A., O’rourke K., Patterson C., Mcdonnell G., Hawkins S., Vandenbroeck K. (2005) Pharmacogenomics of responsiveness to interferon IFN-β treatment in multiple sclerosis: a genetic screen of 100 type I interferon-inducible genes. Clin. Pharmacol. Ther. 78, 635–646.
Torbati S., Karami F., Ghaffarpour M., Zamani M. (2015) Association of CD58 polymorphism with multiple sclerosis and response to interferon ss therapy in a subset of Iranian population. Cell J. 16, 506–513.
Martinez A., De Las Heras V., Mas Fontao A., Bartolome M., De La Concha E.G., Urcelay E., Arroyo R. (2006) An IFNG polymorphism is associated with interferon-beta response in Spanish MS patients. J. Neuroimmunol. 173, 196–199.
Ristic S., Starcevic Cizmarevic N., Lavtar P., Lov-recic L., Perkovic O., Sepcic J., Sega Jazbec S., Kapovic M., Peterlin B. (2017) Angiotensin-converting enzyme insertion/deletion gene polymorphism and interferon-beta treatment response in multiple sclerosis patients: a preliminary report. Pharmacogenet. Genomics. 27, 232–235.
Sayad A., Ghafouri-Fard S., Omrani M.D., Noroozi R., Taheri M. (2017) Myxovirus resistance protein A (MxA) polymorphism is associated with IFNβ response in Iranian multiple sclerosis patients. Neurol. Sci. 38, 1093–1099.
Sriram U., Barcellos L.F., Villoslada P., Rio J., Baranzini S.E., Caillier S., Stillman A., Hauser S.L., Montalban X., Oksenberg J.R. (2003) Pharmacogenomic analysis of interferon receptor polymorphisms in multiple sclerosis. Genes Immun. 4, 147–152.
Malhotra S., Morcillo-Suarez C., Nurtdinov R., Rio J., Sarro E., Moreno M., Castillo J., Navarro A., Montalban X., Comabella M. (2013) Roles of the ubiquitin peptidase USP18 in multiple sclerosis and the response to interferon-beta treatment. Eur. J. Neurol. 20, 1390–1397.
Villoslada P., Barcellos L.F., Rio J., Begovich A.B., Tintore M., Sastre-Garriga J., Baranzini S.E., Casquero P., Hauser S.L., Montalban X., Oksenberg J.R. (2002) The HLA locus and multiple sclerosis in Spain. Role in disease susceptibility, clinical course and response to interferon-beta. J. Neuroimmunol. 130, 194–201.
Fernandez O., Fernandez V., Mayorga C., Guerrero M., Leon A., Tamayo J.A., Alonso A., Romero F., Leyva L., Alonso A., Luque G., De Ramon E. (2005) HLA class II and response to interferon-beta in multiple sclerosis. Acta Neurol. Scand. 112, 391–394.
Comabella M., Fernandez-Arquero M., Rio J., Guinea A., Fernandez M., Cenit M.C., De La Concha E.G., Montalban X. (2009) HLA class I and II alleles and response to treatment with interferon-beta in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 210, 116–119.
Comabella M., Craig D.W., Morcillo-Suarez C., Rio J., Navarro A., Fernandez M., Martin R., Montalban X. (2009) Genome-wide scan of 500,000 single-nucleotide polymorphisms among responders and nonresponders to interferon beta therapy in multiple sclerosis. Arch. Neurol. 66, 972–978.
O’doherty C., Favorov A., Heggarty S., Graham C., Favorova O., Ochs M., Hawkins S., Hutchinson M., O’Rourke K., Vandenbroeck K. (2009) Genetic polymorphisms, their allele combinations and IFN-beta treatment response in Irish multiple sclerosis patients. Pharmacogenomics. 10, 1177–1186.
Platanias L.C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386.
Weinstock-Guttman B., Tamano-Blanco M., Bhasi K., Zivadinov R., Ramanathan M. (2007) Pharmacogenetics of MXA SNPs in interferon-beta treated multiple sclerosis patients. J. Neuroimmunol. 182, 236–239.
Tamura T., Yanai H., Savitsky D., Taniguchi T. (2008) The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584.
Shusta E.V., Zhu C., Boado R.J., Pardridge W.M. (2002) Subtractive expression cloning reveals high expression of CD46 at the blood-brain barrier. J. Neuropathol. Exp. Neurol. 61, 597–604.
Astier A.L., Meiffren G., Freeman S., Hafler D.A. (2006) Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J. Clin. Invest. 116, 3252–3257.
Falschlehner C., Schaefer U. Walczak H. (2009) Following TRAIL’s path in the immune system. Immunology. 127, 145–154.
Hoffmann O., Zipp F., Weber J.R. (2009) Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) in central nervous system inflammation. J. Mol. Med. (Berl.). 87, 753–763.
Stegbauer J., Lee D.H., Seubert S., Ellrichmann G., Manzel A., Kvakan H., Muller D.N., Gaupp S., Rump L.C., Gold R., Linker R.A. (2009) Role of the renin-angiotensin system in autoimmune inflammation of the central nervous system. Proc. Natl. Acad. Sci. USA. 106, 14942–14947.
Platten M., Youssef S., Hur E.M., Ho P.P., Han M.H., Lanz T.V., Phillips L.K., Goldstein M.J., Bhat R., Raine C.S., Sobel R.A., Steinman L. (2009) Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl. Acad. Sci. USA. 106, 14948–14953.
Constantinescu C.S., Goodman D.B., Grossman R.I., Mannon L.J., Cohen J.A. (1997) Serum angiotensin-converting enzyme in multiple sclerosis. Arch. Neurol. 54, 1012–1015.
Kawajiri M., Mogi M., Higaki N., Matsuoka T., Ohyagi Y., Tsukuda K., Kohara K., Horiuchi M., Miki T., Kira J.I. (2009) Angiotensin-converting enzyme (ACE) and ACE2 levels in the cerebrospinal fluid of patients with multiple sclerosis. Mult. Scler. 15, 262–265.
Leyva L., Fernandez O., Fedetz M., Blanco E., Fernandez V.E., Oliver B., Leon A., Pinto-Medel M.J., Mayorga C., Guerrero M., Luque G., Alcina A., Matesanz F. (2005) IFNAR1 and IFNAR2 polymorphisms confer susceptibility to multiple sclerosis but not to interferon-beta treatment response. J. Neuroimmunol. 163, 165–171.
Malhotra S., Morcillo-Suarez C., Brassat D., Goertsches R., Lechner-Scott J., Urcelay E., Fernandez O., Drulovic J., Garcia-Merino A., Martinelli Boneschi F., Chan A., Vandenbroeck K., Navarro A., Bustamante M.F., Rio J., Akkad D.A., Giacalone G., Sanchez A.J., Leyva L., Alvarez-Lafuente R., Zettl U.K., Oksenberg J., Montalban X., Comabella M. (2011) IL28B polymorphisms are not associated with the response to interferon-beta in multiple sclerosis. J. Neuroimmunol. 239, 101–104.
Malhotra S., Rio J., Urcelay E., Nurtdinov R., Bustamante M.F., Fernandez O., Oliver B., Zettl U., Brassat D., Killestein J., Lechner-Scott J., Drulovic J., Chan A., Martinelli-Boneschi F., Garcia-Merino A., Montalban X., Comabella M. (2015) NLRP3 inflammasome is associated with the response to IFN-β in patients with multiple sclerosis. Brain. 138, 644–652.
Malhotra S., Sorosina M., Rio J., Peroni S., Midaglia L., Villar L.M., Alvarez-Cermeno J.C., Schroeder I., Esposito F., Clarelli F., Zettl U.K., Lechner-Scott J., Spataro N., Navarro A., Comi G., Montalban X., Martinelli-Boneschi F., Comabella M. (2018) NLRP3 polymorphisms and response to interferon-beta in multiple sclerosis patients. Mult. Scler. 24, 1507–1510.
Favorov A.V., Andreewski T.V., Sudomoina M.A., Favorova O.O., Parmigiani G., Ochs M.F. (2005) A Markov chain Monte Carlo technique for identification of combinations of allelic variants underlying complex diseases in humans. Genetics. 171, 2113–2121.
Lvovs D., Фаворова О.О., Фаворов А.В. (2012) Полигенный подход к исследованиям полигенных заболеваний. Acta Naturae. 4, 62–75.
Byun E., Caillier S.J., Montalban X., Villoslada P., Fernandez O., Brassat D., Comabella M., Wang J., Barcellos L.F., Baranzini S.E., Oksenberg J.R. (2008) Genome-wide pharmacogenomic analysis of the response to interferon beta therapy in multiple sclerosis. Arch. Neurol. 65, 337–344.
Mahurkar S., Moldovan M., Suppiah V., Sorosina M., Clarelli F., Liberatore G., Malhotra S., Montalban X., Antiguedad A., Krupa M., Jokubaitis V.G., Mckay F.C., Gatt P.N., Fabis-Pedrini M.J., Martinelli V., Comi G., Lechner-Scott J., Kermode A.G., Slee M., Taylor B.V., Vandenbroeck K., Comabella M., Boneschi F.M., Australian, New Zealand Multiple Sclerosis Genetics C., King C. (2017) Response to interferon-beta treatment in multiple sclerosis patients: a genome-wide association study. Pharmacogenomics J. 17, 312–318.
Esposito F., Sorosina M., Ottoboni L., Lim E.T., Replogle J.M., Raj T., Brambilla P., Liberatore G., Guaschino C., Romeo M., Pertel T., Stankiewicz J.M., Martinelli V., Rodegher M., Weiner H.L., Brassat D., Benoist C., Patsopoulos N.A., Comi G., Elyaman W., Martinelli Boneschi F., De Jager P.L. (2015) A pharmacogenetic study implicates SLC9a9 in multiple sclerosis disease activity. Ann. Neurol. 78, 115–127.
Clarelli F., Liberatore G., Sorosina M., Osiceanu A.M., Esposito F., Mascia E., Santoro S., Pavan G., Colombo B., Moiola L., Martinelli V., Comi G., Martinelli-Boneschi F. (2017) Pharmacogenetic study of long-term response to interferon-beta treatment in multiple sclerosis. Pharmacogenomics J. 17, 84–91.
Ross C.J., Towfic F., Shankar J., Laifenfeld D., Thoma M., Davis M., Weiner B., Kusko R., Zeskind B., Knappertz V., Grossman I., Hayden M.R. (2017) A pharmacogenetic signature of high response to Copaxone in late-phase clinical-trial cohorts of multiple sclerosis. Genome Med. 9, 50.
Фаворова О.О., Башинская В.В., Кулакова О.Г., Фаворов А.В., Бойко А.Н. (2014) Полногеномный поиск ассоциаций как метод анализа генетической архитектуры полигенных заболеваний (на примере рассеянного склероза). Молекуляр. биология. 48, 573–586.
Ohgaki R., Van I.S.C., Matsushita M., Hoekstra D., Kanazawa H. (2011) Organellar Na+/H+ exchangers: novel players in organelle pH regulation and their emerging functions. Biochemistry. 50, 443–450.
Beydoun R., Hamood M.A., Gomez Zubieta D.M., Kondapalli K.C. (2017) Na(+)/H(+) exchanger 9 regulates iron mobilization at the blood-brain barrier in response to iron starvation. J. Biol. Chem. 292, 4293–4201.
Liu G., Zhang F., Hu Y., Jiang Y., Gong Z., Liu S., Chen X., Jiang Q. Hao J. (2017) Genetic variants and multiple sclerosis risk gene SLC9A9 expression in distinct human brain regions. Mol. Neurobiol. 54, 6820–6826.
Australia and New Zealand Multiple Sclerosis Genetics C. (2009) Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 41, 824–828.
International Multiple Sclerosis Genetics C., Wellcome Trust Case Control C., Sawcer S., Hellenthal G., Pirinen M., Spencer C.C., Patsopoulos N.A., Moutsianas L., Dilthey A., Su Z., Freeman C., Hunt S.E., Edkins S., Gray E., Booth D.R., Potter S.C., Goris A., Band G., Oturai A.B., Strange A., Saarela J., Bellenguez C., Fontaine B., Gillman M., Hemmer B., Gwilliam R., Zipp F., Jayakumar A., Martin R., Leslie S., Hawkins S., Giannoulatou E., D’Alfonso S., Blackburn H., Martinelli Boneschi F., Liddle J., Harbo H.F., Perez M.L., Spurkland A., Waller M.J., Mycko M.P., Ricketts M., Comabella M., Hammond N., Kockum I., Mccann O.T., Ban M., Whittaker P., Kemppinen A., Weston P., Hawkins C., Widaa S., Zajicek J., Dronov S., Robertson N., Bumpstead S.J., Barcellos L.F., Ravindrarajah R., Abraham R., Alfredsson L., Ardlie K., Aubin C., Baker A., Baker K., Baranzini S.E., Bergamaschi L., Bergamaschi R., Bernstein A., Berthele A., Boggild M., Bradfield J.P., Brassat D., Broadley S.A., Buck D., Butzkueven H., Capra R., Carroll W.M., Cavalla P., Celius E.G., Cepok S., Chiavacci R., Clerget-Darpoux F., Clysters K., Comi G., Cossburn M., Cournu-Rebeix I., Cox M.B., Cozen W., Cree B.A., Cross A.H., Cusi D., Daly M.J., Davis E., De Bakker P.I., Debouverie M., D’hooghe M B., Dixon K., Dobosi R., Dubois B., Ellinghaus D., Elovaara I., Esposito F., Fontenille C., Foote S., Franke A., Galimberti D., Ghezzi A., Glessner J., Gomez R., Gout O., Graham C., Grant S.F., Guerini F.R., Hakonarson H., Hall P., Hamsten A., Hartung H.P., Heard R.N., Heath S., Hobart J., Hoshi M., Infante-Duarte C., Ingram G., Ingram W., Islam T., Jagodic M., Kabesch M., Kermode A.G., Kilpatrick T.J., Kim C., Klopp N., Koivisto K., Larsson M., Lathrop M., Lechner-Scott J.S., Leone M.A., Leppa V., Liljedahl U., Bomfim I.L., Lincoln R.R., Link J., Liu J., Lorentzen A.R., Lupoli S., Macciardi F., Mack T., Marriott M., Martinelli V., Mason D., Mccauley J.L., Mentch F., Mero I.L., Mihalova T., Montalban X., Mottershead J., Myhr K.M., Naldi P., Ollier W., Page A., Palotie A., Pelletier J., Piccio L., Pickersgill T., Piehl F., Pobywajlo S., Quach H.L., Ramsay P.P., Reunanen M., Reynolds R., Rioux J.D., Rodegher M., Roesner S., Rubio J.P., Ruckert I.M., Salvetti M., Salvi E., Santaniello A., Schaefer C.A., Schreiber S., Schulze C., Scott R.J., Sellebjerg F., Selmaj K.W., Sexton D., Shen L., Simms-Acuna B., Skidmore S., Sleiman P.M., Smestad C., Sorensen P.S., Sondergaard H.B., Stankovich J., Strange R.C., Sulonen A.M., Sundqvist E., Syvanen A.C., Taddeo F., Taylor B., Blackwell J.M., Tienari P., Bramon E., Tourbah A., Brown M.A., Tronczynska E., Casas J.P., Tubridy N., Corvin A., Vickery J., Jankowski J., Villoslada P., Markus H.S., Wang K., Mathew C.G., Wason J., Palmer C.N., Wichmann H.E., Plomin R., Willoughby E., Rautanen A., Winkelmann J., Wittig M., Trembath R.C., Yaouanq J., Viswanathan A.C., Zhang H., Wood N.W., Zuvich R., Deloukas P., Langford C., Duncanson A., Oksenberg J.R., Pericak-Vance M.A., Haines J.L., Olsson T., Hillert J., Ivinson A.J., De Jager P.L., Peltonen L., Stewart G.J., Hafler D.A., Hauser S.L., Mcvean G., Donnelly P.,Compston A. (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 476, 214–219.
Dhib-Jalbut S., Valenzuela R.M., Ito K., Kaufman M., Ann Picone M., Buyske S. (2013) HLA DR and DQ alleles and haplotypes associated with clinical response to glatiramer acetate in multiple sclerosis. Mult. Scler. Relat. Disord. 2, 340–348.
Алифирова В.М., Орлова Ю.Ю., Бабенко С.А., Рудко А.А., Пузырев В.П. (2006) Полиморфизм 1188 А/С гена IL12B у больных рассеянным склерозом в Томской области и возможности оценки эффективности иммуномодулирующей терапии. Журнал неврологии и психиатрии им. С.С. Корсакова. 3, 130–135.
Царева Е.Ю., Кулакова О.Г., Макарычева О.Ю., Бойко А.Н., Щур С.Г., Лащ Н.Ю., Попова Н.Ф., Гусев Е.И., Башинская В.В., Львов Д.В., Фаворов А.В., Ochs M.F., Фаворова О.О. Фармакогеномика рассеянного склероза: ассоциация полиморфизма генов иммунного ответа с эффективностью лечения копаксоном. Молекуляр. биология. 45, 963–972.
Kulakova O., Bashinskaya V., Kiselev I., Baulina N., Tsareva E., Nikolaev R., Kozin M., Shchur S., Favo-rov A., Boyko A., Favorova O. (2017) Pharmacogenetics of glatiramer acetate therapy for multiple sclerosis: the impact of genome-wide association studies identified disease risk loci. Pharmacogenomics. 18, 1563–1574.
Tsareva E.Y., Kulakova O.G., Boyko A.N., Shchur S.G., Lvovs D., Favorov A.V., Gusev E.I., Vandenbroeck K., Favorova O.O. (2012) Allelic combinations of immune-response genes associated with glatiramer acetate treatment response in Russian multiple sclerosis patients. Pharmacogenomics. 13, 43–53.
Barsova R.M., Lvovs D., Titov B.V., Matveeva N.A., Shakhnovich R.M., Sukhinina T.S., Kukava N.G., Ruda M.Y., Karamova I.M., Nasibullin T.R., Mustafina O.E., Osmak G.J., Tsareva E.Y., Kulakova O.G., Favorov A.V., Favorova O.O. (2015) Variants of the coagulation and inflammation genes are replicably associated with myocardial infarction and epistatically interact in Russians. PLoS One. 10, e0144190.
Kulakova O.G., Tsareva E.Y., Lvovs D., Favorov A.V., Boyko A.N., Favorova O.O. (2014) Comparative pharmacogenetics of multiple sclerosis: IFN-β versus glatiramer acetate. Pharmacogenomics. 15, 679–685.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология