

Молекулярная биология, 2019, T. 53, № 6, стр. 1012-1019
Ниши гемопоэтических стволовых клеток в костном мозге
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: abelyavs@yahoo.com
Поступила в редакцию 03.12.2018
После доработки 04.04.2019
Принята к публикации 08.04.2019
Аннотация
Гемопоэтические стволовые клетки (ГСК) существуют в тесном контакте со своим специфическим микроокружением, называемом нишей, которое поддерживает функционирование ГСК и оказывает значительное влияние на их свойства. Существование ниши ГСК, выдвинутое в 1978 г. как чисто теоретическое понятие, находит все больше экспериментальных подтверждений и в настоящее время признано специалистами в области гемопоэза. В настоящем обзоре приведено краткое описание различных клеточных компонентов ниши ГСК в костном мозге, метаболического состояния ниши и ГСК, а также рассмотрены другие аспекты биологии ниши. Можно предполагать, что углубление наших знаний о нише ГСК позволит создать клеточные in vitro системы, моделирующие нишу, манипулировать свойствами ГСК и достигать их многократного размножения в культуре с целью дальнейшего применения в терапевтической практике.
ВВЕДЕНИЕ
Представление о гемопоэтической стволовой клетке (ГСК) как о предшественнике, общем для всех клеток крови, впервые было предложено А.А. Максимовым в начале XX века на основе гистологического анализа [1]. Однако экспериментально существование ГСК было подтверждено много позднее – в 1963 году [2]. Понятие ниши ГСК, как специфических клеток, которые находятся в ассоциации с ГСК, определяют их поведение и, прежде всего, ингибируют их дифференцировку и обеспечивают самовоспроизведение, впервые было выдвинуто R. Schofield в 1978 г. [3]. При этом постулировалось, что те дочерние клетки, которые вытесняются из ниши, вступают на путь дифференцировки.
МЕЗЕНХИМАЛЬНЫЙ КОМПОНЕНТ НИШИ
Схематическое изображение организации ниши ГСК в костном мозге (КМ), соответствующее современным представлениям, показано на рис. 1. Первым кандидатом на роль ниши ГСК в КМ стали остеобласты эндостальных областей кости. Такой вывод, сделанный в двух одновременно вышедших в 2003 году публикациях, опирался на корреляции между увеличением числа остеобластов и числом клеток с фенотипом ГСК, выявляемых при инактивации рецептора морфогенетического белка кости (BMPRIA) [4] или обработке паратгормоном [5]. В следующем году увидела свет работа [6], согласно которой селективная элиминация остеобластов приводила не только к прогрессирующей дегенерации костной ткани, но и к уменьшению числа ГСК при одновременном увеличении экстрамедуллярного кроветворения в селезенке и печени [6]. Позднее показали, что остеобласты необходимы и достаточны для дифференцировки В-лимфоцитов в КМ [7].
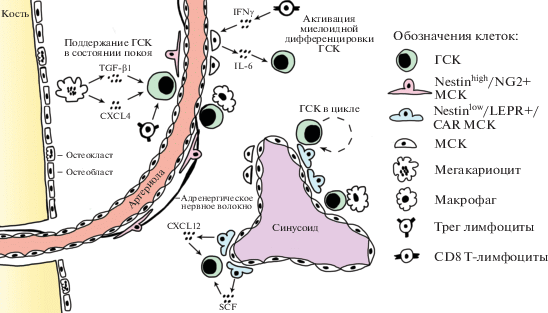
Рис. 1.
Клеточные и молекулярные компоненты ниши ГСК в костном мозге. Ниши ГСК в костном мозге мышей локализованы в области кровеносных сосудов двух главных типов – артериол и синусоидов. ГСК в состоянии покоя находятся в контакте с периартериолярными мезенхимальными стволовыми клетками (МСК) Nestinhigh/NG2+. Мегакариоциты поддерживают состояние покоя ГСК за счет секреции TGF-β1 и CXCL4. Адренергические нервные волокна также могут поддерживать состояние покоя ГСК за счет секреции TGF-β1, кроме того, они регулируют мобилизацию ГСК из ниш. Делящиеся ГСК находятся в контакте с перисинусоидными Nestinlow/LEPR+/CAR МСК. Эти клетки продуцируют факторы SCF и CXCL12, необходимые для поддержания ГСК и удержания их в нише. Макрофаги, подобно мегакариоцитам, поддерживают состояние покоя ГСК и способствуют их удержанию в нише за счет продукции простагландина Е2, онкостатина М и CXCL12. Эффекторные CD8 Т-лимфоциты стимулируют миелоидную дифференцировку ГСК, воздействуя на МСК. Регуляторные Т-лимфоциты (Трег) в составе ниши защищают ГСК от иммунной атаки.
Хотя эти данные в то время выглядели вполне убедительными, они не были вполне прямыми, поскольку отсутствовало подтверждение прямого контакта ГСК и остеобластов или их непосредственной близости, что, в свою очередь, обусловлено отсутствием на то время клеточных маркеров, относительно прямо характеризующих ГСК. Ситуация изменилась с открытием того, что маркерами, достаточно адекватно определяющими фенотип ГСК в КМ, могут служить рецепторы семейства SLAM. ГСК позитивны по SLAM-рецептору CD150 и негативны по CD48 [8]. Кроме того, весьма значительную роль в прояснении идентичности ниши сыграли две группы трансгенных линий мышей, в одной из которых ГСК относительно специфично экспрессировали флуоресцентные белки, а в другой предполагаемые клеточные компоненты ниши содержали маркеры, позволяющие проводить селективную абляцию этих компонентов.
В дальнейшем роль остеобластов в нише ГСК была поставлена под сомнение. На авансцену выдвинулись мезенхимальные стволовые/стромальные клетки (МСК) и эндотелиальные клетки. МСК, открытые А.Я. Фриденштейном [9], способны к организации эктопических очагов кроветворения, в которые могут мигрировать кроветворные и эндотелиальные клетки [10, 11]. В частности, клетки КМ, идентифицируемые по экспрессии гена белка промежуточных филаментов нестина (Nestin-GFP), представляют собой периваскулярно локализованные МСК и являются интегральным компонентом ниши ГСК [12]. Nestin+ МСК пространственно ассоциированы с ГСК и адренергическими нервными волокнами, которые, как показано ранее, регулируют выход ГСК из КМ [13]. В этих МСК на высоком уровне экспрессируются гены, участвующие в поддержании ГСК, под действием паратгормона их численность возрастает вдвое, что, по-видимому, объясняет полученные ранее результаты [5], указавшие на остеобласты, как на нишу ГСК. При деплеции этих клеток in vivo наблюдается значительное снижение хоуминга ГСК в КМ.
Роль хемокина CXCL12, одного из ключевых игроков механизма хоуминга и удержания ГСК нишей, хорошо известна. В дальнейшем с использованием мышей, в локус гена CXCL12 которых встроен репортерный ген Gfp, идентифицировали ретикулярные клетки, характеризующиеся высокой экспрессией CXCL12 (так называемые CAR-клетки). Селективная абляция таких клеток приводила к резкому снижению потенциала остеогенной и адипогенной дифференцировки клеток КМ и уменьшению числа ГСК [14]. Кроме Nestin+ и CAR МСК, идентифицированы также периваскулярные МСК LepR+ [15, 16] и NG2+ [17], ассоциированные с ГСК и входящие в состав ниши. Вполне вероятно, что популяции этих МСК в значительной степени перекрываются, однако существуют различия в их локализации в КМ, особенно относительно сосудов КМ (более подробно рассмотрено в следующем разделе).
ЭНДОТЕЛИАЛЬНЫЙ КОМПОНЕНТ НИШИ
Впервые роль эндотелиального компонента в нише ГСК выявили после того, как удалось идентифицировать маркеры CD150 и CD48, позволяющие достаточно адекватно визуализировать ГСК в КМ [8]. Значительная часть ГСК обнаружена в ассоциации с синусоидальным эндотелием, хотя некоторые находились вблизи эндостальной области. В дальнейшем МСК идентифицировали в качестве компонента ниши и показали в целом периваскулярное расположение МСК (характерного места локализации МСК как класса клеток).
Тем не менее, существуют, по-видимому, объективные различия между периваскулярными нишами. На данный момент, видимо, можно говорить как минимум о двух классах ниш. Покоящиеся ГСК ассоциированы с малыми артериолами, преимущественно находящимися в эндостальных областях КМ, с которыми связаны NG2+ МСК. Выход ГСК в цикл приводит к перераспределению их из NG2+ периартериолярных ниш в LEPR+ перисинусоидальные ниши [17]. Более того, специфическое истощение NG2+ клеток вызывает пролиферацию ГСК, что приводит к снижению числа длительно репопулирующих ГСК.
Кроме того, эффекты цитокинов, продуцируемых этими классами ниш, различаются. Показано, что делеция CXCL12 или SCF (stem cell factor) во всех периваскулярных нишах, маркированных Nestin-GFP, приводит к резкой потере ГСК в КМ [18]. В то же время, к потере ГСК приводила делеция CXCL12 только в периартериолярных NG2+ клетках, но не в синусоидальных LEPR+ клетках. Наоборот, потерю ГСК вызывала делеция SCF только в LEPR+ клетках, но не в NG2+ клетках.
Хотя основная роль в регуляции ГСК отводилась стромальному компоненту, роль эндотелиальных клеток вряд ли ограничивается только предоставлением поверхности для прикрепления МСК. Так, почти одновременно с выявлением роли МСК в нише показано, что зависимая от VEGFR2 регенерация эндотелиальных клеток необходима для восстановления гемопоэза [19]. Возможно, что обнаруженное свойство связано с плейотрофином (PTN) – цитокином, важным для поддержания ГСК, хоуминга и удержания ГСК в нише, а также для восстановления гемопоэза после миелоабляции [20]. Недавно установлено [21], что основным источником PTN при нормальном гемопоэзе являются МСК, поскольку его делеция в этих клетках уменьшает пул ГСК. В то же время, при миелосупрессии, вызванной облучением, экспрессия PTN возрастает в эндотелиальных клетках. Важно, что специфическая делеция PTN в эндотелиальных клетках подавляла восстановление гемопоэза [21], в то время как делеция PTN в МСК не вызывала такого эффекта. По-видимому, экспрессию PTN в эндотелиальных клетках можно считать одним из основных механизмов контроля восстановления гемопоэза эндотелием КМ.
ОБРАТНАЯ СВЯЗЬ В РЕГУЛЯЦИИ ГСК НИШЕЙ
Учитывая распространенность отрицательных обратных связей в биологических системах, которые позволяют поддерживать динамический баланс в системе и отвечать на изменения среды, а также (см. выше) необходимость кроветворной системы отвечать на различные “вызовы”, можно предполагать, что потомство ГСК играет собственную роль в регуляции функционирования ГСК. Эту точку зрения подтверждает ряд исследований, описанных ниже, однако, по всей видимости, взаимодействия между ГСК и зрелыми кроветворными клетками носят сложный характер и не всегда укладываются в модель отрицательной обратной связи.
Прежде всего, стоит отметить роль макрофагов. Истощение макрофагов КМ, в частности CD169-позитивных, приводит к снижению уровня хемокина CXCL12 [22], который, как обсуждалось выше, способствует удержанию ГСК в нишах, снижению уровня экспрессии других генов, участвующих в удерживании ГСК, в Nestin(+) клетках, и вызывают мобилизацию ГСК. Впоследствии установили, что продуцируемый макрофагами онкостатин М способствует удерживанию ГСК в нише, стимулируя продукцию CXCL12, что, вероятно, и вызывает эффекты, наблюдаемые при истощении макрофагов [23]. Кроме того, популяции макрофагов, экспрессирующих на высоком уровне циклооксигеназу-2, могут, вероятно, участвовать в поддержании ГСК за счет продукции простагландина Е2 [24]. Утверждается, что популяция эндостальных макрофагов также способствует удержанию ГСК, а деплеция этих клеток приводит к мобилизации ГСК [25]. С другой стороны, остеокласты, происходящие из макрофагов, способны, наоборот, вызывать в стрессовых ситуациях мобилизацию ГСК [26].
Другой важный компонент кроветворной системы, способный оказывать значительное воздействие на ГСК – мегакариоциты. ГСК в КМ часто находятся вблизи мегакариоцитов, деплеция которых вызывает выход ГСК в клеточный цикл и экспансию их пула [27, 28]. Негативная регуляция пролиферации ГСК мегакариоцитами обусловлена экспрессией ими хемо/цитокинов CXCL4 [27] и TGF-β [28]. Впрочем, получены также данные [29], которые хотя и подтверждают, что истощение мегакариоцитов вызывает выход ГСК в клеточный цикл, но показывают также, что это сопровождается потерей ГСК за счет, как считают авторы, снижения в КМ уровня тромбопоэтина, необходимого для выживания ГСК.
В целом, можно считать, что в норме мегакариоциты функционируют, как негативные регуляторы размножения ГСК. При этом весьма интересно, что в условиях стресса, вызванного химиотерапией, мегакариоциты, наоборот, не подавляют, а стимулируют размножение ГСК за счет продукции фактора роста фибробластов 1 (FGF1) [28]. По-видимому, роль мегакариоцитов в регуляции состояния и функций ГСК может в значительной степени зависеть от биологического контекста.
Стоит отметить роль Т-лимфоцитов в функционировании ниши. Показано, что аллогенные ГСК после введения мышам, не подвергшимся облучению, способны выживать в течение 30 дней с такой же вероятностью, как и сингенные ГСК, т.е. ниша ГСК представляет собой иммунопривилегированный локус [30]. При этом деплеция FoxP3 регуляторных Т-лимфоцитов (Трег) приводит к потере аллогенных ГСК. Трег обнаружены в поверхностных зонах эндостальных областей. Таким образом, ниша ГСК в КМ способна защищать стволовые клетки от иммунной атаки, как это происходит в некоторых других нишах.
Совсем недавно в КМ обнаружена популяция Трег, которые характеризовались высоким уровнем экспрессии CD150 ‒ маркера ГСК, и локализовались в нише ГСК [31]. За счет CD39 эти лимфоциты продуцировали значительные количества аденозина, который способствовал сохранению ГСК в состоянии покоя. Более того, совместная трансплантация таких нишевых Т-лимфоцитов в значительно большей степени способствует приживлению трансплантированных аллогенных ГСК, чем трансплантация других видов Трег.
Весьма интересно исследование, согласно которому при острой вирусной инфекции эффекторные CD8 Т-лимфоциты продуцируют интерферон-γ (IFNγ), который стимулирует миелопоэз для борьбы с инфекцией. При этом IFNγ действует не непосредственно на ГСК, а на МСК ниши, которые, в свою очередь, продуцируют цитокины, в том числе интерлейкин-6 (IL-6), активирующие миелоидную дифференцировку ранних кроветворных предшественников [32].
МЕТАБОЛИЧЕСКИЕ СОСТОЯНИЯ НИШИ И ГСК
Ряд данных указывает на то, что ГСК в КМ находятся в областях с низкой концентрацией кислорода [33]. В частности, это показано с использованием пимонидазола ‒ индикатора гипоксии [34]. Кроме того, в ГСК повышен уровень ключевого сенсора гипоксии HIF-1α, что характерно для клеток, находящихся в условиях низкого парциального давления кислорода [35]. Повышенный уровень HIF-1α при этом важен для поддержания ГСК в покое. Впрочем, необходимость HIF-1α для ГСК остается неясной, поскольку другими авторами показано нормальное функционирование ГСК у мышей с инактивированным HIF-1α [36].
Установлено, что для поддержания энергетического баланса ГСК используют аэробный гликолиз [37]. Прямой анализ уровня кислорода в различных участках КМ выявил в них весьма низкий общий уровень кислорода ‒ самый низкий в центральных областях и, наоборот, наиболее высокий в эндостальных областях вблизи артериол, а также вблизи сосудов [38]. Таким образом, эти данные свидетельствуют против ранних представлений, согласно которым нишу ГСК составляют остеобласты эндостальных областей [4, 5].
Пребывание ГСК в условиях гипоксии, скорее всего, влияет на уровень активных форм кислорода (АФК) в них. Повышенные уровни АФК, как хорошо известно, могут вызывать повреждения биомолекул и, прежде всего, ДНК [39]. Поэтому как гипоксия, так и снижение уровня дыхания должны работать, как механизмы, обеспечивающие адекватную защиту генетического аппарата ГСК от повреждений.
С другой стороны, АФК являются важным компонентом системы передачи сигналов в клетке. В этой связи важно упомянуть, что снижение уровня АФК ниже критического путем инактивации киназ AKT1 и AKT2 способно вызвать ингибирование дифференцировки ГСК за счет удержания клеток в фазе G0 [40]. Это, в свою очередь, препятствует выполнению ГСК своей основной функции – поддержанию клеточного гомеостаза кроветворной системы. Таким образом, поддержание АФК на оптимальном уровне (или же на пониженном, но с возможностью транзиентного повышения при необходимости инициации дифференцировки) необходимо для сохранения интактности и функциональности ГСК в организме.
Кроме киназ AKT1 и AKT2, в регуляции уровня АФК в ГСК участвуют также АТМ [41], киназа р38 и транскрипционный фактор Foxo3a [42]. Последний, кроме того, участвует в регуляции процесса аутофагии, необходимого для поддержания функции ГСК в течение жизни [43]. Можно предположить, что в регуляции уровня и работы этих факторов и уровня АФК в ГСК в целом могут участвовать клеточные компоненты ниши. Эту гипотезу частично подтверждают некоторые данные, полученные к настоящему времени. Так, при мобилизации ГСК с помощью гранулоцитарного колониестимулирующего фактора (G-CSF) происходит заметное повышение уровня АФК в КМ [44]. Такое повышение необходимо, поскольку уровень мобилизации ГСК снижается в присутствии ингибиторов АФК [45]. Возможно, мобилизация обусловлена активацией металлопротеаз под действием АФК [46].
ЛЕЙКОЗНЫЕ НИШИ
Ниша ГСК поддерживает нормальный гемопоэз, при котором пролиферация клеток тщательно контролируется, однако все больше данных указывает на то, что нарушения генотипа компонентов ниши способны вызывать миелопролиферативные заболевания. Так, инактивация гена RARγ в КМ может приводить к аномальной миелопролиферации, вызванной, по-видимому, в том числе повышенной продукцией TNFα [47]. Показано также, что нокаут гена Mind bomb-1, играющего важную роль в эндоцитозе лигандов рецептора Notch, также приводит к миелопролиферативным нарушениям и накоплению незрелых гранулоцитов [48]. При этом трансплантация нормальных кроветворных клеток в дефектное по Mind bomb-1 микроокружение КМ также вызывает нарушения, что указывает на инструктивную роль микроокружения в возникновении аномальной миелопролиферации.
Значительную роль микроокружение КМ играет в прогрессии лейкоза. Лейкозные клоны после их начального размножения в КМ распространяются на другие участки КМ в тех же и других костях. Показано, что лейкозные клетки способны к хоумингу в специфические участки эндотелия КМ, экспрессирующие CXCL12 и Е-селектин [49]. При этом специфическая делеция CXCL12 в эндотелиальных, но не периваскулярных клетках приводила к торможению острого Т-лимфобластного лейкоза (T-ALL) [50]. К сходным результатам приводила также делеция CXCR4 (рецептора CXCL12) в клетках T-ALL.
Важно, что сами лейкозные клетки способны значительно модифицировать свойства компонентов ниши КМ. Так, при хроническом лимфоцитарном лейкозе МСК в КМ содержат значительно меньше КОЕ-Ф (колониеобразующие единицы ‒ фибробласты), медленнее пролиферируют, а их свойства сходны со свойствами стареющих клеток [51]. Кроме того, в in vitro модели лейкозной ниши также наблюдается старение МСК и, что важно для понимания патологического процесса при лейкозе, нормальные ГСК, помещенные на такую лейкозную нишу, входят в клеточный цикл и интенсивно делятся [52]. В целом, совокупность приведенных данных свидетельствует о взаимном влиянии компонентов ниши и лейкозных клеток, способном, с одной стороны, приводить к патологическим изменениям нормальных ГСК под действием измененной ниши, а с другой, вызывать патологическое ремоделирование нормальной ниши под действием лейкозных клеток.
СПЕЦИФИЧНОСТЬ НИШИ ГСК
Важным в биологии ниш ГСК является следующий вопрос: насколько специфичны идентифицированные ниши в КМ в отношении ГСК, в том числе, способно ли их потомство, вступившее на путь дифференцировки, находиться в тех же нишах, и если нет, то какова в таком случае их локализация в КМ. Совсем недавно показано, что маркированные экспрессией фактора Виллебранда (vWF+ ГСК) ГСК, имеющие сдвиг в сторону миелоидной/мегакариоцитарной дифференцировки, ассоциированы с нишами, в которых присутствуют мегакариоциты. В то же время, с артериолярными NG2+ нишами ассоциированы vWF-ГСК, которые имеют сдвиг в сторону лимфоидной дифференцировки [53]. Истощение NG2+ клеток селективно снижает число лимфоидных ГСК, тогда как деплеция мегакариоцитов, наоборот, способствует экспансии пула миелоидных ГСК. Однако функциональность последних при этом снижается, поскольку нарушается их способность к долговременному самоподдержанию.
Согласно [54] делеция CXCL12 из эндотелиальных клеток приводит к элиминации ГСК но не миелоэритроидных или лимфоидных предшественников, в то же время делеция CXCL12 из остеобластов элиминирует лимфоидные предшественники, но не ГСК или миелоэритроидные предшественники. На основании полученных результатов сделан вывод, что ГСК и лимфоидные предшественники занимают разные ниши в КМ: периваскулярную и эндостальную соответственно. Впрочем, данный вывод вряд ли можно считать доказанным, поскольку согласно [55] делеция CXCL12 в остеобластах такого эффекта не оказывала.
В интересном исследовании, проведенном в 2017 г., изучено распределение гранулоцитарно-макрофагальных предшественников (ГМП) в КМ [56]. Выяснилось, что при нормальном гемопоэзе индивидуальные ГМП рассеяны случайным образом по КМ. При этом во время регенерации ГМП сосредоточены в кластерах, образующихся, очевидно, в результате ограниченного размножения/воспроизведения, при этом параллельно происходит локальная дифференцировка в зрелые гранулоциты. Размножение ГМП контролируется SCF, IL-1β, G-CSF, однако после выполнения задач регенерации размножение ГМП прекращается под действием негативных регуляторов TGF-β и CXCL4, и кластеры исчезают. В лейкозных клетках кластеры ГМП образуются постоянно, так как отсутствует негативный контроль.
Недавно провели весьма интересное исследование, в котором мышам, не подвергавшимся миелоабляции, ввели большие количества ГСК и выявили в КМ большое число вакантных ниш для ГСК [57]. Это противоречит устоявшимся воззрениям, согласно которым число ниш ГСК в КМ ограничено и примерно соответствует числу ГСК.
С другой стороны, встречаемость МСК, участвующих в образовании периваскулярных ниш ГСК, таких как Nestin+, LEPR+ или NG2+ клетки, значительно выше (не менее, чем в 20 раз), чем самих ГСК в КМ. Вполне возможно, что существенная часть этих клеток образует настоящие вакантные ниши ГСК.
Впрочем, альтернативное объяснение можно получить в результате анализа работ, в которых показана значительная инструктивная роль лейкозных клеток по отношению к нишам [51, 52]. Если лейкозные клетки способны ремоделировать компонент ниши, то логично предположить, что и нормальные ГСК могут действовать подобным образом. В таком случае ниши, занимаемые трансплантированными клетками, не предсуществуют в КМ, а возникают под воздействием ГСК на клетки КМ, обладающие некоторыми, но, возможно, не всеми характеристиками “зрелой” ниши стволовой клетки.
Работа выполнена при поддержке Российского научного фонда (грант № 18-14-00300).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Автор заявляет об отсутствии конфликта интересов.
Список литературы
Maximov A.A. (1909) Der Lymphozyt als gemeinsame Stammzelle der verschiedenen Blutelemente in der embryonalen Entwicklung und im postfetalen Leben der Säugetiere. Folia Haematologica. 8, 125–134.
Becker A.J., McCulloch E.A., Till J.E. (1963) Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature. 197, 452–454.
Schofield R. (1978) The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 4(1–2), 7–25.
Zhang J., Niu C., Ye L., Huang H., He X., Tong W.G., Ross J., Haug J., Johnson T., Feng J.Q., Harris S., Wiedemann L.M., Mishina Y., Li L. (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 425(6960), 836—841.
Calvi L.M., Adams G.B., Weibrecht K.W., Weber J.M., Olson D.P., Knight M.C., Martin R.P., Schipani E., Divieti P., Bringhurst F.R., Milner L.A., Kronenberg H.M., Scadden D.T. (2003) Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 425(6960), 841–846.
Visnjic D., Kalajzic Z., Rowe D.W., Katavic V., Lorenzo J., Aguila H.L. (2004) Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 103(9), 3258–3264.
Zhu J., Garrett R., Jung Y., Zhang Y., Kim N., Wang J., Joe G.J., Hexner E., Choi Y., Taichman R.S., Emerson S.G. (2007) Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 109(9), 3706—3712.
Kiel M.J., Yilmaz O.H., Iwashita T., Yilmaz O.H., Terhorst C., Morrison S.J. (2005) SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 121(7), 1109–1121.
Friedenstein A.J., Deriglasova U.F., Kulagina N.N., Panasuk A.F., Rudakowa S.F., Luriá E.A., Ruadkow I.A. (1974) Precursors for fibroblasts in different populations of hematopoietic cells as detected by the in vitro colony assay method. Exp. Hematol. 2(2), 83–92.
Chan C.K., Chen C.C., Luppen C.A., Kim J.B., DeBoer A.T., Wei K., Helms J.A., Kuo C.J., Kraft D.L., Weissman I.L. (2009) Endochondral ossification is required for hematopoietic stem-cell niche formation. Nature. 457(7228), 490–494.
Sacchetti B., Funari A., Michienzi S., Di Cesare S., Piersanti S., Saggio I., Tagliafico E., Ferrari S., Robey P.G., Riminucci M., Bianco P. (2007) Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 131(2), 324–336.
Méndez-Ferrer S., Michurina T.V., Ferraro F., Mazloom A.R., Macarthur B.D., Lira S.A., Scadden D.T., Ma’ayan A., Enikolopov G.N., Frenette P.S. (2010) Mesenchymal and hematopoietic stem cells form a unique bone marrow niche. Nature. 466(7308), 829–834.
Katayama Y., Battista M., Kao W.M., Hidalgo A., Peired A.J., Thomas S.A., Frenette P.S. (2006) Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 124(2), 407–421.
Omatsu Y., Sugiyama T., Kohara H., Kondoh G., Fujii N., Kohno K., Nagasawa T. (2010) The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 33(3), 387–399.
Ding L., Saunders T.L., Enikolopov G., Morrison S.J. (2012) Endothelial and perivascular cells maintain hematopoietic stem cells. Nature. 481(7382), 457–462.
Zhou B.O., Yue R., Murphy M.M., Peyer J.G., Morrison S.J. (2014) Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 15(2), 154–168.
Kunisaki Y., Bruns I., Scheiermann C., Ahmed J., Pinho S., Zhang D., Mizoguchi T., Wei Q., Lucas D., Ito K., Mar J.C., Bergman A., Frenette P.S. (2013) Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 502(7473), 637–643.
Asada N., Kunisaki Y., Pierce H., Wang Z., Fernandez N.F., Birbrair A., Ma’ayan A., Frenette P.S. (2017) Differential cytokine contributions of perivascular hematopoietic stem cell niches. Nat. Cell. Biol. 19(3), 214–223.
Hooper A.T., Butler J.M., Nolan D.J., Kranz A., Iida K., Kobayashi M., Kopp H.G., Shido K., Petit I., Yanger K., James D., Witte L., Zhu Z., Wu Y., Pytowski B., Rosenwaks Z., Mittal V., Sato T.N., Rafii S. (2009) Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 4(3), 263—274.
Himburg H.A., Harris J.R., Ito T., Daher P., Russell J.L., Quarmyne M., Doan P.L., Helms K., Nakamura M., Fixsen E., Herradon G., Reya T., Chao N.J., Harroch S., Chute J.P. (2012) Pleiotrophin regulates the retention and self-renewal of hematopoietic stem cells in the bone marrow vascular niche. Cell Rep. 2(4), 964–975.
Himburg H.A., Termini C.M., Schlussel L., Kan J., Li M., Zhao L., Fang T., Sasine J.P., Chang V.Y., Chute J.P. (2018) Distinct bone marrow sources of pleiotrophin control hematopoietic stem cell maintenance and regeneration. Cell Stem Cell. 23(3), 370–381.
Chow A., Lucas D., Hidalgo A., Méndez-Ferrer S., Hashimoto D., Scheiermann C., Battista M., Leboeuf M., Prophete C., van Rooijen N., Tanaka M., Merad M., Frenette P.S. (2011) Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 208(2), 261–271.
Albiero M., Poncina N., Ciciliot S., Cappellari R., Menegazzo L., Ferraro F., Bolego C., Cignarella A., Avogaro A., Fadini G.P. (2015) Bone marrow macrophages contribute to diabetic stem cell mobilopathy by producing oncostatin M. Diabetes. 64(8), 2957–2968.
Ludin A., Itkin T., Gur-Cohen S., Mildner A., Shezen E., Golan K., Kollet O., Kalinkovich A., Porat Z., D’Uva G., Schajnovitz A., Voronov E., Brenner D.A., Apte R.N., Jung S., Lapidot T. (2012) Monocytes-macrophages that express α-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat. Immunol. 13(11), 1072–1082.
Winkler I.G., Sims N.A., Pettit A.R., Barbier V., Nowlan B., Helwani F., Poulton I.J., van Rooijen N., Alexander K.A., Raggatt L.J., Lévesque J.P. (2010) Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 116(23), 4815–4828.
Kollet O., Dar A., Shivtiel S., Kalinkovich A., Lapid K., Sztainberg Y., Tesio M., Samstein R.M., Goichberg P., Spiegel A., Elson A., Lapidot T. (2006) Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 12(6), 657–664.
Bruns I., Lucas D., Pinho S., Ahmed J., Lambert M.P., Kunisaki Y., Scheiermann C., Schiff L., Poncz M., Bergman A., Frenette P.S. (2014) Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 20(11), 1315–1320.
Zhao M., Perry J.M., Marshall H., Venkatraman A., Qian P., He X.C., Ahamed J., Li L. (2014) Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 20(11), 1321–1326.
Nakamura-Ishizu A., Takubo K., Fujioka M., Suda T. (2014) Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem. Biophys. Res. Commun. 454(2), 353–357.
Fujisaki J., Wu J., Carlson A.L., Silberstein L., Putheti P., Larocca R., Gao W., Saito T.I., Lo Celso C., Tsuyuzaki H., Sato T., Côté D., Sykes M., Strom T.B., Scadden D.T., Lin C.P. (2011) In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 474(7350), 216–219.
Hirata Y., Furuhashi K., Ishii H., Li H.W., Pinho S., Ding L., Robson S.C., Frenette P.S., Fujisaki J. (2018) CD150high bone marrow Tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell. 22(3), 445–453.
Schürch C.M., Riether C., Ochsenbein A.F. (2014) Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell. 14(4), 460–472.
Suda T., Takubo K., Semenza G.L. (2011) Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 9(4), 298–310.
Parmar K., Mauch P., Vergilio J.A., Sackstein R., Down J.D. (2007) Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA. 104(13), 5431–5436.
Takubo K., Goda N., Yamada W., Iriuchishima H., Ikeda E., Kubota Y., Shima H., Johnson R.S., Hirao A., Suematsu M., Suda T. (2010) Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 7(3), 391–402.
Vukovic M., Sepulveda C., Subramani C., Guitart A.V., Mohr J., Allen L., Panagopoulou T.I., Paris J., Lawson H., Villacreces A., Armesilla-Diaz A., Gezer D., Holyoake T.L., Ratcliffe P.J., Kranc K.R. (2016) Adult hematopoietic stem cells lacking Hif-1α self-renew normally. Blood. 127(23), 2841–2846.
Takubo K., Nagamatsu G., Kobayashi C.I., Nakamura-Ishizu A., Kobayashi H., Ikeda E., Goda N., Rahimi Y., Johnson R.S., Soga T., Hirao A., Suematsu M., Suda T. (2013) Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 12(1), 49–61.
Spencer J.A., Ferraro F., Roussakis E., Klein A., Wu J., Runnels J.M., Zaher W., Mortensen L.J., Alt C., Turcotte R., Yusuf R., Côté D., Vinogradov S.A., Scadden D.T., Lin C.P. (2014) Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 508(7495), 269–273.
Zuo L., Zhou T., Pannell B.K., Ziegler A.C., Best T.M. (2015) Biological and physiological role of reactive oxygen species – the good, the bad and the ugly. Acta Physiol. (Oxf.). 214(3), 329–348
Juntilla M.M., Patil V.D., Calamito M., Joshi R.P., Birnbaum M.J., Koretzky G.A. (2010) AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 115(20), 4030–4038.
Ito K., Hirao A., Arai F., Matsuoka S., Takubo K., Hamaguchi I., Nomiyama K., Hosokawa K., Sakurada K., Nakagata N., Ikeda Y., Mak T.W., Suda T. (2004) Regulation of oxidative stress by ATM is required for self-renewal of hematopoietic stem cells. Nature. 431(7011), 997–1002.
Miyamoto K., Araki K.Y., Naka K., Arai F., Takubo K., Yamazaki S., Matsuoka S., Miyamoto T., Ito K., Ohmura M., Chen C., Hosokawa K., Nakauchi H., Nakayama K., Nakayama K.I., Harada M., Motoyama N., Suda T., Hirao A. (2007) Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 1(1), 101–112.
Warr M.R., Binnewies M., Flach J., Reynaud D., Garg T., Malhotra R., Debnath J., Passegué E. (2013) FOXO3A directs a protective autophagy program in hematopoietic stem cells. Nature. 494(7437), 323–327.
Ludin A., Gur-Cohen S., Golan K., Kaufmann K.B., Itkin T., Medaglia C., Lu X.J., Ledergor G., Kollet O., Lapidot T. (2014) Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 21(11), 160–1619.
Tesio M., Golan K., Corso S., Giordano S., Schajnovitz A., Vagima Y., Shivtiel S., Kalinkovich A., Caione L., Gammaitoni L., Laurenti E., Buss E.C., Shezen E., Itkin T., Kollet O., Petit I., Trumpp A., Christensen J., Aglietta M., Piacibello W., Lapidot T. (2011) Enhanced c-Met activity promotes G-CSF-induced mobilization of hematopoietic progenitor cells via ROS signaling. Blood. 117(2), 419–428.
Zalba G., Fortuño A., Orbe J., San José G., Moreno M.U., Belzunce M., Rodríguez J.A., Beloqui O., Páramo J.A., Díez J. (2007) Phagocytic NADPH oxidase-dependent superoxide production stimulates matrix metalloproteinase-9: implications for human atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 27(3), 587–593.
Walkley C.R., Olsen G.H., Dworkin S., Fabb S.A., Swann J., McArthur G.A., Westmoreland S.V., Chambon P., Scadden D.T., Purton L.E. (2007) A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 129(6), 1097–1110.
Kim Y.W., Koo B.K., Jeong H.W., Yoon M.J., Song R., Shin J., Jeong D.C., Kim S.H., Kong Y.Y. (2008) Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood. 112(12), 4628–4638.
Sipkins D.A., Wei X., Wu J.W., Runnels J.M., Côté D., Means T.K., Luster A.D., Scadden D.T., Lin C.P. (2005) In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 435(7044), 969–973.
Pitt L.A., Tikhonova A.N., Hu H., Trimarchi T., King B., Gong Y., Sanchez-Martin M., Tsirigos A., Littman D.R., Ferrando A.A., Morrison S.J., Fooksman D.R., Aifantis I., Schwab S.R. (2015) CXCL12-Producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell. 27(6), 755–768.
Janel A., Dubois-Galopin F., Bourgne C., Berger J., Tarte K., Boiret-Dupré N., Boisgard S., Verrelle P., Déchelotte P., Tournilhac O., Berger M.G. (2014) The chronic lymphocytic leukemia clone disrupts the bone marrow microenvironment. Stem Cells Dev. 23(24), 2972–2982.
Vanegas N.P., Vernot J.P. (2017) Loss of quiescence and self-renewal capacity of hematopoietic stem cell in an in vitro leukemic niche. Exp. Hematol. Oncol. 6, 2.
Pinho S., Marchand T., Yang E., Wei Q., Nerlov C., Frenette P.S. (2018) Lineage-biased hematopoietic stem cells are regulated by distinct niches. Dev Cell. 44(5), 634–641.
Ding L., Morrison S.J. (2013) Hematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 495(7440), 231–235.
Greenbaum A., Hsu Y.M., Day R.B., Schuettpelz L.G., Christopher M.J., Borgerding J.N., Nagasawa T., Link D.C. (2013) CXCL12 in early mesenchymal progenitors is required for hematopoietic stem-cell maintenance. Nature. 495(7440), 227—230.
Hérault A., Binnewies M., Leong S., Calero-Nieto F.J., Zhang S.Y., Kang Y.A., Wang X., Pietras E.M., Chu S.H., Barry-Holson K., Armstrong S., Göttgens B., Passegué E. (2017) Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature. 544(7648), 53–58.
Shimoto M., Sugiyama T., Nagasawa T. (2017) Numerous niches for hematopoietic stem cells remain empty during homeostasis. Blood. 129(15), 2124–2131.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология