

Молекулярная биология, 2019, T. 53, № 5, стр. 755-773
Гуманизированные мыши со сверхэкспрессией генов человека как инструмент изучения функций провоспалительных цитокинов
Е. А. Горшкова a, b, *, Р. В. Зварцев a, М. С. Друцкая a, Е. О. Губернаторова a, b, **
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук,
Лаборатория молекулярных механизмов иммунитета
119991 Москва, Россия
b Кафедра иммунологии биологического факультета Московского государственного университета
им. М.В. Ломоносова
119234 Москва, Россия
* E-mail: gorshsama@gmail.com
** E-mail: ekaterina.gubernatorova412@gmail.com
Поступила в редакцию 10.04.2019
После доработки 24.04.2019
Принята к публикации 25.04.2019
Аннотация
Нарушение регуляции экспрессии провоспалительных цитокинов может приводить к развитию серьезных патологий: ревматоидного артрита, псориаза, нейродегенеративных заболеваний. В последние годы в исследованиях молекулярных механизмов, лежащих в основе развития заболеваний, активно используют трансгенных мышей, в том числе при моделировании регулируемого системного усиления экспрессии различных цитокинов. Важно отметить, что многие из этих моделей относятся к гуманизированным, то есть используемые мыши несут гены цитокинов человека, а эндогенный мышиный ген или оставляют, или удаляют. Гуманизированные модели представляют особую ценность для биомедицины, в частности, для разработки терапии, основанной на блокировке патогенетической активности цитокина, или для определения функциональной значимости геномных полиморфизмов. В обзоре обсуждаются существующие модели сверхэкспрессии основных плейотропных провоспалительных цитокинов: TNF, IL-1β, IL-6, ‒ а также воспалительных цитокинов с более специфическими функциями: IL-8, IL-17, IL-32 ‒ и их значение для фундаментальных и клинических исследований.
ВВЕДЕНИЕ
Цитокины играют центральную роль в регуляции иммунной системы, и нарушение их экспрессии может приводить к развитию серьезных патологий. Так, повышение уровня фактора некроза опухоли (TNF) и интерлейкина-6 (IL-6) наблюдают у пациентов с ревматоидным артритом; сверхэкспрессия IL-1β характерна для острого прогрессирующего панкреатита, а экспрессия IL-6, IL-17 и других цитокинов значительно повышена при воспалительных заболеваниях нервной системы. Разработка технологий геномного редактирования с целью получения мышей с дефицитом в цитокиновых сигнальных каскадах, а также трансгенных мышей, в геном которых искусственно вставлены гены, в том числе человека, кодирующие цитокины, стала прорывом в иммунологии. Кроме того, применение методов обратной генетики позволило установить молекулярные механизмы передачи сигналов, функции цитокинов и их рецепторов в норме и при развитии воспаления и ряда аутоиммунных заболеваний [1]. Несколько десятилетий назад, благодаря открытию общих механизмов регуляции экспрессии генов, стало возможным получение трансгенных животных, в которых привнесенный ген конститутивно экспрессируется на высоком уровне. При изучении трансгенных мышей со сверхэкспрессией целевых генов был сделан ряд важных открытий относительно молекулярных механизмов патогенеза заболеваний, ассоциированных с повышенной экспрессией провоспалительных цитокинов [2‒4].
Мышиные модели заболеваний человека уже многие десятилетия позволяют исследовать молекулярные механизмы, лежащие в основе патогенеза заболеваний. С другой стороны, ортологичные белки человека и грызунов обладают различной аминокислотной последовательностью, что создает серьезные ограничения для разработки и доклинических испытаний на мышах лекарственных препаратов на основе антител. Гуманизация мышей, то есть введение в геном мыши гена, кодирующего белок человека, частично нивелирует недостатки мышиных моделей. Часто для сохранения работоспособности системы передачи сигнала через мышиный рецептор цитокина достаточно гуманизации лиганда (собственно, цитокина); именно такие примеры и будут рассмотрены в настоящем обзоре. В противном случае необходима двойная гуманизация пары лиганд‒рецептор. Гуманизированные мыши, в которых ген человека, кодирующий определенный белок, заменяет соответствующий мышиный ген или экспрессируется параллельно с мышиным геном, представляют собой уникальный организм, в котором сочетаются преимущества животных моделей и молекулярной имитации реальных патофизиологических процессов, происходящих в организме человека. Для получения такого организма используют методы трансгенеза, то есть внесения в геном одного или нескольких генов другого вида.
Методы получения трансгенных животных могут быть разделены на два типа по специфичности доставки целевой ДНК в геном. В первом случае итоговая локализация трансгена случайна, так как встраивание происходит либо в результате инфицирования эмбриона ретровирусом [5], либо в результате случайной интеграции, связанной с геномными микрогомологиями вектора, введенного в пронуклеус [6] (рис. 1а, б). Большинство трансгенных мышей, рассмотренных в обзоре, получено с помощью случайной интеграции, которая позволяет доставлять последовательность гена человека в геном мыши целиком в составе вектора, несущего собственные регуляторные элементы (рис. 1б). Важная особенность классического трансгенеза состоит в том, что при микроинъекции в геном может встраиваться разное число копий ДНК-конструкции. По этой причине первичные трансгенные организмы и клетки из этих организмов могут сильно отличаться друг от друга по уровню экспрессии трансгена и, следовательно, по интенсивности проявления фенотипа. Существенный недостаток вышеописанного метода заключается в непредсказуемости локуса интеграции трансгена.
Рис. 1.
Технологии внесения чужеродного гена для получения гуманизированных мышей. а ‒ Введение целевого гена с помощью рекомбинантного ретровируса, заражающего 8-клеточные эмбрионы, приводит к случайному встраиванию трансгена. б ‒ Микроинъекция экзогенной ДНК в мужской пронуклеус оплодотворенной яйцеклетки приводит к случайному встраиванию трансгена. в ‒ Для направленного введения целевого гена с помощью системы CRISPR/Cas9 (Easi-CRISPR) могут быть использованы как оплодотворенные яйцеклетки, так и линии эмбриональных стволовых клеток (ЭСК). г ‒ Технология “knock-in” заключается в направленном геномном редактировании линии ЭСК, которую затем вносят в предымплантированный эмбрион, из которого развивается химерный организм. МЭФ – мышиные эмбриональные фибробласты; хн – химерная направляющая.
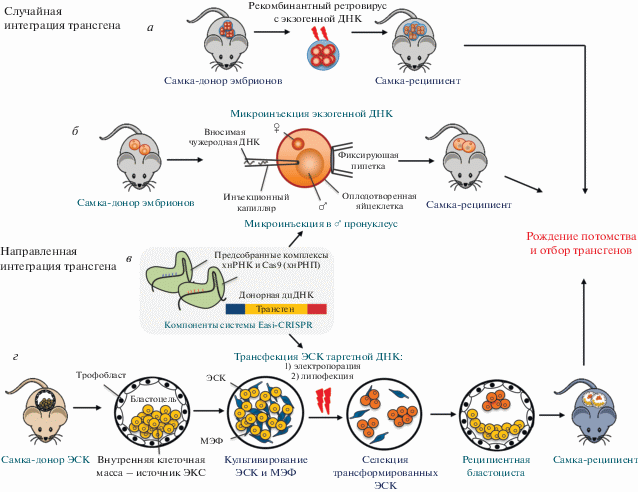
Более совершенным способом редактирования генома мышей считается трансфекция линии эмбриональных стволовых клеток (ЭСК) вектором, приспособленным для гомологичной рекомбинации, при которой локус доставки трансгена строго определяется последовательностями плеч гомологии (рис. 1г). Традиционно мышей, полученных при помощи этого метода, называют “нокинными” (от англ. knock-in), но не трансгенными, несмотря на то, что они могут быть сконструированы для экспрессии чужеродных генов. Часто в качестве сайта встраивания вектора используется локус ROSA26, промотор которого активен в большинстве тканей взрослой мыши и обеспечивает стабильную экспрессию привнесенного гена [7]. Несмотря на то, что ЭСК-технология считается “золотым стандартом” в обратной генетике, производство линий мышей при помощи этого метода занимает годы ‒ начиная с разработки дизайна геномного редактирования и заканчивая длительным переводом мутации на генетическую основу чистой линии мышей.
В последнее время стремительное развитие получил метод геномного редактирования с использованием системы CRISPR/Cas9. В контексте получения гуманизированных мышей некоторые вариации этого метода, например, Easi-CRISPR [8], могут объединять достоинства классического трансгенеза (скорость) и ЭСК-технологии (направленность модификации). Так, введение компонентов системы, необходимых для специфического внесения разрывов (двух РНП-комплексов нуклеазы Cas9 и химерной направляющей РНК), и ДНК-конструкции для гомологичной рекомбинации, несущей желаемую вставку, может осуществляться на этапе одноклеточного эмбриона, т.е. не требует обязательного использования ЭСК (рис. 1в) [9]. Однако до сих пор публикаций по моделям гуманизированных мышей, полученных с помощью CRISPR/Cas9, не было.
Вне зависимости от способа получения мыши, несущие трансген, обладают рядом особенностей. Так, к серьезным недостаткам мышей со сверхэкспрессией генов человека, кодирующих провоспалительные цитокины, относится невозможность самоподдержания линии в гомозиготном состоянии ‒ системно или локально возникающие аномалии и воспалительные синдромы не позволяют трансгенным мышам эффективно размножаться. Для поддержания таких мышиных линий, как правило, необходимо либо скрещивать мышь, несущую трансген в гетерозиготном состоянии, с мышью дикого типа, либо часто обновлять размножающиеся (бридинг) пары, состоящие из гетерозигот по трансгену.
Как уже отмечалось, число копий трансгена, встроившихся в геном, может варьировать. В то же время число интегрированных копий (или копийность трансгена) может напрямую влиять на интенсивность проявления искомого фенотипа у первичных трансгенов (фаундеров, или основателей линии). Например, трансгенная линия мышей, содержащая 5 копий гена TNF человека, имеет гораздо более выраженный фенотип, чем линия, содержащая одну копию гена [2]. Такие различия в эффективности трансгенеза могут стать причиной ошибочного вывода о непричастности исследуемого фактора к патогенезу изучаемых патологических процессов. Если ДНК-конструкция встроилась в несколько участков генома, то со временем, при возвратном скрещивании мышей, применяющемся для перевода трансгенной линии на чистую генетическую основу (бэкграунд), будет происходить потеря некоторых копий трансгена и, как следствие, снижение копийности. Наконец, необходимо помнить о том, что выбор генетической линии, на основе которой будут созданы трансгенные мыши, также имеет большое значение, поскольку разные линии обладают различной чувствительностью к развитию определенных заболеваний [10]. В представленном обзоре рассмотрены фенотипические особенности полученных к настоящему моменту линий трансгенных мышей, сверхэкспрессирующих провоспалительные цитокины человека.
ФЕНОТИПИЧЕСКИЕ ОСОБЕННОСТИ ТРАНСГЕННЫХ МЫШЕЙ СО СВЕРХЭКСПРЕССИЕЙ И ГУМАНИЗАЦИЕЙ ПО ГЕНАМ, КОДИРУЮЩИМ ЦИТОКИНЫ
Фенотипические особенности трансгенных мышей, гуманизированных по TNF, IL-1, IL-6, IL-8, IL-17E и IL-32, представлены в соответствующих разделах ниже.
TNF
Фактор некроза опухоли (TNF) получил свое название благодаря способности индуцировать гибель опухолевых клеток [11]. Теперь известно, что спектр его функций и активностей огромен ‒ это центральный регулятор иммунитета в норме и при патологических состояниях [12‒14]. В зависимости от контекста TNF может осуществлять диаметрально противоположные функции. У этого цитокина есть два высокоаффинных рецептора. TNFR1 (p55) экспрессируется практически на всех типах клеток, в то время как TNFR2 (p75) имеет более узкий профиль экспрессии, ограниченный клетками гемопоэтического происхождения, нейронами и олигодендроцитами. Контроль экспрессии TNF осуществляется на многих уровнях, включая посттранскрипционный [15]. Нарушение продукции TNF приводит к развитию ряда заболеваний, таких как ассоциированная с раком кахексия [16], отторжение трансплантатов [17], эндотоксический шок [18], ревматоидный артрит [19] и псориаз [20]. Мышиные модели заболеваний человека, предполагающие использование трансгенных мышей со сверхэкспрессией TNF человека (hTNF)22*, по большей части связаны с патологиями опорно-двигательного аппарата и центральной нервной системы (ЦНС), такими как ревматоидный артрит, атаксия и паралич (табл. 1).
Таблица 1.
Фенотипические особенности трансгенных мышей со сверхэкспрессией TNF человека
| Специфичность экспрессии | Линия мышей | Фенотип трансгенных мышей | Источник | |
|---|---|---|---|---|
| в наивном состоянии | в мышиной модели | |||
| Системно | B6D2F1 (BDF1) – потомки скрещивания самки C57BL/6 и самца DBA/2 | Тяжелый полиартрит через 3‒4 недели после рождения и прогрессирующая потеря веса | Введение блокаторов hTNF предотвращало развитие симптомов | [2] |
| Разрушение и деформация межпозвоночных дисков | [21] | |||
| Системная, индуцируемая доксициклином сверхэкспрессия | Не указана | Псориатический артрит с костной эрозией, воспалением кожи и дистрофией ногтевых пластин | Исчезновение симптомов через 6 недель после отмены доксициклина | [22] |
| Системно | FVB/J | Тяжелый артрит, развивающийся через 18‒20 недель после рождения | Введение блокаторов hTNF предотвращало развитие симптомов | [10] |
| Астроциты | B6D2F1 (BDF1) – потомки скрещивания самки C57BL/6 и самца DBA/2 | Летальное нейровоспаление с гидроцефалией, атаксия, потеря веса, нарушение ориентации, судороги и паралич | Введение блокаторов hTNF увеличивало продолжительность жизни, но не предотвращало развитие симптомов. | [3] |
| Астроциты (только трансмембранный TNF) | Тяжелое нейровоспаление, полный паралич задних конечностей, нормальная масса тела | |||
| Нейроны | Тяжелое нейровоспаление, паралич, прогрессирующая демиелинизация | Введение блокаторов hTNF предотвращало развитие симптомов | ||
| Нейроны (только трансмембранный TNF) | Развитие без фенотипических аномалий | |||
| Т-клетки | Летальная прогрессирующая потеря веса, тромбоз сосудов, некроз тканей, нарушение структуры лимфоидных органов | Введение блокаторов hTNF предотвращало развитие симптомов | [23] | |
При ревматоидном артрите воспаление локализуется в синовиальной прослойке и характеризуется утолщением слоя синовиальных клеток за счет их пролиферации, а также активной инфильтрацией иммунных клеток. Эта пролиферирующая масса, называемая паннусом, за счет давления разрушает суставной хрящ и кость, что приводит к необратимому нарушению структуры и функциональности сустава [24]. Впервые мыши со сверхэкспрессией hTNF были охарактеризованы в 1991 году [2]. Через 3–4 недели после рождения у них развивался тяжелый полиартрит с нарушением функциональности суставов вплоть до полного обездвиживания конечностей, отеком суставов и прилегающих тканей, гиперплазией синовиальной мембраны, разрастанием фиброзной ткани и полиморфным лимфоцитарным инфильтратом в синовиальном слое. Интересно, что, несмотря на все характерные проявления артрита у пациентов, в крови трансгенных мышей отсутствовали антитела к коллагену [2]. В связи с тем, что полиартрит развивался у мышей вследствие сверхэкспрессии цитокина, введение блокаторов hTNF предотвращало заболевание. Кроме того, мыши со сверхэкспрессией hTNF отличались от контрольных прогрессирующей потерей веса, не связанной с нарушением подвижности.
У мышей, трансгенных по hTNF, снижена фертильность, что затрудняет их содержание в гомозиготном состоянии. В связи с этим была разработана более совершенная модель hTNF-зависимого ревматоидного артрита ‒ с использованием индуцируемой доксициклином системной сверхэкспрессией hTNF [22]. После курса антибиотика у этих мышей развивались признаки псориатического артрита с воспалением суставов и нарушением их подвижности, эрозией костей, вызванной активацией остеокластов, а также воспалением кожи, деформацией матрикса ногтей, активацией кожных кератиноцитов и продукцией аларминов S100A8 и S100A9 в коже и суставах. Интересно, что, в отличие от вышеописанной модели полиартрита у мышей с конститутивной экспрессией hTNF, в доксициклининдицируемой модели у животных не наблюдали воспаления голеностопных суставов, а передние конечности были поражены псориатическим артритом значительно сильнее задних. Отмена антибиотика в течение 6 недель приводила к исчезновению большинства дефектов.
Повышенный уровень TNF в сыворотке и спинномозговой жидкости наблюдается у пациентов с воспалительными заболеваниями нервной системы [25‒27]. Исследование фенотипа мышей со сверхэкспрессией hTNF в нервной системе позволило выявить некоторые аспекты участия этого цитокина в патогенезе нейровоспаления. Известно, что в ЦНС TNF продуцируется как нейронами, так и астроцитами. При исследовании трансгенных мышей со сверхэкспрессией hTNF в астроцитах или нейронах было показано, что нарушение выработки hTNF обоими типами клеток приводит к развитию тяжелой нейропатологии, характеризующейся параличом, судорогами и прогрессирующей демиелинизацией [3]. Фармакологическая блокировка hTNF у мышей со сверхэкспрессией цитокина в нейронах предотвращала развитие заболевания, в то время как у мышей с астроцитарной сверхэкспрессией hTNF на фоне анти-hTNF-терапии лишь увеличивалась продолжительность жизни, а развитие нейровоспаления продолжалось. С целью выяснить, какая именно форма hTNF, растворимая или трансмембранная, задействована в индукции нейровоспаления, получили трансгенных мышей, у которых астроциты или нейроны продуцировали только трансмембранную форму hTNF. Установили, что только трансмембранный hTNF из астроцитов индуцирует развитие нейровоспаления, в то время как у мышей со сверхэкспрессией трансмембранной формы цитокина на нейронах патологии не развивались [3]. Таким образом, с использованием трансгенных мышей со сверхэкспрессией hTNF в клетках ЦНС удалось установить главный клеточный источник патогенного TNF при нейровоспалении. В более сложных мышиных моделях выявлен существенный вклад и других клеточных источников TNF при нейровоспалении (моноцитов и микроглии), однако эти результаты неоднозначны и обсуждаются в следующей главе обзора.
При исследовании эффектов Т-клеточной сверхэкспрессии hTNF также выявлены новые функции этого цитокина. Показано, что у трансгенных мышей со сверхэкспрессией hTNF в Т‑лимфоцитах развивается летальная прогрессирующая потеря веса, но, в отличие от мышей с системной сверхэкспрессией, не развивается артрит [23]. Помимо потери веса у этих трансгенных мышей развивался множественный тромбоз мелких сосудов с последующим некрозом прилегающих тканей, а также наблюдались аномалии в структуре лимфоидных органов. Так, тимус этих трансгенных мышей был существенно редуцирован и не имел различимых кортикальной и медуллярной зон. Мезентериальные лимфоузлы были увеличены в размере, но практически не содержали лимфоцитов, а периферические лимфоузлы и селезенка, хотя и без существенных макроскопических аномалий, на микроскопическом уровне имели редуцированные лимфоидные фолликулы и гиперплазию ретикулярных клеток. Наконец, в перитонеальной полости мышей с Т-клеточной сверхэкспрессией hTNF обнаружили высокое содержание иммунных клеток. Как и в случае с системной сверхэкспрессией hTNF, введение анти-hTNF-антител предотвращало развитие симптомов у мышей с Т-клеточной сверхэкспрессией hTNF. Таким образом, системная и тканеспецифическая сверхэкспрессия hTNF в трансгенных мышах позволила установить и подтвердить патогенную роль сигнала, передаваемого через TNFR1, при некоторых аутоиммунных и воспалительных заболеваниях.
IL-1β
IL-1β ‒ мощный провоспалительный цитокин, регуляция которого контролируется внутриклеточным сигнальным каскадом, включающим инфламмасомы и каспазу-1. Системное повышение уровня IL-1β происходит во время иммунного ответа и влияет как на развитие лихорадки, так и на продукцию белков острой фазы. Хронические аутовоспалительные синдромы, например, семейная средиземноморская лихорадка и криопиринассоциированные периодические синдромы, также характеризуются повышенным уровнем этого цитокина [28].
Тканеспецифическая сверхэкспрессия IL-1β человека (hIL-1β) у мышей приводит к развитию признаков локального хронического воспаления (табл. 2). Так, при индукции сверхэкспрессии hIL-1β в головном мозге наблюдалась значительная инфильтрация нейтрофилов в очаг воспаления, которая не приводила к гибели нейронов [31], но влияла на поведение мышей [32]. При сверхэкспрессии hIL-1β в желудке у трансгенных мышей развивался гастрит, а у некоторых особей наблюдали спонтанное развитие пренеоплазии или аденокарциномы желудка, причем доля последних увеличивалась при инфицировании Helicobacter felis. Также у этих мышей наблюдали экспансию миелоидных супрессоров, которые рассматриваются в качестве медиаторов раннего канцерогенеза [33]. Сверхэкспрессия hIL-1β в поджелудочной железе приводила к локальному воспалительному ответу – у таких мышей развивался панкреатит с характерными, в том числе и для человека, признаками: инфильтрацией Т-клеток, атрофией тканей и стенозом канальцев. В легких на фоне повышенного уровня hIL-1β наблюдали сочетание признаков хронической обструктивной болезни легких и нейтрофильной астмы [35]. При сверхпродукции hIL-1β в лимфоидных органах у мышей развивалась спленомегалия, а лимфоузлы увеличивались до 7–10 мм в диаметре за счет накопления Т- и В-клеток [36]. Таким образом, мышей со сверхэкспрессией hIL-1β можно использовать как модель хронического воспаления и его вовлечения в канцерогенез.
Таблица 2.
Фенотипические особенности трансгенных мышей со сверхэкспрессией IL-1β человека
| Цитокин | Специфичность экспрессии | Линия мышей | Фенотип трансгенных мышей | Источник | |
|---|---|---|---|---|---|
| в наивном состоянии | в мышиной модели | ||||
| IL-1α | Системно | Повышенная эмбриональная и неонатальная смертность; смерть в результате острой сердечной недостаточности; задержка роста, увеличение суставов, потеря шерсти, гипертрофия сердечной мышцы | [4] | ||
| Сердце | С57BL/6 | Гипертрофия левого желудочка сердца | [29] | ||
| Системно | C3H/HeJ | Прогрессирующая потеря веса, потеря шерсти, тяжелый симметричный полиартрит суставов, нейтрофилия и повышенная пролиферация клеток моноцитарно-макрофагальной линии | [30] | ||
| IL-1β | Астроциты | С57BL/6 | Тяжелое нейровоспаление | Нарушение долговременной и кратковременной памяти в поведенческих тестах | [31, 32] |
| Желудок | С57BL/6 | Спонтанное развитие гастрита, рака желудка | Развитие рака желудка ускорено на фоне инфекции Helicobacter felis | [33] | |
| Поджелудочная железа | C57BL/6J | Хронический панкреатит с Т-клеточным инфильтратом | [34] | ||
IL-6
Интерлейкин-6 (IL-6) относится к важнейшим провоспалительным цитокинам с широким спектром иммунорегуляторных функций, как гомеостатических, так и связанных с защитным ответом организма. В ранних работах IL-6 охарактеризован как фактор роста B-клеток [37]. В дальнейшем показано, что IL-6 также важен для клеток врожденного иммунитета и Т-клеток [38], синтеза белков острой фазы и контроля уровня свободного железа в организме при инфекции [39]. Кроме того, IL-6 осуществляет регуляцию процессов, не связанных напрямую с иммунным ответом, таких как метаболизм и поведение [40], а также регенерация [41].
В одной из первых линий трансгенных мышей с системной сверхэкспрессией IL-6 человека (hIL-6) (табл. 3) наблюдали симптомы болезни Кастлемана: плазмацитоз иммунных органов, а также легких, печени и почек; прогрессирующий гломерулонефрит; повышение уровня IgG в сыворотке крови [42]. Кроме того, у мышей развивалась атрофия мышц ‒ в результате лизосомального протеолиза тканей, вызванного повышенной активностью катепсинов B и L [44]. При переводе трансгена на генетическую основу линии BALB/c у животных развивались перевиваемые плазмацитомы [42].
Таблица 3.
Фенотипические особенности трансгенных мышей со сверхэкспрессией IL-6 человека
| Специфичность экспрессии | Линия мышей | Фенотип трансгенных мышей | Источник | |
|---|---|---|---|---|
| в наивном состоянии* | в мышиной модели | |||
| Системно | С57BL/6J | Поликлональный плазмацитоз повышенная продукция IgG, гломерулонефрит в возрасте 18 недель; мышечная атрофия, связанная с повышенной экспрессией катепсинов B и L; нейтрофилия костного мозга, анемия | Введение антитела против IL-6R мыши снижало выраженность симптомов болезни Кастлемана (плазмацитоз, гломерулонефрит, атрофия, анемия) | [42‒46] |
| BALB/c | Развитие пересаживаемых плазмацитом, повышенная продукция IgG и IgA | [42] | ||
| В-клетки | С57BL/6 | Поликлональный плазмацитоз иммунных органов и инфильтрация плазмацитов в печень, почки и легкие | [47] | |
| Нервная ткань | Не указана | Увеличение количества астроцитов и клеток микроглии; задержка роста, снижение уровня IGF-1, остеопения, нарушение регуляции остеобластов и остеокластов | [48‒50] | |
| Печень | С57BL/6 | Плазмацитоз только в иммунных органах; дисфункция почек, приводящая к гибели мышей на 4 месяц жизни; мегакариоцитоз селезенки | [51] | |
| Мозг, легкие | FVB/N | Высокий уровень hIL-6 в крови, задержка развития и роста, ассоциированная со снижением экспрессии IGF-1 и GH | [52] | |
| Легкие | C57BL/6 | Утолщение дыхательных путей с субэпителиальным фиброзом, инфильтрация иммунных клеток в легкое | Сниженная чувствительность к метахолину | [53, 54] |
| Лизоцимпродуцирующие миелоидные клетки | C57BL6/CBA гибриды | Неонатальная смертность более 80% мышей, задержка развития, нейтрофилия и снижение количества эритроцитов селезенки, анемия | [55] | |
Для созданных позднее трансгенных мышей с тканеспецифичной сверхэкспрессией hIL-6 была характерна существенная задержка роста по сравнению с контрольными мышами. Интересно, что аналогичной фенотипической особенностью обладали также мыши со сверхэкспрессией IL-6 мыши в астроцитах или кератиноцитах [56, 57]. Вероятно, на формирование этого признака влияло именно системное повышение уровня IL-6. Задержку роста мышей со сверхэкспрессией hIL-6 объясняли несколькими механизмами: во-первых, снижением системного уровня инсулиноподобного фактора роста 1 (IGF-1) [52]; во-вторых, нарушением роста костей [58, 59]. В опытах in vitro было показано, что остеобласты hIL-6-трансгенных мышей обладают меньшим пролиферативным потенциалом и способностью к минерализации. Аналогичные эффекты наблюдали при добавлении рекомбинантного hIL-6 к остеобластам мышей дикого типа. Интересно, что для остеокластов трансгенных мышей, наоборот, регистрировали высокий уровень активации и повышенное содержание в костной ткани [45]. Еще одним фенотипическим признаком, характерным для мышей с системной сверхэкспрессией hIL-6, была выраженная нейтрофилия костного мозга. В опытах, где к культурам костномозговых клеток-предшественников в полужидкой среде добавляли ростовые факторы миелоидных клеток (GM-CSF, M-CSF и G-CSF), было установлено, что у трансгенных мышей формируется значимо большее число колониеобразующих единиц GM-CFU, G-CFU и M-CFU, чем у мышей дикого типа. У мышей со сверхэкспрессией hIL-6 в мозге наблюдали увеличение количества астроцитов и разветвленности клеток микроглии [48], а при сверхэкспрессии hIL-6 в легких у животных развивался фиброз ткани легкого, но они были менее подвержены бронхоспазму по сравнению с контрольными мышами [53, 54].
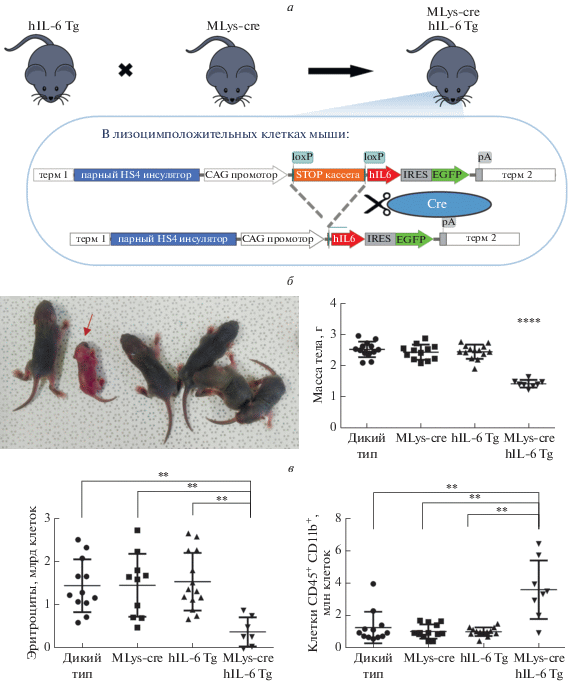
В нашей лаборатории разработана новая система трансгенной сверхэкспрессии hIL-6 [60]. В отличие от предшествующих работ, в которых кДНК IL6 экспрессировалась под убиквитарным или тканеспецифичным промотором, генетическая конструкция для трансгенеза содержала loxP-фланкированную стоп-кассету между CAG-промотором и кДНК IL6. Таким образом, тип ткани, в котором происходила сверхэкспрессия цитокина, зависел от специфичности рекомбиназы Cre, которая попадала в организм потомков при скрещивании трансгенов с делитерными мышами (в их геном был встроен ген рекомбиназы Cre под тканеспецифичным промотором). Кроме того, для детектирования клеток, сверхэкспрессирующих hIL-6, в рамку считывания hIL-6 была вставлена последовательность, кодирующая флуоресцентный белок EGFP. Аналогичная конструкция (рис. 2а) использована и для получения линии трансгенных мышей со сверхэкспрессией hIL-6 в лизоцимположительных миелоидных клетках (MLys-cre hIL-6 Tg) [55] и резидентных макрофагах (Cx3cr1CreER hIL-6 Tg) [61]. Оказалось, что 85% трансгенных мышей, сверхэкспрессирующих hIL-6 в лизоцимположительных миелоидных клетках, погибали в первые две недели жизни [55]. Кроме того, новорожденные отставали в росте и наборе веса (рис. 2б). Единичные особи, доживавшие до 4 недель, обладали выраженным воспалительным фенотипом, характеризующимся увеличением лимфоузлов и селезенки, воспалением кожи и множественной инфильтрацией иммунных клеток в печени. В сыворотке крови 3-дневных мышей содержалось 0.6–1.0 нг/мл hIL-6, а в надклеточной среде первичных культур макрофагов костного мозга и спленоцитов, полученных из этих мышей, уровень продуцируемого hIL-6 достигал 1.0–2.5 нг/мл. Кроме того, у 3-дневных Mlys-cre hIL-6 Tg трансгенных мышат достоверно снижено содержание эритроцитов (Ter119+CD45– клеток) и увеличено количество миелоидных клеток (CD45+CD11b+) селезенки (рис. 2в). EGFP-положительные клетки были представлены нейтрофилами (CD45+CD11b+Gr-1+). По всей видимости, среди популяций лизоцимположительных клеток именно нейтрофилы были важным источником hIL-6 в мутантных мышах [62]. Таким образом, изучение фенотипов мышей со сверхэкспрессией hIL-6 показало важную роль этого цитокина в онтогенезе организма. Системное увеличение концентрации цитокина влияло не только на иммунную систему, но и на пролиферацию и метаболизм клеток других тканей, например, костной, что замедляло рост мышей. Помимо этого, сверхэкспрессия hIL-6 в миелоидных клетках приводила к нежизнеспособности всего организма.
IL-8
Рис. 2.
Мыши со сверхэкспрессией hIL-6 в миелоидных клетках погибают неонатально на фоне задержки роста, анемии и воспаления. а ‒ Схема получения мышей MLys-cre hIL-6 Tg. б ‒ Задержка роста и отставание в наборе веса у мышей MLys-cre hIL-6 Tg (отмечена стрелкой) как характерная черта системной сверхэкспрессии цитокина. в ‒ В возрасте 3 суток в селезенке мышей MLys-cre hIL-6 Tg снижено абсолютное содержание эритроцитов и увеличено количество миелоидных клеток CD45+CD11b+ по сравнению с мышатами других генотипов из того же потомства (литтермейтами). Данные представлены в виде среднего ± SEM. **P ≤ 0.01, ****P ≤ 0.0001.
IL-8 (CXCL-8, NAP-1) представляет собой провоспалительный хемотаксический цитокин (хемокин) человека, основная функция которого заключается в привлечении нейтрофилов в очаг воспаления [63], его классификация как интерлейкина устарела. IL-8 стимулирует миграцию нейтрофилов в ткани и органы, активируя их адгезию к эндотелиальным клеткам путем интенсификации экспрессии L-селектина и β2-интегрина на поверхности [64]. У мышей нет собственного функционального гена IL-8, что затрудняет изучение функций этого белка человека в мышиной модели. В этом контексте интересна способность IL-8 человека (hIL-8) активировать нейтрофилы не только человека, но и других видов, например, обезьян, крыс, собак и мышей [65]. Трансгенные по hIL-8 мыши (табл. 4) экспрессировали hIL-8 в печени под контролем промотора АpoE и HCR-энхансера, специфичного для печени человека [66]. Показано, что у трансгенных по hIL-8 мышей, несмотря на повышенное содержание нейтрофилов крови, спонтанное воспаление не развивается. Интересно, что у hIL-8-трансгенных мышей не обнаружено нейтрофильного инфильтрата в тканях, в том числе и в печени, которая была источником hIL-8, а экспрессия L-селектина на поверхности нейтрофилов была ниже, чем у мышей дикого типа. Установлено, что внутрибрюшинное введение hIL-8 трансгенным мышам не вызывало инфильтрации нейтрофилов, в то время как у контрольных мышей после введения hIL-8 в перитонеальной полости детектировали обильный нейтрофильный инфильтрат. Таким образом, системная сверхэкспрессия hIL-8 у мышей приводит к экспансии нейтрофилов, не отвечающих на активирующие стимулы из-за сниженной экспрессии молекул адгезии.
Таблица 4.
Фенотипические особенности трансгенных мышей со сверхэкспрессией IL-8, IL-17E, IL-32β человека
| Цитокин | Специфичность экспрессии | Линия мышей | Фенотип трансгенных мышей | Источник | |
|---|---|---|---|---|---|
| в наивном состоянии* | в мышиной модели | ||||
| IL-8 | Печень | B6D2F1 (BDF1) – потомки скрещивания самки C57BL/6 и самца DBA/2 | Увеличение числа нейтрофилов в крови с уменьшением экспрессии L-селектина и b-интегрина на нейтрофилах | Снижение способности нейтрофилов мигрировать в ответ на введение hIL-8 | [66] |
| IL-17E | Печень | B6D2F1 (BDF1) – потомки скрещивания самки C57BL/6 и самца DBA/2 | Увеличение числа лейкоцитов в крови, особенно эозинофилов; высокий уровень IL-4, IL-5, GM-CSF, IFNγ и эотаксина, IgM, IgG и IgE в сыворотке крови; спленомегалия | [67] | |
| IL-32β | Системно | C57BL/6 | Снижение тяжести симптомов артрита, индуцированного антителами к коллагену | [68] | |
| Системно | C57BL/6 | Более активное накопление амилоида в модели болезни Альцгеймера по сравнению с диким типом, интенсивное нейровоспаление | [69, 70] | ||
IL-17E
IL-17E, также известный как IL-25, ‒ член подсемейства IL-17 провоспалительных цитокинов, в которое входят шесть молекул: IL-17 A, B, C, D, E, F, ‒ обладающих высокой степенью гомологии, но выполняющих различные функции. В то время как эффекты сверхэкспрессии мышиных членов семейства IL-17 изучены достаточно детально [71‒73], работ по определению фенотипа мышей со сверхэкспрессией цитокинов семейства IL-17 человека крайне мало. Наиболее интересный фенотип найден у мышей со сверхэкспрессией IL-17Е человека (hIL-17E) в печени под контролем промотора ApoE (табл. 4) [67]. Установлено, что мыши, трансгенные по hIL-17E, имеют значительные отличия в гемопоэтическом компартменте: их костный мозг характеризовался повышенным содержанием эозинофилов и их предшественников, а также CD5+CD19+ и CD34+CD19+ В-клеток. Кроме того, у мышей с экспрессией hIL-17E в печени развивалась спленомегалия и лимфаденопатия ‒ с преобладанием эозинофилов и В-клеток в лимфоузлах и селезенке. Число эозинофилов в крови и бронхоальвеолярной жидкости у этих мышей было также в несколько раз выше по сравнению с контрольными мышами. Наконец, в сыворотке трансгенных по hIL-17E мышей обнаружен высокий уровень IL-4, IL-5, GM-CSF, IFNγ и эотаксина, а также десятикратное превышение концентраций IgM, IgG и IgE. Таким образом, по результатам изучения фенотипа мышей со сверхэкспрессией hIL-17E пришли к выводу, что этот цитокин может воздействовать на предшественники эозинофилов и В-клеток в костном мозге, опосредуя экспансию эозинофилов и В-клеток на периферии и системную активацию иммунного ответа по Th2-типу, для которого характерна продукция IL-4, IL-5, эотаксина, IgE, IgG и эозинофилия.
IL-32β
IL-32, впервые описанный только в 2005 году, представляет собой цитокин с плейотропными функциями, способный индуцировать выработку TNF и IL-1β [74]. Благодаря способности активировать воспалительные факторы IL-32 участвует в защите от инфекций, вызванных вирусами иммунодефицита [75], гриппа [76], а также Mycobacterium tuberculosis [77], однако, IL-32 также вовлечен в патогенез ряда воспалительных заболеваний [74]. Не так давно обнаружено, что ген IL32 несет несколько вариантов сплайсинга первичного транскрипта: α, β, γ, δ, ε и τ. Так, наиболее биологически активной формой, обладающей провоспалительной и противоинфекционной активностью, оказался IL-32γ, в то время как наиболее распространенный IL-32β ассоциирован с положительным прогнозом течения воспалительных заболеваний [78]. В исследовании на трансгенных мышах со сверхэкспрессией hIL-32β показано, что в модели ревматоидного артрита, индуцированного коктейлем антител к коллагену, у трансгенных мышей со сверхэкспрессией hIL-32β симптомы заболевания менее выражены, чем у мышей контрольного генотипа (табл. 4) [68]. У этих трансгенных мышей, наряду с избыточной экспрессией β-варианта человеческого IL-32, на нормальном уровне экспрессировались все сплайс-формы мышиного IL-32. Установлено, что у hIL-32β-трансгенных мышей значительно меньше выражен отек конечностей, гиперплазия синовиоцитов, костная эрозия и разрушение суставов. Кроме того, у них снижена системная воспалительная реакция, проявляющаяся в накоплении лейкоцитов в крови, а также высоком уровне IgG и IgM. Уровни экспрессии провоспалительных факторов, таких как индуцибельная NO-синтаза (iNOS), циклоокигеназа 2 (COX-2), металлопротеиназы 3 и 9 (MMP-3 и MMP-9 соответственно), а также провоспалительных цитокинов TNF, IL-6 и IL-1β, были значительно выше в суставах контрольных мышей по сравнению с трансгенными [68]. Следует отметить, что для hIL-32β выявлена и провоспалительная модальность. Так, в моделях болезней Альцгеймера и Паркинсона у трансгенных мышей со сверхэкспрессией hIL-32β заболевание протекало в тяжелой форме со стремительной потерей когнитивных навыков [69, 70]. Таким образом, на модели мышей, трансгенных по hIL-32β, показано, что IL-32β, в отличие от наиболее биологически активной сплайс-формы IL-32γ с провоспалительными функциями, обладает как про-, так и противовоспалительными свойствами ‒ в зависимости от иммунологического контекста.
ИСПОЛЬЗОВАНИЕ СИСТЕМ С ГУМАНИЗИРОВАННЫМИ МЫШАМИ В КОНТЕКСТЕ ИЗУЧЕНИЯ ФУНКЦИЙ ПРОВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ И ОЦЕНКИ ЭФФЕКТИВНОСТИ КЛИНИЧЕСКИ ПРИМЕНЯЕМЫХ ПРЕПАРАТОВ
Для современной экспериментальной иммунологии самой удобной редуцированной системой считаются генетически модифицированные мыши ‒ с удаленными или привнесенными целевыми генами. Действительно, технология редактирования генома для других млекопитающих разработана значительно позже, когда уже были созданы и изучены многие сотни нокаутных и трансгенных мышей. Изучение фенотипа таких мышей в контексте различных экспериментальных моделей заболеваний позволяет сделать вывод о роли выбранного гена и его белкового продукта в иммунном ответе (методология обратной генетики). В контексте изучения свойств провоспалительных цитокинов мыши со сверхэкспрессией трансгена представляют собой модельную систему с высокой продукцией цитокина, что характерно для целого ряда аутоиммунных и аутовоспалительных заболеваний человека. Зачастую получаемый фенотип гипертрофирован, благодаря чему удается выявить новые, ранее не установленные, вторичные функции провоспалительных молекул.
Так, TNF относится к ключевым провоспалительным цитокинам, играющим важную роль как в поддержании гомеостаза иммунной системы, так и в защите от патогенов, в контроле и индукции воспалительных и онкологических заболеваний. Повышенный уровень TNF в сыворотке регистрируют у пациентов с такими заболеваниями, как порок сердца [79], бронхиальная астма [80], миома матки [81], диабет [82], депрессия и биполярные расстройства [83] и остеоартрит [84]. Однако понимание причинно-следственной связи между повышением уровня TNF и протекающим патологическим процессом не представлялось возможным без детальных исследований с использованием мышиных моделей.
TNF продуцируется макрофагами, Т- и В-клетками, нейтрофилами, эндотелиальными клетками и фибробластами и может быть представлен в двух формах: мембраносвязанной (26 кДа) и растворимой (17 кДа). Получается, что эффекты TNF могут различаться в зависимости от клеточного источника цитокина и от того, в какой биологически активной форме он предъявлен акцепторной клетке [85]. Синтез TNF инициируется транскрипционной активацией гена TNF, которая в большинстве случаев происходит при связывании NF-kB с соответствующими элементами в 5'-промоторной области. Экспрессия TNF также регулируется на посттранскрипционном уровне через AU-богатый элемент (AU rich element, ARE) в 3'-нетранслируемой области (3'-UTR) мРНК. В неактивированных клетках ARE вызывает быструю деградацию транскриптов TNF, в то время как в активированных клетках эти транскрипты стабилизированы, что приводит к 200-кратному увеличению концентрации мРНК с последующей трансляцией TNF. Одновременно с ростом концентрации мРНК TNF синтезируется тристетрапролин, который по механизму отрицательной обратной связи индуцирует деградацию транскриптов TNF [15]. Благодаря такой жесткой посттранскрипционной регуляции TNF может быть быстро синтезирован клеткой, причем в больших количествах, при получении активирующего сигнала. Именно нарушение посттранскрипционной программы контроля продукции TNF используется для создания мышей со сверхэкспрессией этого цитокина [86].
Как отмечено выше, TNF может связываться с двумя высокоаффинными рецепторами: TNFR1 (p55) и TNFR2 (p75). Следует также заметить, что мышиный TNFR2 характеризуется строго видоспецифичным связыванием лиганда, то есть только c TNF мыши, и не может быть активирован hTNF [87]. Учитывая этот факт, все эффекты hTNF в опубликованных трансгенных мышах можно рассматривать как результат передачи сигнала через TNFR1 в отсутствие потенциально защитного эффекта TNFR2, не взаимодействующего с hTNF [88]. Таким образом, эффекты TNF зависят также от вида распознающего рецептора, а дифференциальный вклад каждого из рецепторов TNF в патогенез заболеваний можно установить при сравнении фенотипов мышей со сверхэкспрессией hTNF и мышей, сверхэкспрессирующих свой TNF.
Исследование трансгенных мышей со сверхэкспрессией hTNF внесло огромный вклад в понимание механизмов патогенеза ревматоидного артрита. Конститутивная системная сверхэкспрессия hTNF в трансгенных мышах, полученных на генетической основе линии BDF1, приводила к развитию тяжелого полиартрита, а также к прогрессирующей потере веса [2, 10]. Кроме того, блокировка цитокина клинически применяемыми лекарственными препаратами предотвращала развитие симптомов у трансгенных мышей. Из этого следует, что наблюдаемые эффекты были результатом нарушенной экспрессии именно hTNF. Таким образом, мыши с системной сверхэкспрессией могут стать удобным инструментом для изучения эффективности терапевтических блокаторов hTNF, а также для исследований детальных механизмов участия TNF и TNFR1 (но не TNFR2) в патогенезе ревматоидного артрита. Модели спонтанного артрита, основанные на использовании трансгенных мышей, имеют одну особенность: у них развивается артрит без аутоиммунного компонента (образования патогенных В‑клеток, продуцирующих артритогенные антитела к коллагену), ‒ поэтому модель можно использовать для изучения патологии, но не для исследования механизмов аутоиммунитета [89]. Использование мышей с индуцируемой сверхэкспрессией имеет ряд преимуществ, включающих контролируемое и синхронное развитие заболевания у экспериментальных животных, а также возможность изучения ранних стадий патогенеза. Так, индуцируемая доксициклином сверхэкспрессия hTNF в мышах представляет собой удобную модель для исследования механизмов развития ревматоидного артрита на любой стадии заболевания [22]. Кроме того, интенсивность проявления симптомов зависит от дозы доксициклина, что позволяет изучать артрит разной степени тяжести. Наконец, эта модель имитирует псориатическую форму артрита, при которой поражаются не только суставы, но и кожа, и матрикс ногтей, что важно для изучения патогенеза этой формы артрита. Однако и эта модель не лишена определенных недостатков ‒ в свете недавно выявленной связи между дисбиозом и ревматоидным артритом [90, 91]. Дело в том, что при индуцируемой антибиотиком экспрессии трансгена происходит нарушение в составе микробиоты мышей, что может влиять на интенсивность проявления симптомов артрита.
В ряде патологий ЦНС задействованы специфические механизмы активации иммунной системы. В частности, нарушение продукции TNF может рассматриваться как одна из причин развития нейровоспаления, поскольку терапевтическое введение блокаторов TNF успешно останавливает развитие ряда патологий ЦНС в животных моделях [92]. Вклад конкретных клеточных источников TNF в воспаление ЦНС, а также функции растворимой и трансмембранной формы TNF частично установлены с использованием трансгенных мышей со сверхэкспрессией hTNF астроцитами и нейронами [3]. Показано, что конститутивная экспрессия hTNF как нейронами, так и астроцитами приводит к развитию летального нейровоспаления. Напротив, сверхэкспрессии трансмембранного TNF достаточно для запуска нейровоспаления у трансгенных мышей только тогда, когда он продуцируется астроцитами, но не нейронами. Однако в ЦНС, кроме астроцитов и нейронов, есть и другие источники TNF. Так, TNF из моноцитов, но не из микроглии, важен для развития нейровоспаления [92], в то время как удаление TNFR2 с CX3CR1+-клеток микроглии усиливало инфильтрацию лимфоцитов в ЦНС, а удаление TNFR2 с моноцитов приводило к ослаблению нейровоспаления [92]. Таким образом, в зависимости от клеточного контекста, участие TNF в нейровоспалении может играть как патогенетическую, так и протективную роль.
Изучение последствий нарушенной экспрессии TNF в определенных типах клеток также имеет важное значение для определения клеточных источников “патогенного” цитокина. Так, специфическая сверхэкспрессия hTNF Т-клетками у трансгенных мышей давала лишь частично пересекающийся с системной сверхэкспрессией фенотип: развивалась летальная прогрессирующая потеря веса, тромбоз, нарушение структуры лимфоидных органов, при отсутствии признаков артрита [23]. Таким образом, мыши с тканеспецифичной сверхэкспрессией hTNF в Т-клетках предоставляют уникальную модель для изучения таких заболеваний, как кахексия, тромбоз и прогрессирующий атеросклероз, в патогенезе которых важную роль играет передача сигнала через TNFR1 [93, 94]. По-видимому, причиной этих заболеваний может быть нарушение экспрессии TNF T-лимфоцитами [95, 96].
При исследовании трансгенных мышей со сверхэкспрессией другого классического провоспалительного цитокина – IL-1β ‒ также выявили важные особенности его функционирования в различных тканях и органах. Например, сверхэкспрессия hIL-1β в астроцитах приводила к нейровоспалению с нарушением долговременной и кратковременной памяти. Таким образом, эта модель позволяет исследовать патогенез воспалительных процессов в ЦНС, а также нейродегенерации [31, 32]. Как упоминалось выше, hIL-1β, продуцирующийся в больших количествах в желудке мышей, помимо воспаления, может вызывать спонтанное развитие рака желудка, ассоциированного с присутствием Helicobacter felis [33]. Наконец, сверхэкспрессия hIL-1β в поджелудочной железе может использоваться как модельная система спонтанно развивающегося Т-клеточнозависимого хронического панкреатита [34]. Таким образом, мыши со сверхэкспрессией hIL-1β представляют востребованные и актуальные экспериментальные модели спонтанно развивающихся хронических воспалительных заболеваний различных органов.
IL-6 ‒ ключевой провоспалительный цитокин с плейотропными функциями. Концентрация hIL-6 в сыворотке крови человека колеблется в широких пределах, поэтому в ней содержится так называемый “буфер IL-6” ‒ растворимые рецепторы к IL-6 (sIL-6R и sgp130), в количестве значительно большем, чем необходимо для связывания цитокина в тех концентрациях, которые наблюдаются в норме [97]. Таким образом, трансгенные мыши со сверхэкспрессией этого цитокина представляют уникальную экспериментальную модель, позволяющую изучать эффекты от нарушенной экспрессии hIL-6. Следует отметить, что рецептор IL-6RA мыши может связывать hIL-6, поэтому для сохранения работоспособности системы передачи сигнала и получения модели сверхэкспрессии достаточно гуманизации лиганда (hIL-6). Интересно, что “в обратном направлении” эта закономерность не сохраняется: IL-6 мыши не связывается с рецептором человека [98].
С помощью трансгенных мышей, сверхэкспрессирующих hIL-6 только в лизоцимположительных миелоидных клетках, установлены новые функции IL-6, не выявленные ранее при изучении трансгенных мышей с системной сверхэкспрессией этого цитокина. Так, задержка роста и экспансия миелоидных клеток в различных тканях была показана еще на уровне системной сверхпродукции hIL-6, однако, только сверхэкспрессия hIL-6 в миелоидных клетках приводила к неонатальной гибели мышей, что может быть связано с выраженной анемией и системной нейтрофилией, проявляющейся уже на третий день жизни. Таким образом, с использованием мышей с тканеспецифичной сверхэкспрессией hIL-6 показано, что этот цитокин может играть ключевую роль в регуляции гемопоэза на ранних этапах постнатального развития.
Кроме того, мыши, трансгенные по hIL-6, могут служить важным инструментом для тестирования потенциальных терапевтических блокаторов этого цитокина. Так, лечение мышей с системной сверхэкспрессией hIL-6, моделирующих развитие болезни Кастлемана, антителами, специфичными к hIL-6, приводило к ослаблению клинических симптомов заболевания [46]. В настоящее время использование моноклональных антител против IL-6 (силтуксимаба) входит в общепринятую терапию болезни Кастлемана [99].
Изучение мышей, трансгенных по генам цитокинов человека, не всегда дает однозначный результат, особенно в том случае, когда отсутствует прямой мышиный аналог человеческого цитокина. У мышей нет собственного функционального гена IL8, хотя hIL-8, будучи экспрессированным в организме мыши, способен работать как хемокин, привлекающий нейтрофилы. В исследованиях на hIL-8-трансгенных мышах показано, что избыток этого человеческого цитокина в мышах приводит к увеличению числа нейтрофилов на периферии. Однако эти нейтрофилы оказались нефункциональными, так как недостаточная экспрессия молекул адгезии приводила к неспособности клеток проникать в ткани. Вследствие этого такую мышиную модель нельзя рассматривать как релевантную для нейтрофилзависимых заболеваний.
Цитокины семейства IL-17 ‒ важные активаторы воспаления при ряде заболеваний: ревматоидном артрите [100], рассеянном склерозе [101] и псориазе [102]. Кроме того, IL-17 участвует в реакции отторжения трансплантированных органов [103, 104]. Таким образом, IL-17 задействован, в первую очередь, в патологиях, носящих аутоиммунный характер. Исследование фенотипа мышей со сверхэкспрессией hIL-17E позволило выявить новые для цитокинов семейства IL-17 функции: воздействие на гемопоэтические предшественники в костном мозге и сдвиг иммунного ответа в сторону реакции по Th2-типу. Мыши со сверхэкспрессией hIL-17E используются в качестве релевантной модели при исследовании состояний, опосредованных системной эозинофилией [105‒107], а ингибиторы IL-17E могут рассматриваться как потенциальные препараты для таргетной терапии Th2-зависимых воспалительных заболеваний, например, аллергии, астмы, дерматита и псориаза [108‒110].
Изучение мышей, сверхэкспрессирующих цитокины человека, в некоторых случаях позволило выявить новые модальности в функциях того или иного цитокина. Например, при изучении мышей, трансгенных по hIL-32β, установлено, что эта сплайс-форма классического провоспалительного цитокина, IL-32, обладает противовоспалительной активностью в модели ревматоидного артрита. Исследования in vitro противовоспалительных функций IL-32β выявляли дозозависимую индукцию экспрессии IL-10 [111] ‒ мощного супрессора воспаления; но только in vivo исследования в модели артрита окончательно доказали наличие противовоспалительной модальности у IL-32β. В дальнейшем в исследованиях in vivo показано, что про- или противовоспалительная активность IL-32β зависит от контекста. Так, в модели болезни Альцгеймера у мышей, трансгенных по hIL-32β, накопление амилоидных конгломератов и снижение когнитивных способностей происходило быстрее, чем у контрольных животных [69, 70]. Таким образом, с помощью трансгенных мышей со сверхэкспрессией hIL-32β удалось продемонстрировать диаметрально противоположные функции этой сплайс-формы цитокина IL-32.
Рассмотренные в работе мышиные системы со сверхэкспрессией провоспалительных цитокинов человека еще раз доказывают ценность технологии обратной генетики для биомедицинской науки. Правильно сконструированные трансгенные мыши позволяют не только исследовать патогенез заболеваний с воспалительным компонентом, выявляя клеточные источники цитокина, ответственные за развитие конкретных патологий, но и представляют собой уникальные модели для разработки и доклинического тестирования терапевтических препаратов.
Список литературы
Gama Sosa M.A., De Gasperi R., Elder G.A. (2010) Animal transgenesis: an overview. Brain Struct. Funct. 214, 91–109.
Keffer J., Probert L., Cazlaris H., Georgopoulos S., Kaslaris E., Kioussis D., Kollias G. (1991) Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10, 4025–4031.
Akassoglou K., Probert L., Kontogeorgos G., Kollias G. (1997) Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J. Immunol. 158, 438–445.
Isoda K., Kamezawa Y., Tada N., Sato M., Ohsuzu F. (2001) Myocardial hypertrophy in transgenic mice overexpressing human interleukin 1alpha. J. Card. Fail. 7, 355–364.
Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. (2003) Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 31, 1007–1014.
Yan B.W., Zhao Y.F., Cao W.G., Li N., Gou K.M. (2013) Mechanism of random integration of foreign DNA in transgenic mice. Transgenic Res. 22, 983–992.
Soriano P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71.
Quadros R.M., Miura H., Harms D.W., Akatsuka H., Sato T., Aida T., Redder R., Richardson G.P., Inagaki Y., Sakai D., Buckley S.M., Seshacharyulu P., Batra S.K., Behlke M.A., Zeiner S.A., Jacobi A.M., Izu Y., Thoreson W.B., Urness L.D., Mansour S.L., Ohtsuka M., Gurumurthy C.B. (2017) Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 18, 92.
Scott G.J., Gruzdev A. (2019) Genome editing in mouse embryos with CRISPR/Cas9. Methods Mol. Biol. 1960, 23–40.
Li G., Wu Y., Jia H., Tang L., Huang R., Peng Y., Zhang Y. (2016) Establishment and evaluation of a transgenic mouse model of arthritis induced by overexpressing human tumor necrosis factor alpha. Biol. Open. 5, 418–423.
Carswell E.A., Old L.J., Kassel R.L., Green S., Fiore N., Williamson B. (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA. 72, 3666–3670.
Grivennikov S.I., Kuprash D.V., Liu Z.G., Nedospasov S.A. (2006) Intracellular signals and events activated by cytokines of the tumor necrosis factor superfamily: from simple paradigms to complex mechanisms. Int. Rev. Cytol. 252, 129–161.
Kruglov A.A., Kuchmiy A., Grivennikov S.I., Tumanov A.V., Kuprash D.V., Nedospasov S.A. (2008) Physiological functions of tumor necrosis factor and the consequences of its pathologic overexpression or blockade: mouse models. Cytokine Growth Factor Rev. 19, 231–244.
Old L.J. (1988) Tumor necrosis factor. Sci. Am. 258, 59–60, 69–75.
Carballo E., Lai W.S., Blackshear P.J. (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 281, 1001–1005.
Patel H.J., Patel B.M. (2017) TNF-alpha and cancer cachexia: molecular insights and clinical implications. Life Sci. 170, 56–63.
Zhao Y., Wang Y., Zhu M.S., Han W.M., Li Z., Hong S.F., Yin P., Zhuang G.H., Qi Z.Q. (2018) Expression pattern of tumor necrosis factor-alpha-induced protein 8-like 2 in acute rejection of cardiac allograft. Transplant. Proc. 50, 293–298.
Beutler B.A. (1989) Orchestration of septic shock by cytokines: the role of cachectin (tumor necrosis factor). Prog. Clin. Biol. Res. 286, 219–235.
Sfikakis P.P., Kollias G. (2003) Tumor necrosis factor biology in experimental and clinical arthritis. Curr. Opin. Rheumatol. 15, 380–386.
Yamauchi P.S., Bissonnette R., Teixeira H.D., Valdecantos W.C. (2016) Systematic review of efficacy of anti-tumor necrosis factor (TNF) therapy in patients with psoriasis previously treated with a different anti-TNF agent. J. Am. Acad. Dermatol. 75, 612–618. e6.
Gorth D.J., Shapiro I.M., Risbud M.V. (2018) Transgenic mice overexpressing human TNF-alpha experience early onset spontaneous intervertebral disc herniation in the absence of overt degeneration. Cell Death Dis. 10, 7.
Retser E., Schied T., Skryabin B.V., Vogl T., Kanczler J.M., Hamann N., Niehoff A., Hermann S., Eisenblatter M., Wachsmuth L., Pap T., van Lent P.L., Loser K., Roth J., Zaucke F., Ludwig S., Wixler V. (2013) Doxycycline-induced expression of transgenic human tumor necrosis factor alpha in adult mice results in psoriasis-like arthritis. Arthritis Rheum. 65, 2290–2300.
Probert L., Keffer J., Corbella P., Cazlaris H., Patsavoudi E., Stephens S., Kaslaris E., Kioussis D., Kollias G. (1993) Wasting, ischemia, and lymphoid abnormalities in mice expressing T cell-targeted human tumor necrosis factor transgenes. J. Immunol. 151, 1894–1906.
Друцкая М.С., Ефимов Г.А., Зварцев Р.В., Чащина А.А., Чудаков Д.М., Тиллиб С.В., Круглов А.А., Недоспасов С.А. (2014) Экспериментальные модели артрита, патогенез которых связан с экспрессией фактора некроза опухолей (TNF). Биохимия. 79, 1648–1656.
Franciotta D.M., Grimaldi L.M., Martino G.V., Piccolo G., Bergamaschi R., Citterio A., Melzi d’Eril G.V. (1989) Tumor necrosis factor in serum and cerebrospinal fluid of patients with multiple sclerosis. Ann. Neurol. 26, 787–789.
Kim Y.S., Lee K.J., Kim H. (2017) Serum tumour necrosis factor-alpha and interleukin-6 levels in Alzheimer’s disease and mild cognitive impairment. Psychogeriatrics. 17, 224–230.
Zimmerman A.W., Jyonouchi H., Comi A.M., Connors S.L., Milstien S., Varsou A., Heyes M.P. (2005) Cerebrospinal fluid and serum markers of inflammation in autism. Pediatr. Neurol. 33, 195–201.
Dinarello C.A. (2018) Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 281, 8–27.
Nishikawa K., Yoshida M., Kusuhara M., Ishigami N., Isoda K., Miyazaki K., Ohsuzu F. (2006) Left ventricular hypertrophy in mice with a cardiac-specific overexpression of interleukin-1. Am. J. Physiol. Heart Circ. Physiol. 291, H176–183.
Niki Y., Yamada H., Seki S., Kikuchi T., Takaishi H., Toyama Y., Fujikawa K., Tada N. (2001) Macrophage- and neutrophil-dominant arthritis in human IL-1 alpha transgenic mice. J. Clin. Invest. 107, 1127–1135.
Shaftel S.S., Carlson T.J., Olschowka J.A., Kyrkanides S., Matousek S.B., O’Banion M.K. (2007) Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J. Neurosci. 27, 9301–9309.
Hein A.M., Stasko M.R., Matousek S.B., Scott-McKean J.J., Maier S.F., Olschowka J.A., Costa A.C., O’Banion M.K. (2010) Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav. Immun. 24, 243–253.
Tu S., Bhagat G., Cui G., Takaishi S., Kurt-Jones E.A., Rickman B., Betz K.S., Penz-Oesterreicher M., Bjorkdahl O., Fox J.G., Wang T.C. (2008) Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 14, 408–419.
Marrache F., Tu S.P., Bhagat G., Pendyala S., Osterreicher C.H., Gordon S., Ramanathan V., Penz-Osterreicher M., Betz K.S., Song Z., Wang T.C. (2008) Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology. 135, 1277–1287.
Lappalainen U., Whitsett J.A., Wert S.E., Tichelaar J.W., Bry K. (2005) Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol. 32, 311–318.
Bjorkdahl O., Akerblad P., Gjorloff-Wingren A., Leanderson T., Dohlsten M. (1999) Lymphoid hyperplasia in transgenic mice over-expressing a secreted form of the human interleukin-1beta gene product. Immunology. 96, 128–137.
Gauldie J., Richards C., Harnish D., Lansdorp P., Baumann H. (1987) Interferon beta 2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc. Natl. Acad. Sci. USA. 84, 7251–7255.
Crotty S. (2014) T follicular helper cell differentiation, function, and roles in disease. Immunity. 41, 529–542.
Nemeth E., Rivera S., Gabayan V., Keller C., Taudorf S., Pedersen B.K., Ganz T. (2004) IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest. 113, 1271–1276.
Irwin M.R. (2011) Inflammation at the intersection of behavior and somatic symptoms. Psychiatr. Clin. North Am. 34, 605–620.
Behm B., Babilas P., Landthaler M., Schreml S. (2012) Cytokines, chemokines and growth factors in wound healing. J. Eur. Acad. Dermatol. Venereol. 26, 812–820.
Suematsu S., Matsusaka T., Matsuda T., Ohno S., Miyazaki J., Yamamura K., Hirano T., Kishimoto T. (1992) Generation of plasmacytomas with the chromosomal translocation t(12;15) in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA. 89, 232–235.
Ebisui C., Tsujinaka T., Morimoto T., Kan K., Iijima S., Yano M., Kominami E., Tanaka K., Monden M. (1995) Interleukin-6 induces proteolysis by activating intracellular proteases (cathepsins B and L, proteasome) in C2C12 myotubes. Clin. Sci. (Lond.). 89, 431–439.
Tsujinaka T., Ebisui C., Fujita J., Kishibuchi M., Morimoto T., Ogawa A., Katsume A., Ohsugi Y., Kominami E., Monden M. (1995) Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin-6 transgenic mouse. Biochem. Biophys. Res. Commun. 207, 168–174.
Kitamura H., Kawata H., Takahashi F., Higuchi Y., Furuichi T., Ohkawa H. (1995) Bone marrow neutrophilia and suppressed bone turnover in human interleukin-6 transgenic mice. A cellular relationship among hematopoietic cells, osteoblasts, and osteoclasts mediated by stromal cells in bone marrow. Am. J. Pathol. 147, 1682–1692.
Katsume A., Saito H., Yamada Y., Yorozu K., Ueda O., Akamatsu K., Nishimoto N., Kishimoto T., Yoshizaki K., Ohsugi Y. (2002) Anti-interleukin 6 (IL-6) receptor antibody suppresses Castleman’s disease like symptoms emerged in IL-6 transgenic mice. Cytokine. 20, 304–311.
Suematsu S., Matsuda T., Aozasa K., Akira S., Nakano N., Ohno S., Miyazaki J., Yamamura K., Hirano T., Kishimoto T. (1989) IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA. 86, 7547–7551.
Fattori E., Lazzaro D., Musiani P., Modesti A., Alonzi T., Ciliberto G. (1995) IL-6 expression in neurons of transgenic mice causes reactive astrocytosis and increase in ramified microglial cells but no neuronal damage. Eur. J. Neurosci. 7, 2441–2449.
De Benedetti F., Alonzi T., Moretta A., Lazzaro D., Costa P., Poli V., Martini A., Ciliberto G., Fattori E. (1997) Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I. A model for stunted growth in children with chronic inflammation. J. Clin. Invest. 99, 643–650.
De Benedetti F., Meazza C., Oliveri M., Pignatti P., Vivarelli M., Alonzi T., Fattori E., Garrone S., Barreca A., Martini A. (2001) Effect of IL-6 on IGF binding protein-3: a study in IL-6 transgenic mice and in patients with systemic juvenile idiopathic arthritis. Endocrinology. 142, 4818–4826.
Fattori E., Della Rocca C., Costa P., Giorgio M., Dente B., Pozzi L., Ciliberto G. (1994) Development of progressive kidney damage and myeloma kidney in interleukin-6 transgenic mice. Blood. 83, 2570–2579.
Lieskovska J., Guo D., Derman E. (2002) IL-6-overexpression brings about growth impairment potentially through a GH receptor defect. Growth Horm. IGF Res. 12, 388–398.
DiCosmo B.F., Geba G.P., Picarella D., Elias J.A., Rankin J.A., Stripp B.R., Whitsett J.A., Flavell R.A. (1994) Airway epithelial cell expression of Interleukin-6 in transgenic mice. Uncoupling of airway inflammation and bronchial hyperreactivity. J. Clin. Invest. 94, 2028–2035.
Kuhn C., 3rd, Homer R.J., Zhu Z., Ward N., Flavell R.A., Geba G.P., Elias J.A. (2000) Airway hyperresponsiveness and airway obstruction in transgenic mice. Morphologic correlates in mice overexpressing interleukin (IL)-11 and IL-6 in the lung. Am. J. Respir. Cell Mol. Biol. 22, 289–295.
Zvartsev R.V., Korshunova D.S., Gorshkova E.A., Nosenko M.A., Korneev K.V., Maksimenko O.G., Korobko I.V., Kuprash D.V., Drutskaya M.S., Nedospasov S.A., Deikin A.V. (2018) Neonatal lethality and inflammatory phenotype of the new transgenic mice with overexpression of human Interleukin-6 in myeloid cells. Dokl. Biochem. Biophys. 483, 344–347.
Campbell I.L., Abraham C.R., Masliah E., Kemper P., Inglis J.D., Oldstone M.B., Mucke L. (1993) Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA. 90, 10061–10065.
Turksen K., Kupper T., Degenstein L., Williams I., Fuchs E. (1992) Interleukin 6: insights to its function in skin by overexpression in transgenic mice. Proc. Natl. Acad. Sci. USA. 89, 5068–5072.
Lieskovska J., Guo D., Derman E. (2003) Growth impairment in IL-6-overexpressing transgenic mice is associated with induction of SOCS3 mRNA. Growth Horm. IGF Res. 13, 26–35.
De Benedetti F., Rucci N., Del Fattore A., Peruzzi B., Paro R., Longo M., Vivarelli M., Muratori F., Berni S., Ballanti P., Ferrari S., Teti A. (2006) Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 54, 3551–3563.
Корнеев К.В., Свиряева Е.Н., Друцкая М.С., Купраш Д.В., Недоспасов С.А. (2016) Разработка системы для индуцируемой Cre-зависимой продукции интерлейкина-6 человека в клетках мыши и человека. Рос. иммунологический журнал. 10, 188–192.
Друцкая М.С., Гоголева В.С., Атретханы К.-С.Н., Губернаторова Е.О., Зварцев Р.В., Носенко М.А., Недоспасов С.А. (2018) Провоспалительные и иммунорегуляторные функции интерлейкина-6, выявленные с помощью технологий обратной генетики Молекуляр. биология. 52, 963–974.
Горшкова Е.А., Зварцев Р.В., Носенко M.A., Корнеев К.В., Купраш Д.В., Друцкая М.С., Дейкин А.В., Недоспасов С.А. (2018) Tрансгенная мышь со сверхэкспрессией интерлейкина-6 человека в миелоидных клетках. V Международная конференция “ПОСТГЕНОМ’2018. В поисках моделей персонализированной медицины”. Сб. тезисов, Издательство Казанского университета, Казань, с. 269.
Zeilhofer H.U., Schorr W. (2000) Role of interleukin-8 in neutrophil signaling. Curr. Opin. Hematol. 7, 178–182.
Elliott M.J., Finn A.H. (1993) Interaction between neutrophils and endothelium. Ann. Thorac. Surg. 56, 1503–1508.
Rot A. (1991) Chemotactic potency of recombinant human neutrophil attractant/activation protein-1 (interleukin-8) for polymorphonuclear leukocytes of different species. Cytokine. 3, 21–27.
Simonet W.S., Hughes T.M., Nguyen H.Q., Trebasky L.D., Danilenko D.M., Medlock E.S. (1994) Long-term impaired neutrophil migration in mice overexpressing human interleukin-8. J. Clin. Invest. 94, 1310–1319.
Kim M.R., Manoukian R., Yeh R., Silbiger S.M., Danilenko D.M., Scully S., Sun J., DeRose M.L., Stolina M., Chang D., Van G.Y., Clarkin K., Nguyen H.Q., Yu Y.B., Jing S., Senaldi G., Elliott G., Medlock E.S. (2002) Transgenic overexpression of human IL-17E results in eosinophilia, B-lymphocyte hyperplasia, and altered antibody production. Blood. 100, 2330–2340.
Park M.H., Yoon D.Y., Ban J.O., Kim D.H., Lee D.H., Song S., Kim Y., Han S.B., Lee H.P., Hong J.T. (2015) Decreased severity of collagen antibody and lipopolysaccharide-induced arthritis in human IL-32beta overexpressed transgenic mice. Oncotarget. 6, 38566–38577.
Yun H.M., Kim J.A., Hwang C.J., Jin P., Baek M.K., Lee J.M., Hong J.E., Lee S.M., Han S.B., Oh K.W., Choi D.Y., Yoon D.Y., Hong J.T. (2015) Neuroinflammatory and amyloidogenic activities of IL-32 beta in Alzheimer’s disease. Mol. Neurobiol. 52, 341–352.
Jung Y.Y., Katila N., Neupane S., Shadfar S., Ojha U., Bhurtel S., Srivastav S., Son D.J., Park P.H., Yoon D.Y., Hong J.T., Choi D.Y. (2017) Enhanced dopaminergic neurotoxicity mediated by MPTP in IL-32 beta transgenic mice. Neurochem. Int. 102, 79–88.
Haak S., Croxford A.L., Kreymborg K., Heppner F.L., Pouly S., Becher B., Waisman A. (2009) IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J. Clin. Invest. 119, 61–69.
Croxford A.L., Karbach S., Kurschus F.C., Wortge S., Nikolaev A., Yogev N., Klebow S., Schuler R., Reissig S., Piotrowski C., Brylla E., Bechmann I., Scheller J., Rose-John S., Thomas Wunderlich F., Munzel T., von Stebut E., Waisman A. (2014) IL-6 regulates neutrophil microabscess formation in IL-17A-driven psoriasiform lesions. J. Invest. Dermatol. 134, 728–735.
Karbach S., Croxford A.L., Oelze M., Schuler R., Minwegen D., Wegner J., Koukes L., Yogev N., Nikolaev A., Reissig S., Ullmann A., Knorr M., Waldner M., Neurath M.F., Li H., Wu Z., Brochhausen C., Scheller J., Rose-John S., Piotrowski C., Bechmann I., Radsak M., Wild P., Daiber A., von Stebut E., Wenzel P., Waisman A., Munzel T. (2014) Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler. Thromb. Vasc. Biol. 34, 2658–2668.
Kim S.H., Han S.Y., Azam T., Yoon D.Y., Dinarello C.A. (2005) Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 22, 131–142.
Nold M.F., Nold-Petry C.A., Pott G.B., Zepp J.A., Saavedra M.T., Kim S.H., Dinarello C.A. (2008) Endogenous IL-32 controls cytokine and HIV-1 production. J. Immunol. 181, 557–565.
Li W., Yang F., Liu Y., Gong R., Liu L., Feng Y., Hu P., Sun W., Hao Q., Kang L., Wu J., Zhu Y. (2009) Negative feedback regulation of IL-32 production by iNOS activation in response to dsRNA or influenza virus infection. Eur. J. Immunol. 39, 1019–1024.
Bai X., Ovrutsky A.R., Kartalija M., Chmura K., Kamali A., Honda J.R., Oberley-Deegan R.E., Dina-rello C.A., Crapo J.D., Chang L.Y., Chan E.D. (2011) IL-32 expression in the airway epithelial cells of patients with Mycobacterium avium complex lung disease. Int. Immunol. 23, 679–691.
Heinhuis B., Koenders M.I., van de Loo F.A., Netea M.G., van den Berg W.B., Joosten L.A. (2011) Inflammation-dependent secretion and splicing of IL-32 gamma in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 108, 4962–4967.
Zhang S., Guo G.L., Yang L.L., Sun L.Q. (2017) Elevated serum levels of ghrelin and TNF-alpha in patients with cyanotic and acyanotic congenital heart disease. World J. Pediatr. 13, 122–128.
Jiang X.G., Yang X.D., Lv Z., Zhuang P.H. (2018) Elevated serum levels of TNF-alpha, IL-8, and ECP can be involved in the development and progression of bronchial asthma. J. Asthma. 55, 111–118.
Ciebiera M., Wlodarczyk M., Wrzosek M., Wojtyla C., Blazej M., Nowicka G., Lukaszuk K., Jakiel G. (2018) TNF-alpha serum levels are elevated in women with clinically symptomatic uterine fibroids. Int. J. Immunopathol. Pharmacol. 32, 2058738418779461.
Chen Y.L., Qiao Y.C., Xu Y., Ling W., Pan Y.H., Huang Y.C., Geng L.J., Zhao H.L., Zhang X.X. (2017) Serum TNF-alpha concentrations in type 2 diabetes mellitus patients and diabetic nephropathy patients: A systematic review and meta-analysis. Immunol. Lett. 186, 52–58.
Luo Y., He H., Zhang M., Huang X., Fan N. (2016) Altered serum levels of TNF-alpha, IL-6 and IL-18 in manic, depressive, mixed state of bipolar disorder patients. Psychiatry Res. 244, 19–23.
Ozler K., Aktas E., Atay C., Yilmaz B., Arikan M., Gungor S. (2016) Serum and knee synovial fluid matrixmetalloproteinase-13 and tumor necrosis factor-alpha levels in patients with late stage osteoarthritis. Acta Orthop. Traumatol. Turc. 50, 670–673.
Georgopoulos S., Plows D., Kollias G. (1996) Transmembrane TNF is sufficient to induce localized tissue toxicity and chronic inflammatory arthritis in transgenic mice. J. Inflamm. 46, 86–97.
Kontoyiannis D., Pasparakis M., Pizarro T.T., Cominelli F., Kollias G. (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 10, 387–398.
Lewis M., Tartaglia L.A., Lee A., Bennett G.L., Rice G.C., Wong G.H., Chen E.Y., Goeddel D.V. (1991) Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl. Acad. Sci. USA. 88, 2830–2834.
Atretkhany K.N., Mufazalov I.A., Dunst J., Kuchmiy A., Gogoleva V.S., Andruszewski D., Drutskaya M.S., Faustman D.L., Schwabenland M., Prinz M., Kruglov A.A., Waisman A., Nedospasov S.A. (2018) Intrinsic TNFR2 signaling in T regulatory cells provides protection in CNS autoimmunity. Proc. Natl. Acad. Sci. USA. 115, 13051–13056.
Li P., Schwarz E.M. (2003) The TNF-alpha transge-nic mouse model of inflammatory arthritis. Springer Semin. Immunopathol. 25, 19–33.
Horta-Baas G., Romero-Figueroa M.D.S., Montiel-Jarquin A.J., Pizano-Zarate M.L., Garcia-Mena J., Ramirez-Duran N. (2017) Intestinal dysbiosis and rheumatoid arthritis: a link between gut microbiota and the pathogenesis of rheumatoid arthritis. J. Immunol. Res. 2017, 4835189.
Maeda Y., Takeda K. (2017) Role of gut microbiota in rheumatoid arthritis. J. Clin. Med. 6,
Ruddle N.H., Bergman C.M., McGrath K.M., Lingenheld E.G., Grunnet M.L., Padula S.J., Clark R.B. (1990) An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J. Exp. Med. 172, 1193–1200.
Xanthoulea S., Gijbels M.J., van der Made I., Mujcic H., Thelen M., Vergouwe M.N., Ambagts M.H., Hofker M.H., de Winther M.P. (2008) P55 tumour necrosis factor receptor in bone marrow-derived cells promotes atherosclerosis development in low-density lipoprotein receptor knock-out mice. Cardiovasc. Res. 80, 309–318.
Jung I.H., Choi J.H., Jin J., Jeong S.J., Jeon S., Lim C., Lee M.R., Yoo J.Y., Sonn S.K., Kim Y.H., Choi B.K., Kwon B.S., Seoh J.Y., Lee C.W., Kim D.Y., Oh G.T. (2014) CD137-inducing factors from T cells and macrophages accelerate the destabilization of atherosclerotic plaques in hyperlipidemic mice. FASEB J. 28, 4779–4791.
Fujii S. (2015) Atherosclerosis, chronic inflammation, and thrombosis: in search of the missing link in laboratory medicine. Rinsho Byori. 63, 605–611.
Glosli H., Prydz H., Roald B. (2004) Involution of thymus and lymphoid depletion in mice expressing the hTNF transgene. APMIS. 112, 63–73.
Rose-John S. (2012) IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 8, 1237–1247.
Ueda O., Tateishi H., Higuchi Y., Fujii E., Kato A., Kawase Y., Wada N.A., Tachibe T., Kakefuda M., Goto C., Kawaharada M., Shimaoka S., Hattori K., Jishage K. (2013) Novel genetically-humanized mouse model established to evaluate efficacy of therapeutic agents to human interleukin-6 receptor. Sci. Rep. 3, 1196.
Sitenga J., Aird G., Ahmed A., Silberstein P.T. (2018) Impact of siltuximab on patient-related outcomes in multicentric Castleman’s disease. Patient Relat. Outcome Meas. 9, 35–41.
van Hamburg J.P., Tas S.W. (2018) Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J. Autoimmun. 87, 69–81.
Waisman A., Hauptmann J., Regen T. (2015) The role of IL-17 in CNS diseases. Acta Neuropathol. 129, 625–637.
Malakouti M., Brown G.E., Wang E., Koo J., Levin E.C. (2015) The role of IL-17 in psoriasis. J. Dermatolog. Treat. 26, 41–44.
Chen X., Zhao S., Tang X., Ge H., Liu P. (2011) Neutralization of mouse interleukin-17 bioactivity inhibits corneal allograft rejection. Mol. Vis. 17, 2148–2156.
Gorbacheva V., Fan R., Li X., Valujskikh A. (2010) Interleukin-17 promotes early allograft inflammation. Am. J. Pathol. 177, 1265–1273.
Schleich F.N., Chevremont A., Paulus V., Henket M., Manise M., Seidel L., Louis R. (2014) Importance of concomitant local and systemic eosinophilia in uncontrolled asthma. Eur. Respir. J. 44, 97–108.
Drago F., Cogorno L., Agnoletti A.F., Parodi A. (2015) Role of peripheral eosinophilia in adverse cutaneous drug reactions. Eur. Rev. Med. Pharmacol. Sci. 19, 2008–2009.
Mitre E. (2013) Eosinophilia: a diagnostic clue for nonbacterial diseases in patients with systemic inflammatory response syndrome. Crit. Care Med. 41, 2464–2465.
Senra L., Mylonas A., Kavanagh R.D., Fallon P.G., Conrad C., Borowczyk-Michalowska J., Wrobel L.J., Kaya G., Yawalkar N., Boehncke W.H., Brembilla N.C. (2019) IL-17E (IL-25) enhances innate immune responses during skin inflammation. J. Invest. Dermatol. pii: S0022-202X(19)30101-0. https://doi.org/10.1016/ j.jid.2019.01.021
Mosolygo T., Spengler G., Endresz V., Laczi K., Perei K., Burian K. (2013) IL-17E production is elevated in the lungs of Balb/c mice in the later stages of Chlamydia muridarum infection and re-infection. In Vivo. 27, 787–792.
Senra L., Stalder R., Alvarez Martinez D., Chizzolini C., Boehncke W.H., Brembilla N.C. (2016) Keratinocyte-derived IL-17E contributes to inflammation in psoriasis. J. Invest. Dermatol. 136, 1970–1980.
Kang J.W., Choi S.C., Cho M.C., Kim H.J., Kim J.H., Lim J.S., Kim S.H., Han J.Y., Yoon D.Y. (2009) A proinflammatory cytokine interleukin-32 beta promotes the production of an anti-inflammatory cytokine interleukin-10. Immunology. 128, e532–540.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология