

Молекулярная биология, 2019, T. 53, № 6, стр. 883-898
Распределение синонимических кодонов в мРНК определяет путь котрансляционного сворачивания белка в клетке
a Center for Gene Regulation in Health and Disease and Department of Biological, Geological and Environmental Sciences, Cleveland State University
44115 Cleveland, Ohio, USA
b Department of Biochemistry and Center for RNA Science and Therapeutics, Case Western Reserve University
44106 Cleveland, Ohio, USA
c Genomic Medicine Institute, Lerner Research Institute, Cleveland Clinic
Cleveland, Ohio, USA
d DAPCEL, Inc.
44106 Cleveland, Ohio, USA
* E-mail: a.komar@csuohio.edu
Поступила в редакцию 21.04.2019
После доработки 07.05.2019
Принята к публикации 14.05.2019
Аннотация
Сворачивание белка в клетке начинается в процессе его синтеза (трансляции) на рибосоме и, таким образом, представляет котрансляционный процесс. Котрансляционное сворачивание белка тесно взаимосвязано с элонгацией трансляции – процессом неравномерным. Хотя существует множество причин, объясняющих неравномерность трансляции, принято считать, что неодинаковая скорость считывания синонимических кодонов ‒ это один из ключевых факторов, модулирующих скорость элонгации трансляции. Частые/оптимальные кодоны, как правило, транслируются быстрее, чем редкие, и наоборот. Более 30 лет назад была выдвинута гипотеза, утверждающая, что замены синонимических кодонов, приводящие к изменению скорости элонгации трансляции, могут влиять на котрансляционное сворачивание белка, и что многие синонимические кодоны размещены в последовательности мРНК стратегически (в определенных местах) таким образом, что они задают специфическую кинетику трансляции, обеспечивающую, в свою очередь, эффективное, поэтапное котрансляционного сворачивание белка. Постулировалось, что именно такие особенности кинетики трансляции (и специфическая локализация трансляционных пауз) могут определять то временнóе окно, в котором происходит локальное сворачивание фрагментов белковой структуры, особенно в тех участках, где процесс далек от равновесия. Таким образом предполагалось, что определенный характер распределения синонимических кодонов в мРНК может служить вторичным кодом для сворачивания белка в клетке. Хотя в основном эта гипотеза сейчас считается общепринятой, доказать ее оказалось не так легко, и многие из ключевых результатов получены сравнительно недавно. В представленном обзоре рассмотрен прогресс в этой области и даны объяснения, почему доказательства этой простой, на первый взгляд, гипотезы, потребовали существенных усилий.
ВВЕДЕНИЕ
Сворачивание белка ‒ это один из наиболее фундаментальных процессов, происходящих в клетке. В результате этого процесса, полипептидная цепь, производимая рибосомой – белоксинтезирующей частицей клетки, ‒ должна принять правильную трехмерную структуру, чтобы соответствующий белок мог выполнять свои функции. Неправильное сворачивание и агрегация белков ‒ ключевая причина ряда заболеваний, включая многие нейродегенеративные протеинопатии, такие как болезнь Альцгеймера, прионные заболевания и болезнь Паркинсона [1‒3]. Следовательно, знание того, каким образом происходит сворачивания белка в трехмерную структуру, крайне важно.
В течение десятилетий механизм, посредством которого полипептидная цепь приобретает нативную структуру, исследовали в основном в экспериментах по денатурации/ренатурации белка in vitro [4, 5]. Основополагающее исследование, выполненное Anfinsen C.B. и его коллегами по обратимой денатурации рибонуклеазы (в 1950‒1960-х годах), позволило им выдвинуть предположение о том, что аминокислотная последовательность белка содержит всю информацию, необходимую для его сворачивания в уникальную трехмерную структуру [6]. Следом за этими первыми экспериментами были выполнены многие другие углубленные исследования процесса разворачивания/сворачивания белка in vitro, а также моделирование этих процессов in silico, которые дали большой объем информации, позволившей утверждать, что сворачиние белка происходит по определенному пути через ряд последовательных промежуточных состояний [4‒8]. Впоследствии была предложена модель “энергетической воронки” (folding funnel), которая постулировала, что укладка белка в нативную конформацию с наименьшей свободной энергией может происходить через несколько таких путей [7, 9]. Следует отметить, что в большинстве этих исследований были использованы относительно небольшие (по размеру) белки, которые можно было успешно ренатурировать в водных растворах, и полученные результаты с успехом подтверждали гипотезу Anfinsen [4, 5, 7‒9]. Тем не менее, множество попыток достичь in vitro 100%-ной ренатурации денатурированных белков были лишь отчасти успешными [4]. Кроме того, в большинстве случаев восстановление трехмерной структуры белка в пробирке оказалось чрезвычайно медленным процессом, не сопоставимым по времени с таковым, ожидаемым для принятия нативной конформации белка внутри клетки [10].
Стало очевидно, что для всестороннего понимания пути сворачивания белка необходимо установить механизм этого процесса в естественных условиях ‒ близких к тем, которые существуют в клетке in vivo [11‒14]. Эти условия, однако, сильно отличаются от тех, что сушествуют при ренатурации белка в пробирке. Во-первых, сворачивание белка in vivo происходит в плотном окружении клеточных компонентов, поэтому считается, что на него влияет ряд факторов, таких как концентрация и активность/аффинность окружающих макромолекул и наличие катализаторов (изомераз и т.д.) и аксессорных белков фолдинга [15, 16]. Показано, что действие этих клеточных компонентов значительно ускоряет процесс сворачивания белков in vivo и предотвращают их агрегацию [15, 16]. Однако также получены сведения, указывающие на то, что, например, шапероны, взаимодействуя со своими субстратами, сдвигают равновесие между правильной укладкой белка и его формой, ведущей к агрегации. Следовательно, они, в основном, влияют на выход правильно свернутого белка, а не на сам механизм сворачивания. Во-вторых, чрезвычайно важным было признание того факта, что сворачивание белка in vivo начинается в ходе его синтеза на рибосоме, то есть представляет собой котрансляционный процесс [17‒25]. В начале 1960–1970-х годов прошлого века, были получаны первые сведения [26‒32], позволившие утверждать, что укладка белка in vivo начинается в то время, когда растущая полипептидная цепь все еще связана с рибосомой, и что процесс этот направленный, т.е. полипептидная цепь синтезируется и складывается преимущественно в направлении от N-конца к C-концу. На основании этих данных предположили, что котрансляционное сворачивание новосинтезированного полипептида есть результат последовательного, поэтапного структурирования отдельных его участков, выходящих из рибосомы в разные моменты времени и поэтому иерархическую конденсацию полипептидной цепи стали рассматривать как один из наиболее вероятных механизмов сворачивания белка в ходе его синтеза in vivo. Ранние исследования по котрансляционному сворачиванию белка и последующие эксперименты, включая те, которые продолжаются и в настоящее время, позволили выявить множество интересных особенностей этого процесса (см. [17‒25] и ссылки).
Стало также понятно, что котрансляционное сворачивание белков начинается практически сразу после полимеризации первых аминокислотных остатков в пептидилтрансферазном центре рибосомы (см. [17‒25] и ссылки) с формированием α-спиралей в рибосомном канале, а также, возможно, некоторых элементов супервторичной и третичной структур, образующихся в нижнем, расширенном участке рибосомного канала (так называемом вестибюле) – на выходе из него [33‒36]. Сворачивание белка продолжается затем в цитозоле, по выходе белка из рибосомного канала, с образованием промежуточных интермедиатов котрансляционного сворачивания, что приводит в конце-концов к формированию нативной структуры, которая, как правило, полностью формируется после высвобождения полипептидной цепи из рибосомы [17‒25]. Термодинамика и кинетика полипептидной цепи в значительной степени управляют процессом сворачивания белка в ходе всего этого процесса.
Следует отметить, что рибосома прочитывает кодоны мРНК один за одним и транслирует их в последовательность аминокислот белка. Однако стало ясно, что не все кодоны читаются с одинаковой скоростью: периоды быстрой трансляции перемежаются с трансляционными паузами [17, 37, 38]. Следовательно, не исключено, что вариации в локальных скоростях трансляции могут влиять или даже способствовать сворачиванию белка за счет упорядоченной (и разделенной по времени) структуризации определенных участков полипептидной цепи, синтезируемой рибосомой, и что кинетика синтеза белка может служить механизмом для тонкой настройки его котрансляционного сворачивания.
В конце 1980-х годов исследователи группы под руководством Alistair Brown в Institute of Genetic at Glasgow University (Великобритания) [39] и нашей группы на кафедре молекулярной биологии биологического факультета МГУ им. М.В. Ломоносова [40‒42] предположили, что последовательные события сворачивания (образование интермедиатов), которые имеют место в ходе котрансляционного фолдинга, могут быть разделены (по времени) трансляционными паузами, и такие участки медленной трансляции могут служить своебразными знаками “препинания” в этом процессе. Эта гипотеза была сформулирована на основе результатов, в которых была выявлена определенная корреляция между локализацией редких (медленно транслируемых) кодонов в мРНК и границами доменов в кодируемых белках [39, 41] или границами более мелких структурных единиц, таких как, например, элементы вторичной и супервторичной структур [40, 42].
Гипотеза основывалась на нескольких допущениях. Во-первых, она была основана на предположении, что неравномерность в использовании синонимических кодонов в мРНК ведет к специфической кинетике трансляции, которая проявляется в замедлении движения рибосомы (паузах в трансляции) на кластерах редких кодонов или в ускорении движения рибосомы на кластерах высокочастотных кодонов (см. обзор [17], [39‒42]). Во-вторых, постулировалось, что синонимические кодоны расположены в мРНК не случайно, а стратегически ‒ чтобы способствовать упорядоченному котрансляционному сворачиванию белка, ‒ и изменения в использовании синонимических кодонов могут привести к изменению скорости трансляции, что, в свою очередь, может повлиять на сворачивание ([17, 39‒42]). В-третьих, постулировалось, что изменение скорости трансляции влияет, прежде всего, на конформацию связанных с рибосомой новосинтезированных полипептидных цепей; впоследствии это может приводить к изменению конечной конформацию высвобождаемого белка и/или менять равновесие между различными конформерами белка (нативными, полунативными и/или ненативными) и, в свою очередь, может привести, например, к агрегации белка и/или к ускоренной деградации (ко- или посттрансляционной) или к изменению его удельной активности [17].
Следует заметить, что эта гипотеза, в общем смысле утверждающая, что специфический характер использования синонимических кодонов в мРНК может служить кинетическим “путеводителем” в котрансляционном сворачивании белка в клетке, сейчас в основном принята научным сообществом, хотя многие ее постулаты остаются предметом острых дискуссий. Тем не менее появление в молекулярной биологии таких новых технологий, как методы секвенирования нового поколения (Next Generation Sequencing, NGS), одномолекулярной флуоресцентной спектроскопии, а также флуоресценции с временным разрешением, позволяют проводить исследования котрансляционного сворачивания белка на ранее недостижимом уровне и получать новые ключевые доказательства в поддержку выдвинутой гипотезы.
Далее приведен обзор ключевых доказательств в поддержку каждого из постулатов вышеизложенной гипотезы.
ТРАНСЛЯЦИЯ – ЭТО НЕРАВНОМЕРНЫЙ ПРОЦЕСС, УПРАВЛЯЕМЫЙ ИСПОЛЬЗОВАНИЕМ СИНОНИМИЧЕСКИХ КОДОНОВ В мРНК
Генетический код вырожден [43]. За исключением двух аминокислот (метионина и триптофана), все остальные аминокислотные остатки кодируются мультиплетными, так называемыми синонимическими, кодонами [37]. Синонимические кодоны, однако, используются с неодинаковой частотой, как в отдельных мРНК, так и в целых геномах [37, 44‒46]. Такой характер неравномерного использования кодонов известен как “предпочтение кодонов” (codon usage bias) [37, 44‒46] (рис. 1a). Предпочтение кодонов варьирует между организмами (в разных организмах выбор частых и редких синонимических кодонов различен) и представляет собой уникальную характеристику каждого организма [47‒50] (рис. 1a). Это позволяет предположить, что выбор кодонов может иметь функциональное значение в процессах, не связанных напрямую с кодированием аминокислотных остатков в полипептидной цепи [37, 44‒46]. Специфичный для данного организма выбор кодона соответствует специфичным для организма различиям в популяциях/концентрациях тРНК, узнающих данные кодоны [44]. В целом, показано, что, как в одноклеточных, так и в многоклеточных организмах, наблюдается сильная положительная корреляция между использованием кодонов и клеточным содержанием соответствующих тРНК [47, 51, 52]. Это означает, что предпочтение кодонов оказывает, по-видимому, прямое влияние на скорость элонгации трансляции. Действительно, обнаружено, что часто используемые кодоны, как правило, транслируются быстрее, чем редко используемые кодоны вследствие большей доступности (в ходе трансляции) соответствующих часто встречающихся тРНК, узнающих эти кодоны, и наоборот (см. [22, 37, 53‒55]) (рис. 1б). Ряд дополнительных результатов подтверждает это: (i) обнаружено, что высокоэкспрессируемые гены обогащены предпочтительными/часто используемыми кодонами по сравнению с низкоэкспрессируемыми генами, которые содержат в основном синонимические редкие кодоны [56‒59]; (ii) замена синонимических часто используемых кодонов на редко используемые (и наоборот) влияет на уровни экспрессии белка (см. обзоры [22, 37, 53‒55]); (iii) изменения в уровне экспрессии/содержании определенных тРНК также изменяют уровни экспрессии белка [60‒64].
Рис. 1.
Вырожденность генетического кода и неравномерное/неслучайное использование кодонов формируют предпочтение кодонов и задают неравномерный ритм трансляции. а ‒ Предпочтение кодонов у Escherichia coli (EC), Saccharomyces cerevisiae (SC) и Homo sapiens (HS) (https://www.kazusa.or.jp/codon/). На этих примерах хорошо видны значительные отличия в использовании кодонов у трех видов. Представлена частота встречаемости кодонов на тысячу. б ‒ Преимущественно используемые (частые) кодоны транслируются быстрее, чем нечасто используемые (редкие) кодоны, из-за большей доступности (в ходе трансляции) соответствующих часто встречающихся тРНК, узнающих частые кодоны. Как правило, редкие кодоны приводят к значительному увеличению времени остановки рибосомы вследствие увеличения времени ожидания соответствующей тРНК. Представлена упрощенная схема (не указан фактор элонгации 1A, который стимулирует GTP-зависимое связывание аминоацил-тРНК в А-сайте рибосомы в ходе синтеза белка). Цвета тРНК/кодонов соответствуют различной частоте их использования.
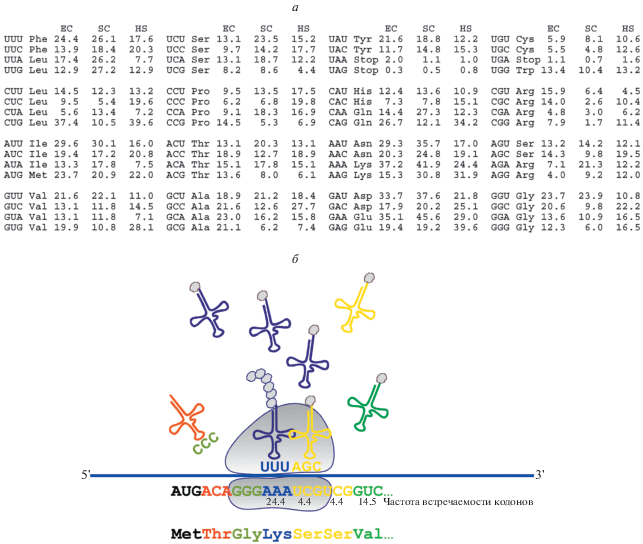
Хотя в целом считается общепринятым, что часто используемые кодоны способствуют ускорению трансляции, а редкие кодоны вызывают паузы в трансляции, доказать тот факт, что конкретный редкий кодон или кластер редких кодонов будет определять появление соответсвующей паузы в трансляции на данном участке в мРНК, несущем эти кодоны, оказалось довольно непросто.
Ранее в экспериментах, направленных на проверку этого постулата, использовали анализ размеров насцентных полипептидных цепей, прикрепленных к рибосоме [65‒68]. Предполагали, что замедления в ходе трансляции, должны увеличивать время нахождения рибосомы на определенных участках мРНК (обогащенных редкими кодонами) и, исходя из этого, ожидали, что концентрация новосинтезированных пептидов соответствующих размеров в составе полирибосом будет тоже возрастать. Исходно накопление новосинтезированных пептидов и их распределение по размерам анализировали с помощью гель-фильтрации [65, 66] и/или электрофореза в геле [67, 68]. И хотя в целом наблюдали, что увеличение числа редких кодонов приводило к более медленному движению рибосомы по мРНК [65‒68], точное определение числа и положения трансляционных пауз оказалось сложным делом из-за низкого разрешения этих методов. Разработка и использование метода защиты РНК от микрококковой нуклеазы [69, 70] и впоследствии технологии рибосомного профайлинга (сочетает рибосомный футпринтинг с глубоким секвенированием фрагментов мРНК, защищенных рибосомами в ходе трансляции) позволили провести прямые эксперименты по анализу распределения рибосом на мРНК с разрешением в один кодон [71‒73]. Однако профайлинг рибосом на первых порах дал неоднозначные результаты. Несмотря на то, что вариации в локальных скоростях трансляции успешно детектировали, исходно не удавалось получить достоверную корреляцию между позицией рибосом и расположением редких кодонов в мРНК [71]. Выяснилось, что определение положения пауз и их связи с редкими кодонами в исходной версии профайлинга рибосом – задача не из легких. Дело в том, что для остановки трансляции использовали антибиотики (типа циклогексимида), которые искажали положение рибосом на матрицах и маскировали повышенную плотность рибосом на неоптимальных кодонах [71, 74]. В систематически пересмотренном методе рибосомного профайлинга (который не включает использования антибиотиков) рибосомные паузы обнаружены на редких кодонах с разрешением в один кодон [75]. Эти новые данные позволили также выявить четкую отрицательную корреляцию между плотностью рибосом и индексом адаптации кодона, что согласуется с предположением о том, что редкие кодоны декодируются мало представленными тРНК медленнее, чем частые кодоны [75].
СИНОНИМИЧЕСКИЕ КОДОНЫ РАСПОЛОЖЕНЫ В ПОСЛЕДОВАТЕЛЬНОСТИ мРНК НЕСЛУЧАЙНЫМ ОБРАЗОМ И НАХОДЯТСЯ В СТРАТЕГИЧЕСКИ ВАЖНЫХ РАЙОНАХ
Согласно нейтральной теории молекулярной эволюции, давление отбора на синонимические кодоны (кодирующие одни и те же аминокислоты) существенно ослаблено, а, значит, их распределение в мРНК и их замены в ходе эволюции происходят случайным образом [76]. Однако оказалось, что менее половины всех синонимических замен соответствует этим “нейтральным” ожиданиям и на замены синонимических кодонов (синонимические мутации) наложены явные ограничения, то есть они находятся под давлением отбора [77, 78]. При детальном анализе последовательностей мРНК выявлены определенные предпочтения в распределении кодонов внутри открытых рамок считывания (open reading frames, ORFs) мРНК ‒ явление, изначально описанное как “кодоновый контекст” [79‒81].
В дополнение к предпочтению в использовании синонимических кодонов относительно соседних кодонов в мРНК (кодоновый контекст [79‒81] и так называемоего предпочтения в использовании пар кодонов [82‒86]) установлено также, что выбор положения синонимических кодонов в определенных участках мРНК (в частности, редких кодонов) происходит не случайным образом, а в соответствии с определенными предпочтениями [87‒94]. Показано, что редкие/нечасто встречающиеся кодоны, как правило, собраны в кластеры, локализующиеся в определенных участках мРНК. В частности, обнаружено, что кластеры редких кодонов часто локализуются на 5'- и 3'-концах ORF [88‒92] и в так называемом кластере “+70” редких кодонов, который расположен на расстоянии 35–40 кодонов от сигнальных пептидных последовательностей (или трансмембранных сегментов) в секретируемых белках [87, 93]. Такие кластеры редких кодонов выявлены и во многих других участках мРНК [88]. Хотя первые исследования, направленные на изучение положения синонимических кодонов в мРНК [39‒42, 94‒97], нельзя назвать исчерпывающими (в частности из-за отсутствия полноценной информации о структуре белков и кодирующих нуклеотидных последовательностях), полученные в них результаты тем не менее позволили сделать вывод, что местоположения кластеров редких кодонов вдоль мРНК эволюционно высоко консервативны, о чем свидетельствует, например, сходство в их расположении в семействах гомологичных белков у различных организмов [17, 41, 42, 88, 94, 98‒100]. Эти данные послужили одним из подтверждений предположения, что такое расположение кластеров редких кодонов может быть связано со структурой белка [17, 88]. Стратегическое размещение кластеров редких кодонов, например, обнаружили в участках мРНК, кодирующих доменные/субдоменные линкеры (или в участках, находящихся в непосредственой близи от этих линкеров) (рис. 2). Предполагали, что такая локализация этих кластеров позволяет разделить во времени сворачивание доменов или субдоменов на рибосоме [17, 88, 94].
Рис. 2.
Кластеры редких кодонов часто находятся в мРНК не в случайных, а в стратегически важных позициях. Вверху – профиль использования кодонов в мРНК, кодирущей белок кристаллина бета-B2 быка (Bos taurus). Видно, что протяженный кластер редких кодонов кодирует часть линкера между доменами и небольшой фрагмент С-концевого домена, следующий за линкером. Внизу – модель третичной структуры кристаллина бета-B2 (PDB 2BB2). N-концевой домен окрашен в синий цвет, С-концевой ‒ в желтый цвет, а линкер, соединяющий оба домена, ‒ в серый цвет. Стрелками указаны остатки Pro80 и Lys89, расположенные в начале и в конце линкерного пептида, соединяющего домены; Asn95 находится на конце β-структуры в начале С-концевого домена кристаллина бета-B2.
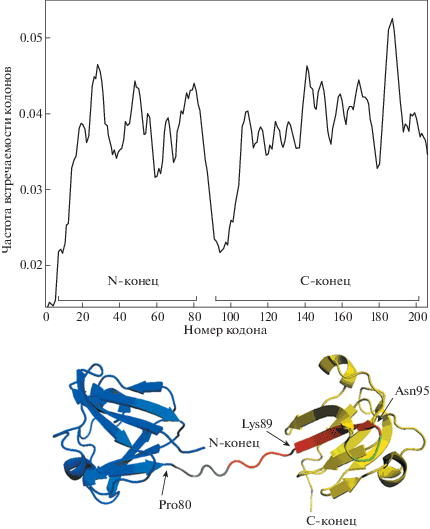
В результате проведенных полногеномных анализов ORFомов прокариотических и эукариотических организмов выявлено, что кластеризация редких кодонов (i) не ограничена определенным набором генов или генотипов, (ii) не зависит от и не имеет отношения к общему GC-составу генома организма и (iii) встречается гораздо чаще, чем можно ожидать, исходя из случайного выбора [88, 98]. Хотя точную функциональную роль многих консервативных кластеров кодонов еще предстоит установить, становится очевидным, что выбор кодонов имеет функциональное значение помимо кодирования аминокислот и поддержания определенного уровня/эффективности трансляции белка, а мРНК (а, значит, и генетический код), действительно, может содержать вторичную информацию, связанную со структурой белка и его сворачиванием.
ИЗМЕНЕНИЯ В ИСПОЛЬЗОВАНИИ СИНОНИМИЧЕСКИХ КОДОНОВ ВЛИЯЮТ НА ЛОКАЛЬНУЮ СКОРОСТЬ ЭЛОНГАЦИИ ТРАНСЛЯЦИИ
Общепризнано, что использование кодонов влияет главным образом на общую скорость трансляции. Для оптимизации экспрессии белка уже получили широкое применение подходы, включающие замену большинства (или ограниченного набора) нечасто используемых кодонов синонимическими, часто используемыми [54, 101‒103]. Использование генов, оптимизированных посредством такой стратегии, во многих случаях позволило увеличить выход рекомбинантных белков. Это подтверждало предположение о том, что на оптимизированных мРНК скорость биосинтеза белка/скорость элонгации трансляции возрастает [54, 101‒103].
Следует отметить, что выбор синонимических кодонов, помимо трансляции, влияет также на время полужизни/стабильность самой мРНК [55, 104‒106]. Время полужизни мРНК ‒ ключевая детерминанта генной экспрессии, и обычно мРНК с большим периодом полужизни продуцирует и больше белка. Недавно обнаружено, что стабильные/долгоживущие мРНК содержат в основном предпочтительные/оптимальные кодоны, в то время как в составе ORF многих нестабильных/короткоживущих мРНК с повышенной частотой встречаются непредпочтительные/неоптимальные/редкие кодоны [104‒106]. Показано, что замена предпочтительных кодонов на синонимические непредпочтительные приводит к существенной дестабилизации мРНК и наоборот [104‒106].
Результаты этих и других экспериментов послужили толчком для понимания научным сообществом влияния использования синонимических кодонов и индекса адаптации кодона (CAI) как меры, определяющей предпочтения синонимических кодонов [107], на эффективность трансляции и синтеза белка [101‒107]. Однако, несмотря на общее принятие идеи, что замена синонимических кодонов в гене может сушественно изменять скорость/эффективность синтеза кодируемого белка и увеличивать или уменьшать его выход, не было четкого понимания, как локальные замены синонимических кодонов влияют на скорость элонгации в тех участках мРНК, где эти замены произошли. И хотя таких исследований было немного, необходимые доказательства все-таки были получены.
Сначала предпринимались попытки определить увеличение/уменьшение времени задержки рибосомы в определенных участках на мРНК после замены синонимических кодонов по увеличению/уменьшению содержания новосинтезированных полипептидов соответствующей длины [68, 108]. Эти попытки недавно возобновлены, уже с использованием технологии профайлинга рибосом и флуоресценции с временным разрешением на уровне единичных молекул, что позволило проанализировать время задержки рибосомы на различных синонимических кодонах [38, 75, 109, 110]. Исследование совокупной флуоресценции в режиме реального времени использовали и для того, чтобы определить, как множественные замены синонимических кодонов влияют на скорость движения рибосомы по мРНК [36, 111]. Результаты этих экспериментов в сочетании с экспериментами, в которых измеряли влияние концентрации тРНК на скорость трансляции, ясно указывали на то, что есть прямая связь между использованием синонимических кодонов и локальными скоростями элонгации трансляции и изменения в использовании синонимических кодонов действительно влияют на кинетику трансляции.
СВОРАЧИВАНИЕ БЕЛКА in vivo – ЭТО КОТРАНСЛЯЦИОННЫЙ ПРОЦЕСС
Для прямой демонстрации влияния использования синонимических кодонов на котрансляционное сворачивание белка необходимо было получить два типа доказательств: (i) показать, что использование кодонов может влиять на окончательную конформацию белка и (ii) что эти конформационные изменения происходят в новосинтезированных цепях, связанных с рибосомой, т.е. котрансляционно. Как уже упоминалось выше, первоначальная гипотеза была выдвинута еще в конце 1980-х годов [39‒42], но в то время сама идея, что сворачивание in vivo происходит котрансляционно, еще не получила широкой поддержки.
Таким образом, сначала необходимо было получить убедительные доказательства в поддержку идеи котрансляционного сворачивания белка. Большинство проведенных ранее экспериментов по этой теме включало выделение/фракционирование комплексов, связанных с рибосомой новосинтезированных пептидов с помощью градиента плотности сахарозы с последующей оценкой структуры и свойств этих полипептидов путем измерения (i) их специфической ферментативной активности, (ii) распознавания их специфическими/конформационными антителами или (iii) формирования правильных дисульфидных связей внутри и/или между новосинтезированными цепями [23, 24]. Впоследствии разработали и другие методы [23, 24], такие как определение устойчивости новосинтезированных цепей, связанных с рибосомой, к протеолитическому расщеплению и/или способности кофакторов и лигандов (таких как гем) связываться с растущей полипептидной цепью (как признак того, что достигнута конформация, компетентная к связыванию).
В нашей группе в сотрудничестве с лабораторией Александра Сергеевича Спирина в Институте белка РАН (Пущино, Россия) в исследованиях по котрансляционному сворачиванию цепей α‑глобина в качестве такого лиганда использовали гем [112, 113]. Трансляцию в системе in vitro проводили в присутствии [3H]-меченого гемина и [35S]-меченого метионина и показали, что полноразмерные новосинтезированных цепи α-глобина, соединенные с рибосомой, эффективно связывают гем [113]. Кроме того, обнаружили, что незавершенные новосинтезированные цепи α-глобина, прикрепленные к рибосоме, также способны к котрансляционному связыванию гема. Это означало, что структура, обеспечивающая связывание гема, формируется в цепи α-глобина еще до завершения его синтеза. Эти результаты стали экспериментальным подтверждением гипотезы котрансляционного сворачивания на примере молекулы α-глобина [113].
Однако в то время одно из самых убедительных доказательств котрансляционного сворачивания белка было получено в работах В.А. Колба, Е.В. Макеева и А.С. Спирина. Они одними из первых начали изучение котрансляционного сворачивания белка с использованием технологий, обеспечивающих измерения в реальном времени [114, 115]. Эти авторы разработали методику, позволяющую наблюдать ферментативную активность вновь синтезированной люциферазы светлячка в бесклеточной системе в люминометрической кювете в реальном времени, и показали, что люциферазная активность (как индикатор правильной укладки белка) может быть обнаружена, как только полноразмерная молекула сформировалась в ходе трансляции [114]. Важно отметить, что столь быстрое проявление ферментивной активности было несовместимо со сценарием посттрансляционного сворачивания [114]. Впоследствии эти же авторы продемонстрировали, что связанная с рибосомами люцифераза энзиматически активна, и пришли к выводу, что сворачивание люциферазы светлячка происходит в ходе трансляции [115].
В последнее время появилось множество современных технологий, таких как ЯМР-спектроскопия, криоэлектронная микроскопия и флуоресцентные методы (резонансный перенос энергии флуоресценции, FRET, и анизотропия флуоресценции/динамическая деполяризация флуоресценции), а также некоторые другие подходы для изучения котрансляционного сворачивания белка [19, 23, 24]. Использование этих новейших технологий позволило получить неопровержимые доказательства в пользу котрансляционного сворачивания белка, и в настоящее время эта концепция получила широкое признание [17‒25]. Однако оставалось неясным, в какой степени путь котрансляционного сворачивания белка уникален и действительно ли на него может влиять кинетика трансляции.
Вот почему многие исследователи сосредоточили свое внимание на анализе влияния кинетики трансляции на котрансляционное сворачивание белка. Эти эксперименты оказались чрезвычайно сложными из-за многочисленных механизмов и систем “контроля качества”, с помощью которых клетка распознает неправильно свернутые белки и обеспечивает их правильное сворачивание или в противном случае направляет их на деградацию [12‒16]. Представлялось вполне возможным, что изменения в котрансляционном фолдинге белка, вызванные заменами синонимических кодонов, могли быть не столь значительными, чтобы преодолеть влияние клеточной системы контроля качества белка и сети шаперонов, для того чтобы их можно было в итоге достоверно детектировать.
ИСПОЛЬЗОВАНИЕ СИНОНИМИЧЕСКИХ КОДОНОВ ВЛИЯЕТ НА КОТРАНСЛЯЦИОННЫЙ ПУТЬ СВОРАЧИВАНИЯ БЕЛКА
Использование бесклеточных систем трансляции, где влияние механизмов контроля качества белка не так четко выражено или может быть направленно нивелировано путем удаления компонентов систем контроля качества, выглядело таким образом вполне логичным в начале и до сих пор служит одним из предпочтительных подходов в анализе влияния использования синонимических кодонов на котрансляционное сворачивание белка. Трансляционные системы in vitro и особенно полностью реконструированные системы in vitro позволяют также легко контролировать многие другие факторы/компоненты, необходимые для синтеза и сворачивания белка и тем самым отвечать на многие ключевые вопросы в этой области.
В 1999 году на примере модельного белка хлорамфениколацетилтрансферазы (CAT) с использованием системы in vitro мы привели одно из первых доказательств, подтверждающих гипотезу, что замены синонимических кодонов влияют на движение рибосом и сворачивание белка [108]. В этом исследовании 16 последовательно расположенных редких кодонов в гене CAT были заменены часто встречающимися, что привело к ускорению движения рибосом через мутированный участок и повлияло на удельную активность фермента (по сравнению с диким типом) [108]. Так как удельная активность фермента может рассматриваться как мера правильности его укладки, мы сделали вывод, что введенные синонимические мутации повлияли на сворачивание CAT. Мы предположили, что повышенная скорость трансляции выбранной области CAT приводит к тому, что соответствующая часть полипептидной цепи синтезируется раньше времени. В результате нарушается взаимодействие этой вновь синтезированной области полипептида с той, которая была синтезирована непосредственно перед ней и еще не успела должным образом уложиться из-за нехватки времени [108]. Так было получено одно из первых экспериментальных подтверждений гипотезы, что кинетика трансляции белка влияет на путь его фолдинга in vivo. Но в то время эта концепция не получила всеобщего признания.
Следующий прорыв произошел 8 лет спустя. В 2007 году Chava Kimchi-Sarfaty, Michael Gottesman и их коллеги [116] показали, что субстратная специфичность P-гликопротеина (P-gp), продукта гена множественной лекарственной устойчивости 1 (MDR1), изменяется под воздействием синонимических однонуклеотидных полиморфизмов (SNP), которые считались нейтральными/“молчащими”. Авторы тщательно исследовали ex vivo (в трансфицированных клетках) влияние встречающихся в природе полиморфных вариантов MDR1 на трансляцию мРНК P-gp, экспрессию белка, его активность и субстратную специфичность. Они пришли к выводу, что единственная синонимическая мутация C3435T (Ile-ATC>ATT), изменяющая относительно частый кодон ATC на более редкий ATT, влияет на скорость элонгации трансляции в этом участке (хотя это и не было продемонстрировано напрямую) и, тем самым, на конформацию P-gp и активность/субстратную специфичность белка [116]. Это исследование имело огромное значение, так как в нем было впервые продемонстрировано, что встречающиеся в природе синонимические/“молчащие” SNP, которые не изменяют первичной структуры синтезируемого полипептида, могут влиять на его структурные и функциональные свойства. На основании этих результатов появилась возможность объяснить различия (наблюдаемые ранее в нескольких клинических исследованиях) в фармакокинетике P-gp у лиц, несущих этот молчащий SNP [116].
Значимость использования синонимических кодонов в укладке белка подверждена в ряде последующих исследований, где показано, что замены синонимических кодонов влияют на чувствительность белков к ограниченному протеолизу [117, 118], на степень их фосфорилирования [118], их спектроскопические характеристики [119], склонность к агрегации [119‒121] и удельную активность [122], что в итоге может привести к ряду заболеваний [123‒128]. Также показано, что выбор синонимических кодонов в случае генов секретируемых белков влияет на эффективное взаимодействие новосинтезированных полипептидов с сигналраспознающей частицей [93], тем самым влияя на секрецию белка.
Особый интерес представляет исследование, проведенное Patricia Clark и коллегами [119], в котором использовали так называемый метод комплементации бимолекулярной флуоресценции (bimolecular fluorescence complementation, BiFC) [129, 130] для создания флуоресцентного белка, состоящего из трех полудоменов. Полудомены N‑ (который при взаймодействии с центральным полудоменом давал желтую флуоресценцию) и C‑концевой (который при взаймодействии с центральным полудоменом давал сине-зеленую флуоресценцию) конкурировали друг с другом за взаимодействие с центральным полудоменом, а результат определял флуоресцентные свойства итоговой белковой структуры. Используя систему клеточной экспрессии и наблюдая за сигналом FRET между метками желтого флуоресцентного белка (YFP) и сине-зеленого флуоресцентного белка (CFP), авторы продемонстрировали, что скорость, с которой новосинтезированный белок выходит из рибосомы (на нее влияли синонимические замены кодонов в междоменном линкере), может определять конечную конформацию свернутого белка [119].
Исследования, приведенные выше, имели, однако, ряд недостатков. Как правило, в них рассматривали только один аспект гипотезы, а также преимущественно анализировали свойства полипептидных цепей, уже высвободившихся из рибосомы. Во всех вышеупомянутых экспериментах для анализа конформации полипептидных цепей использовали косвенные методы (например, определение удельной активности, ограниченного протеолиза), а не прямые подходы (ЯМР, рентгеноструктурный анализ) к исследованию структуры белка. Только в нескольких исследованиях предпринималась попытка исследовать структуру связанных с рибосомой новосинтезированных цепей, продуцируемых из мРНК с синонимическими мутациями, по сравнению с диким типом мРНК. Изменения в кинетике синтеза белка как результат синонимических мутаций обычно не отслеживали и возможные последствия неправильного кодирования (miscoding) в трансляции (которое может происходить в результате изменений синонимических кодонов), как правило, также не принимали во внимание. Наконец, ни в одном из исследований не использовали измерения в реальном времени для того, чтобы продемонстрировать, что синонимические мутации могут влиять в режиме реального времени не только на кинетику синтеза белка, но и на кинетику его котрансляционного сворачивания.
Недавно указанные выше пробелы мы восполнили в сотрудничестве с Harald Schwalbe (The Johann Wolfgang Goethe-Universität in Frankfurt, Германия) и Мариной В. Родниной (The Max Planck Institute for Biophysical Chemistry in Goettingen, Германия) в исследовании, которое призвано было ответить на все эти нерешенные вопросы [111]. С целью понять, как дифференциальное использование синонимических кодонов влияет на кинетику трансляции, ко- и посттрансляционный фолдинг, а также конформацию и стабильность белка, мы проанализировали экспрессию рекомбинантного белка хрусталика крупного рогатого скота кристаллина гамма-В в клетках Escherichia coli in vivo, а также провели такой анализ in vitro в полностью реконструированной высокопроизводительной системе трансляции из E. coli. Выбор пал на кристаллин гамма-B, так как ранее нами показано, что трансляция этого двухдоменного белка происходит неравномерно [68]. По результатам этих экспериментов мы предположили, что использование кодонов и скорость трансляции мРНК кристаллина гамма-B оптимально настроены на синтез и фолдинг этого белка в клетке [68]; однако прямых экспериментальных данных в поддержку этого предположения тогда не было получено. Базируясь на этих предположениях, мы сконструировали два варианта мРНК, кодирующей кристаллин гамма-B: 1) с использованием кодонов, которые оптимальны для трансляции белка в E. coli (с распределением кодонов в мРНК, аналогичным найденному у быка, Bos taurus, которое должно было обеспечивать близкую к природной кинетику трансляции), и 2) с неизмененным составом кодонов, характерным для быка, но не оптимальным для трансляции в E. coli [111].
В ходе анализа влияния использования синонимических кодонов на трансляцию мРНК кристаллина гамма-B нами установлено, что выбор кодонов изменяет как локальную, так и общую скорость трансляции и приводит к образованию альтернативных конформаций белка [111]. С помощью измерений в реальном времени, с использованием флуоресценции и FRET, нами показано, что кинетика синтеза и котрансляционного сворачивания кристаллина гамма-B, действительно, изменяется под воздействием замен синонимических кодонов. Более того, впервые была выявлена значительная структурная гетерогенность очищенных полноразмерных препаратов синонимических вариантов кристаллина гамма-B, что продемонстрировано с использованием метода прямого определения структуры (двумерной ЯМР-спектроскопии). Мы пришли к выводу, что синонимические полиморфизмы изменили распределение различных состояний в ландшафте доступных конформаций белка как на рибосоме, так и после высвобождения полипептидной цепи [111]. Важно отметить, что с помощью масс-спектрометрии и микросеквенирования мы четко подтвердили, что синонимические варианты кристаллина гамма-B, синтезируемые с разных мРНК, обладают идентичной аминокислотной последовательностью. В целом, полученные результаты стали весомым подтверждением гипотезы о роли синонимических кодонов в фолдинге белка и того факта, что специфический характер распределения кодонов вдоль мРНК, действительно, может служить вторичным кодом для сворачивания белка в клетке [111]. Таким образом, мы пришли к выводу, что использование кодонов определяет уникальную кинетику трансляции, которая влияет на распределение интермедиатов фолдинга, как на рибосоме, так и после высвобождения полипептидной цепи, а изменения в использовании кодонов и кинетики трансляции могут привести к кинетически “запертым” интермедиатам фолдинга. Эти интермедиаты могут быть затем переведены с помощью (или без) молекулярных шаперонов (ко- или посттрансляционно) в нативную конформацию. Однако не исключено, что такие кинетически “запертые” интермедиаты могут оставаться вполне стабильными и направлять общее сворачивание белка в ненативное и/или склонное к агрегации состояние. Неправильно свернутые, кинетически “запертые” полипептиды впоследствии могут подвергаться деградации (ко- или посттрансляционно) (рис. 3).
Рис. 3.
Использование синонимических кодонов меняет кинетику синтеза белка и может изменять котрансляционное сворчивание белка. Модель иллюстрирует влияние использования кодонов/кинетики трансляции на конечную конформацию синтезируемого белка. а ‒ Природная (нативная) кинетика трансляции ведет к эффективному формированию нативной структуры через ряд продуктивных котрансляционных интермедиатов. б ‒ Изменения в использовании кодонов/кинетики трансляции могут привести к формированию кинетически “запертых” интермедиатов. Эти интермедиаты затем могут быть конвертированы с помощью (или без) молекулярных шаперонов (ко- или посттрансляционно) в нативный белок. Однако подобные кинетически “запертые” интермедиаты могут оставаться стабильными и направлять весь процесс сворачивания белка в ненативное и/или склонное к агрегации состояние. Непродуктивные, “запертые” в неправильной конформации, молекулы белка могут быть также деградированы (ко- или посттрансляционно). Возникшие из-за измененного использования синонимических кодонов конечные неправильно свернутые формы белка могут привести к развитию различных заболеваний.

Важно отметить, что непродуктивное сворачивание, обусловленное использованием альтернативных синонимических кодонов, может также быть причиной ряда заболеваний [123‒127]. Недавно в сотрудничестве с Chava Kimchi-Sarfaty нами показано, что синонимическая мутация c.459G>A (GTG>GTA) (это положение кодирует Val153 или Val107 (Val107 ‒ соответствует нумерации аминокислотного остатка после отщепления препропептида)), которая ранее была идентифицирована в гене F9 (кодирует фактор свертывания крови FIX) у пациентов с легкой формой гемофилии B [128], изменяет кинетику синтеза FIX и влияет на его конформацию, что приводит к снижению внеклеточного уровня белка [127]. Таким образом, стал понятен молекулярный механизм патогенеза гемофилии B, в основе которого лежит единичная синонимическая мутация в гене F9 [127]. С 2008 года ‒ с момента открытия казавшейся молчащей мутации в гене F9 у больных гемофилией В [128] ‒ развитие этой патологии оставалось необъяснимым. Это связано с тем, что причину заболеваний, ассоциированных с синонимическими мутациями, принято было искать в определенных свойствах самой мРНК, таких как изменение уровня/стабильности мРНК в клетках или изменения характера сплайсинга (пропуск экзонов или сохранение интронов), которые оставались у этих больных неизменными [125, 126].
ВЫВОДЫ И ПЕРСПЕКТИВЫ
Исследование эффектов использования синонимических кодонов, несомненно, углубляет наше понимание процесса сворачивания белка в клетке. Важно, что это действительно помогает объяснить случаи генетических заболеваний, связанных с синонимическими мутациями, ‒ случаи, которые нельзя было объяснить ранее известными эффектами синонимических мутаций на сплайсинг и/или стабильность мРНК [125, 126]. Результаты этих исследований, несомненно, повлияли на восприятие научным сообществом таких случаев синонимических мутаций, влияющих на функию генов и фенотип и связанных со сворачиванием белков и их стабильностью [123‒127]. Наконец, эти исследования дали новый подход для увеличения продукции функционально активных рекомбинантных белков и объяснили давнюю парадигму: почему при замене большинства редко используемых кодонов синонимическими часто используемыми с целью увеличения CAI (этот прием широко используются для оптимизации продукции белков в гетерологичных или гомологичных системах) на выходе часто получают биологически неактивные нерастворимые белковые агрегаты [54]. Теперь стало понятно, что максимизация скорости трансляции может войти в противоречие с продуктвным характером самого процесса биосинтеза белка и привести к неправильному его сворачиванию.
Однако еще много над чем предстоит поработать. Так, пока нет четкого понимания того, как именно синонимические мутации влияют на структуру котрансляционных интермедиатов фолдинга. Использование таких новых технологий, как FRET единичных молекул/или криоэлектронная микроскопия с разрешением по времени [131], потенциально может дать ответы на эти вопросы и пролить свет на то, как именно синонимические мутация вляют на появления котрансляционных интермедиатов сворачивания белка или их конформацию. Однако в настоящее время применение криоэлектронной микроскопии с временным разрешением в анализе динамики новосинтезированных полипептидных цепей сопряжено с определенными трудностями, а FRET сам по себе не позволяет напрямую визуализировать структуру новосинтезированных пептидов.
Тем не менее, уже на сегодняшний день наше углубленное понимание роли синонимических кодонов в процессе сворачивания белка позволяет с уверенностью утверждать, что использование синонимических кодонов вдоль мРНК служит инструкцией к котрансляционному фолдингу белка в клетке.
Эта работа была бы невозможна без исходного участия и вклада Ивана Алексеевича Аджубея и Игоря Александровича Крашенинникова, последовавшей затем поддержки Славы Колба, Айгара Коммера, Льва Павловича Овчинникова и Александра Сергеевича Спирина с последующим сотрудничеством с Rainer Jaenicke, Claude Reiss, а также в ближайшее время с Chava Kimchi-Sarfaty, Harald Schwalbe и Мариной Родниной.
Я бесконечно признателен моим коллегам и сотрудникам за их огромное великодушие, вдохновляющие обсуждения и бесценный вклад в работу.
Приношу извинения тем авторам, чьи работы или оригинальные публикации не были процитированы в этой короткой обзорной статье.
В последние годы работа в моей лаборатории была поддержана грантами Human Frontier Science Program Organization [HFSP grant #RGP0024/2010], the American Heart Association [AHA grant 13GRNT17070025], the National Institutes of Health [NIH grant HL121779], the Center for Gene Regulation in Health and Disease (GRHD) at CSU и биотехнологической компанией “DAPCEL Inc.”, которая уже более 10 лет успешно реализует идеи, полученные в ходе данных исследований, для синонимической оптимизации генов, нацеленной на продукцию (в любом организме-продуценте) правильно свернутых растворимых белков.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Hartl F.U. (2017) Protein misfolding diseases. Annu. Rev. Biochem. 86, 21‒26.
Chiti F., Dobson C.M. (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27‒68.
Soto C., Pritzkow S. (2018) Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 21, 1332‒1340.
Fersht A.R. (2008) From the first protein structures to our current knowledge of protein folding: delights and skepticisms. Nat. Rev. Mol. Cell Biol. 9, 650‒654.
Ferina J., Daggett V. (2019) Visualizing protein folding and unfolding. J. Mol. Biol. 431, 1540–1564.
Anfinsen C.B. (1973) Principles that govern the folding of protein chains. Science. 181, 223‒230.
Finkelstein A.V. (2018) 50+ years of protein folding. Biochemistry (Mosc.). 83 (Suppl. 1), S3‒S18.
Bartlett A.I., Radford S.E. (2009) An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nat. Struct. Mol. Biol. 16, 582‒588.
Abaskharon R.M., Gai F. (2016) Meandering down the energy landscape of protein folding: are we there yet? Biophys. J. 110, 1924‒1932.
Jaenicke R. (1991) Protein folding: local structures, domains, subunits, and assemblies. Biochemistry. 30, 3147‒3161.
Hartl F.U., Hayer-Hartl M. (2009) Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574‒581.
Hingorani K.S. Gierasch L.M. (2014) Comparing protein folding in vitro and in vivo: foldability meets the fitness challenge. Curr. Opin. Struct. Biol. 24, 81‒90.
Balchin D., Hayer-Hartl M., Hartl F.U. (2016) In vivo aspects of protein folding and quality control. Science. 353, aac4354.
Gruebele M., Dave K., Sukenik S. (2016) Globular protein folding in vitro and in vivo. Annu. Rev. Biophys. 45, 233‒251.
Dahiya V., Buchner J. (2019) Functional principles and regulation of molecular chaperones. Adv. Protein Chem. Struct. Biol. 114, 1‒60.
Jayaraj G.G., Hipp M.S., Hartl F.U. (2019) Functio-nal modules of the proteostasis network. Cold Spring Harb. Perspect. Biol. Mar 4, pii: a033951. https://doi.org/10.1101/cshperspect.a033951
Komar A.A. (2009) A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 34, 16‒24.
Kramer G., Boehringer D., Ban N., Bukau B. (2009) The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol. 16, 589‒597.
Cabrita L.D., Dobson C.M., Christodoulou J. (2010) Protein folding on the ribosome. Curr. Opin. Struct. Biol. 20, 33‒45.
Pechmann S., Willmund F., Frydman J. (2013) The ribosome as a hub for protein quality control. Mol. Cell. 49, 411‒421.
Gloge F. Becker A.H. Kramer G., Bukau B. (2014) Co-translational mechanisms of protein maturation. Curr. Opin. Struct. Biol. 24, 24‒33.
Chaney J.L., Clark P.L. (2015) Roles for synonymous codon usage in protein biogenesis. Annu. Rev. Biophys. 44, 143‒166.
Thommen M., Holtkamp W., Rodnina M.V. (2017) Co-translational protein folding: progress and methods. Curr. Opin. Struct. Biol. 42, 83‒89.
Komar A.A. (2018) Unraveling co-translational protein folding: concepts and methods. Methods. 137, 71‒81.
Williams N.K, Dichtl B. (2018) Co-translational control of protein complex formation: a fundamental pathway of cellular organization? Biochem. Soc. Trans. 46, 197‒206.
Cowie D.B., Spiegelman S., Roberts R.B., Duerksen J.D. (1961) Ribosome-bound beta-galactosidase. Proc. Natl. Acad. Sci. USA. 47, 114‒122
Zipser D., Perrin D. (1963) Complementation on ribosomes. Cold Spring Harbor. Symp. Quant. Biol. 28, 533‒537.
Kiho Y., Rich A. (1964) Induced enzyme formed on bacterial polyribosomes. Proc. Natl. Acad. Sci. USA. 51, 111‒118.
Hamlin J., Zabin I. (1972) β-Galactosidase: immunological activity of ribosome-bound, growing polypeptide chains. Proc. Natl. Acad. Sci. USA. 69, 412‒416.
Bergman L.W., Kuehl W.M. (1979) Formation of intermolecular disulfide bonds on nascent immunoglobulin polypeptides. J. Biol. Chem. 254, 5690‒5694.
Bergman L.W., Kuehl W.M. (1979) Formation of an intrachain disulfide bond on nascent immunoglobulin light chains. J. Biol. Chem. 254, 8869‒8876.
Bergman L.W., Kuehl W.M. (1979) Co-translational modification of nascent immunoglobulin heavy and light chains. J. Supramol. Struct. 11, 9‒24.
Gilbert R.J., Fucini P., Connell S., Fuller S.D., Nierhaus K.H., Robinson C.V., Dobson C.M., Stuart D.I. (2004) Three-dimensional structures of translating ribosomes by Cryo-EM. Mol. Cell. 14, 57‒66.
Kosolapov A., Deutsch C. (2009) Tertiary interactions within the ribosomal exit tunnel. Nat. Struct. Mol. Biol. 16, 405‒411.
Tu L., Khanna P., Deutsch C. (2014) Transmembrane segments form tertiary hairpins in the folding vestibule of the ribosome. J. Mol. Biol. 426, 185‒198.
Holtkamp W., Kokic G., Jäger M., Mittelstaet J., Komar A.A., Rodnina M.V. (2015) Cotranslational protein folding on the ribosome monitored in real time. Science. 350, 1104‒1107.
Komar A.A. (2018) The Yin and Yang of codon usage. Hum. Mol. Genet. 25(R2), R77–R85.
Schuller A.P., Green R. (2018) Roadblocks and resolutions in eukaryotic translation. Nat. Rev. Mol. Cell Biol. 19, 526‒541.
Purvis I.J., Bettany A.J., Santiago T.C., Coggins J.R., Duncan K., Eason R., Brown A.J. (1987) The efficiency of folding of some proteins is increased by controlled rates of translation in vivo. A hypothesis. J. Mol. Biol. 193, 413‒417.
Krasheninnikov I.A., Komar A.A., Adzhubeĭ I.A. (1988) Role of the rare codon clusters in defining the boundaries of polypeptide chain regions with identical secondary structures in the process of co-translational folding of proteins. Dokl. Akad. Nauk SSSR. 303, 995‒999.
Krasheninnikov I.A., Komar A.A., Adzhubeĭ I.A. (1989) Frequency of using codons in mRNA and co-ding of the domain structure of proteins. Dokl. Akad. Nauk SSSR. 305, 1006‒1012.
Krasheninnikov I.A., Komar A.A, Adzhubeĭ I.A. (1989) Role of the code redundancy determining cotranslational protein folding. Biokhimiia. 5, 187‒200.
Crick F.H., Barnett L., Brenner S., Watts-Tobin R.J. (1961) General nature of the genetic code for proteins. Nature. 192, 1227‒1232.
Hershberg R., Petrov D.A. (2008) Selection on codon bias. Annu. Rev. Genet. 42, 287‒299.
Sharp P.M., Emery L.R. Zeng K. (2010) Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. London, Ser. B. 365, 1203‒1212.
Behura S.K.m Severson D.W. (2013) Codon usage bias: causative factors, quantification methods and genome-wide patterns: with emphasis on insect genomes. Biol. Rev. Camb. Philos. Soc. 88, 49‒61.
Ikemura T. (1985) Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 2, 13‒34.
Sharp P.M., Cowe E., Higgins D.G., Shields D.C., Wolfe K.H., Wright F. (1988) Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res. 16, 8207‒8211.
Andersson S.G., Kurland C.G. (1990) Codon preferences in free-living microorganisms. Microbiol. Rev., 54, 198‒210.
Nakamura Y., Gojobori T., Ikemura T. (2000) Codon usage tabulated from the international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 28, 292.
Ikemura T. (1981) Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 146, 1‒21.
Ikemura T. (1982) Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in syno-nymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J. Mol. Biol. 158, 573‒597.
Quax T.E., Claassens N.J., Söll D., van der Oost J. (2015) Codon bias as a means to fine-tune gene expression. Mol. Cell. 59, 149‒161.
Komar A.A. (2016) The art of gene redesign and recombinant protein production: approaches and perspectives. In: Protein Therapeutics. Eds Zuben E. Sauna, Chava Kimchi-Sarfaty. Springer, pp. 161‒177.https://doi.org/10.1007/978-3-319-41818-6
Hanson G., Coller J. (2018) Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 19, 20‒30.
Sharp P.M., Tuohy T.M., Mosurski K.R. (1986) Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 14, 5125‒5143.
Shields D.C., Sharp P.M. (1987) Synonymous codon usage in Bacillus subtilis reflects both translational selection and mutational biases. Nucleic Acids Res. 15, 8023‒8040.
Sharp P.M., Devine K.M. (1989) Codon usage and gene expression level in Dictyostelium discoideum: highly expressed genes do ‘prefer’ optimal codons. Nucleic Acids Res. 17, 5029‒5039.
Karlin S, Mrázek J, Campbell AM. (1998) Codon usages in different gene classes of the Escherichia coli genome. Mol. Microbiol. 29, 1341‒1355.
Brinkmann U., Mattes R.E., Buckel P. (1989) High-level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene. 85, 109‒114.
Chen K.S., Peters T.C., Walker J.R. (1990) A minor arginine tRNA mutant limits translation preferentially of a protein dependent on the cognate codon. J. Bacteriol. 172, 2504‒2510.
Chen G.T., Inouye M. (1994) Role of the AGA/AGG codons, the rarest codons in global gene expression in Escherichia coli. Genes Dev. 8, 2641‒2652.
Zahn K., Landy A. (1996) Modulation of lambda integrase synthesis by rare arginine tRNA. Mol. Microbiol. 21, 69‒76.
Del Tito B.J. Jr, Ward J.M., Hodgson J., Gershater C.J., Edwards H., Wysocki L.A., Watson F.A., Sathe G., Kane J.F. (1995) Effects of a minor isoleucyl tRNA on heterologous protein translation in Escherichia coli. J. Bacteriol. 177, 7086‒7091.
Protzel A., Morris A.J. (1974) Gel chromatographic analysis of nascent globin chains. Evidence of nonuniform size distribution. J. Biol. Chem. 249, 4594‒4600.
Chaney W.G., Morris A.J. (1978) Nonuniform size distribution of nascent peptides: the role of messenger RNA. Arch. Biochem. Biophys. 191, 734‒741.
Krasheninnikov I.A. Komar A.A., Adzhubeĭ I.A. (1991) Nonuniform size distribution of nascent globin peptides, evidence for pause localization sites, and a cotranslational protein-folding model. J. Protein Chem. 10, 445–454.
Komar A.A., Jaenicke R. (1995) Kinetics of translation of gamma B crystallin and its circularly permutated variant in an in vitro cell-free system: possible relations to codon distribution and protein folding. FEBS Lett. 376, 195‒198.
Wolin S.L., Walter P. (1988) Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 7, 3559‒3569.
Hollingsworth M.J., Kim J.K., Stollar N.E. (1998) Heelprinting analysis of in vivo ribosome pause sites. Methods Mol. Biol. 77, 153‒165.
Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324, 218‒223.
McGlincy N.J., Ingolia N.T. (2017) Transcriptome-wide measurement of translation by ribosome profiling. Methods. 126, 112‒129.
Ingolia N.T., Hussmann J.A., Weissman J.S. (2018) Ribosome profiling: global views of translation. Cold Spring Harb. Perspect. Biol. 11(5), pii: a032698. https://doi.org/10.1101/cshperspect.a032698
Hussmann J.A., Patchett S., Johnson A., Sawyer S., Press W.H. (2015) Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet. 11, e1005732.
Mohammad F., Green R., Buskirk A.R. (2019) A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. Elife. 8, e42591.
Kimura M. (1984) The Neutral Theory of Molecular Evolution Cambridge. Cambridge, UK: Cambridge University Press, pp. 367.
Zhao Z., Fu Y.X., Hewett-Emmett D., Boerwinkle E. (2003) Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene. 312, 207‒213.
Chamary J.V., Parmley J.L., Hurst L.D. (2006) Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 7, 98‒108.
Yarus M., Folley L.S. (1985) Sense codons are found in specific contexts. J. Mol. Biol. 182, 529‒540.
Buckingham R.H. (1990) Codon context. Experientia. 46, 1126‒1133.
Buckingham, R.H. (1994) Codon context and protein synthesis: enhancements of the genetic code. Biochimie. 76, 351‒354.
Gutman G.A., Hatfield G.W. (1989) Nonrandom utilization of codon pairs in Escherichia coli. Proc. Natl. Acad. Sci. USA. 86, 3699‒3703.
Tats A., Tenson T., Remm M. (2008) Preferred and avoided codon pairs in three domains of life. BMC Genomics. 9, 463.
Diambra L.A. (2017) Differential bicodon usage in lowly and highly abundant proteins. PeerJ. 5, e3081.
Brule C.E., Grayhack E.J. (2017). Synonymous codons: choose wisely for expression. Trends Genet. 33, 283‒297.
Alexaki A., Kames J.M., Holcomb D.D., Athey J., Santana-Quintero L.V., Lam P.V., Hamasaki-Katagiri N., Osipova E., Simonyan V., Bar H., Komar A.A., Kimchi-Sarfaty C. (2019) Codon and Codon-Pair Usage Tables (CoCoPUTs): facilitating genetic variation analyses and recombinant gene design. J. Mol. Biol. 431(13), 2434‒2441. https://doi.org/10.1016/j.jmb.2019.04.021
Képès F. (1996) The “+70 pause”: hypothesis of a translational control of membrane protein assembly. J. Mol. Biol. 262, 77‒86.
Clarke T.F. 4th, Clark P.L. (2008) Rare codons cluster. PLoS One. 3, e3412.
Kudla G., Murray A.W., Tollervey D., Plotkin J.B. (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science. 324, 255‒258.
Clarke T.F. 4th, Clark P.L. (2010) Increased incidence of rare codon clusters at 5' and 3' gene termini: implications for function. BMC Genomics. 11, 118.
Goodman D.B., Church G.M., Kosuri S. (2013) Causes and effects of N-terminal codon bias in bacterial genes. Science. 342, 475‒479.
Bentele K., Saffert P., Rauscher R., Ignatova Z., Bluthgen N. (2013) Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 9, 675.
Pechmann S., Chartron J.W., Frydman J. (2014) Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo. Nat. Struct. Mol. Biol. 21, 1100‒1105.
Thanaraj T.A., Argos P. (1996) Ribosome-mediated translational pause and protein domain organization. Protein Sci. 5, 1594‒1612.
Thanaraj T.A., Argos P. (1996) Protein secondary structural types are differentially coded on messenger RNA. Protein Sci. 5, 1973‒1983.
Adzhubei A.A., Adzhubei I.A., Krasheninnikov I.A., Neidle S. (1996) Non-random usage of ‘degenerate’ codons is related to protein three-dimensional structure. FEBS Lett. 399, 78‒82.
Oresic M., Shalloway D. (1998) Specific correlations between relative synonymous codon usage and protein secondary structure. J. Mol. Biol. 281, 31‒48.
Chartier M., Gaudreault F., Najmanovich R. (2012) Large-scale analysis of conserved rare codon clusters suggests an involvement in co-translational molecular recognition events. Bioinformatics. 28, 1438‒1445.
Widmann M., Clairo M., Dippon J., Pleiss J. (2008) Analysis of the distribution of f unctionally relevant rare codons. BMC Genomics. 9, 207.
McKownvR.L., Raab R.W., Kachelries P., Caldwell S., Laurie G.W. (2013) Conserved regional 3' grouping of rare codons in the coding sequence of ocular prosecretory mitogen lacritin. Invest. Ophthalmol. Vis. Sci. 54, 1979‒1987.
Gustafsson C., Govindarajan S., Minshull J. (2004) Codon bias and heterologous protein expression. Trends Biotechnol. 22, 346–353.
Wu G., Zheng Y., Qureshi I., Zin H.T., Beck T., Bulka B., Freeland S.J. (2007) SGDB: a database of synthetic genes re-designed for optimizing protein over-expression. Nucleic Acids Res. 35, D76–D79.
Quax T.E., Claassens N.J., Soll D., van der Oost J. (2015) Codon bias as a means to fine-tune gene expression. Mol. Cell. 59, 149–161.
Presnyak V., Alhusaini N., Chen Y.H., Martin S., Morris N., Kline N., Olson S., Weinberg, D., Baker K.E., Graveley B.R., Coller J. (2015) Codon optimality is a major determinant of mRNA stability. Cell. 160, 1111‒1124.
Boël G., Letso R., Neely H., Price W.N., Wong K.H., Su M., Luff J.D., Valecha M., Everett J.K., Acton T.B., Xiao R., Montelione G.T., Aalberts D.P., Hunt J.F. (2016) Codon influence on protein expression in E. coli correlates with mRNA levels. Nature. 529, 358‒363.
Mishima Y., Tomari Y. (2016) Codon usage and 3' UTR length determine maternal mRNA stability in zebrafish. Mol. Cell. 61, 874‒885.
Sharp P.M., Li W.H. (1987) The codon adaptation index ‒ a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281‒1295.
Komar A.A., Lesnik T., Reiss C. (1999) Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 462, 387‒391.
Uemura S., Aitken C.E., Korlach J., Flusberg B.A., Turner S.W., Puglisi J.D. (2010) Real-time tRNA transit on single translating ribosomes at codon resolution. Nature. 464, 1012‒1017.
Prabhakar A., Puglisi E.V., Puglisi J.D. (2019) Single-molecule fluorescence applied to translation. Cold Spring Harb. Perspect. Biol. 11, pii: a032714.
Buhr F., Jha S., Thommen M., Mittelstaet J., Kutz F., Schwalbe H., Rodnina M.V., Komar A.A. (2016) Synonymous codons direct cotranslational folding toward different protein conformations. Mol. Cell. 61, 341‒351.
Komar A.A., Kommer A., Krasheninnikov I.A., Spirin A.S. (1993) Cotranslational heme binding to nascent globin chains. FEBS Lett. 326, 261‒263.
Komar A.A., Kommer A., Krasheninnikov I.A., Spirin A.S. (1997) Cotranslational folding of globin. J. Biol. Chem. 272, 10646‒10651.
Kolb V.A., Makeyev E.V., Spirin A.S. (1994). Folding of firefly luciferase during translation in a cell-free system. EMBO J. 13, 3631‒3637.
Makeyev E.V., Kolb V.A., Spirin A.S. (1996) Enzymatic activity of the ribosome-bound nascent polypeptide. FEBS Lett. 378, 166‒170.
Kimchi-Sarfaty C., Oh J.M., Kim I.W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M. (2007) A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 315, 525‒528.
Zhang G., Hubalewska M., Ignatova Z. (2009) Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 16, 274‒280.
Zhou M., Guo J., Cha J., Chae M., Chen S., Barral J.M., Sachs M.S., Liu Y. (2013) Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 495, 111‒115.
Sander I.M., Chaney J.L., Clark P.L. (2014) Expan-ding Anfinsen’s principle: contributions of synonymous codon selection to rational protein design, J. Am. Chem. Soc. 136, 858‒861.
Hu S., Wang M., Cai G., He M. (2013) Genetic code-guided protein synthesis and folding in Escherichia coli. J. Biol. Chem. 288, 30855‒30861.
Kim S.J., Yoon J.S., Shishido H., Yang Z., Rooney L.A., Barral J.M., Skach W.R. (2015) Protein folding. Translational tuning optimizes nascent protein folding in cells. Science. 348, 444‒448.
Yu C.H., Dang Y., Zhou Z., Wu C., Zhao F., Sachs M.S., Liu Y. (2015) Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol. Cell. 59, 744‒754.
Komar A.A. (2007) SNPs, silent but not invisible. Science. 315, 466‒467.
Komar A.A. (2007) Silent SNPs: impact on gene function and phenotype. Pharmacogenomics. 8, 1075‒1080.
Sauna. Z.E., Kimchi-Sarfaty C. (2011) Understan-ding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 12, 683‒691.
Hunt R.C., Simhadri V.L., Iandoli M., Sauna Z.E., Kimchi-Sarfaty,C. (2014) Exposing synonymous mutations. Trends Genet. 30, 308‒321.
Simhadri V.L., Hamasaki-Katagiri N., Lin B.C., Hunt R., Jha S., Tseng S.C., Wu A., Bentley A.A., Zichel R., Lu Q., Zhu L., Freedberg D.I., Monroe D.M., Sauna Z.E., Peters R., Komar A.A., Kimchi-Sarfaty C. (2017) Single synonymous mutation in factor IX alters protein properties and underlies haemophilia B. J. Med. Genet. 54, 338‒345.
Knobe K.E., Sjorin E., Ljung R.C. (2008) Why does the mutation G17736A/Val107Val (silent) in the F9 gene cause mild haemophilia B in five Swedish families? Haemophilia. 14, 723–728.
Shyu Y.J., Liu H., Deng X., Hu C.D. (2006) Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques. 40, 61‒66.
Shyu Y.J., Hu C.D. (2008) Fluorescence complementation: an emerging tool for biological research. Trends Biotechnol. 26, 622‒630.
Chen B., Kaledhonkar S., Sun M., Shen B., Lu Z., Barnard D., Lu T.M., Gonzalez R. L.Jr, Frank J. (2015) Structural dynamics of ribosome subunit association studied by mixing-spraying time-resolved cryogenic electron microscopy. Structure. 23, 1097‒1105.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология