

Молекулярная биология, 2020, T. 54, № 1, стр. 17-28
Генетическое разнообразие лобно-височной деменции
Ю. А. Шпилюкова a, *, Е. Ю. Федотова a, С. Н. Иллариошкин a
a Научный центр неврологии
125367 Москва, Россия
* E-mail: jshpilyukova@gmail.com
Поступила в редакцию 06.05.2019
После доработки 21.06.2019
Принята к публикации 27.06.2019
Аннотация
Лобно-височная деменция ‒ прогрессирующее нейродегенеративное заболевание с большим клиническим, генетическим и патоморфологическим разнообразием, третья по распространенности причина деменции среди всех возрастов и самая распространенная причина деменции с ранним началом (до 65 лет). Несмотря на многофакторную природу лобно-височной деменции, до 40% пациентов имеют отягощенный семейный анамнез, при этом аутосомно-доминантный паттерн наследования прослеживается в четверти случаев. В представленном обзоре описаны ключевые гены, мутации в которых могут приводить к развитию лобно-височной деменции, а также связанные с ними возможные патогенетические механизмы развития дегенеративного процесса, приведена информация о клинических особенностях заболевания, характерных для различных генетических вариантов. Особый акцент сделан на фенотипе лобно-височной деменции, ассоциированной с боковым амиотрофическим склерозом.
ВВЕДЕНИЕ
Термин лобно-височная деменция (ЛВД) используется для описания группы нейродегенеративных расстройств, при которых наблюдается неуклонно прогрессирующее нарушение исполнительных функций, поведения и/или речи [1]. ЛВД считается третьей по распространенности причиной деменции после болезни Альцгеймера (БА) и деменции с тельцами Леви [1].
Термин ЛВД применим для обозначения клинического синдрома, ассоциированного с определенным патоморфологическим процессом, который называют лобно-височной долевой дегенерацией (FTLD – frontotemporal lobar degeneration). ЛВД может проявляться несколькими клиническими синдромами, такими как поведенческий вариант (пвЛВД), семантический вариант первичной прогрессирующей афазии (свППА), аграмматический вариант первичной прогрессирующей афазии (авППА), ЛВД, ассоциированная с боковым амиотрофическим склерозом (ЛВД-БАС) и ЛВД с клиническими проявлениями синдромов атипичного паркинсонизма (прогрессирующий надъядерный паралич – ПНП, и кортикобазальный синдром – КБС). Диагноз первых трех вариантов устанавливается согласно разработанным клиническим критериям. Признаки поражения мотонейрона или атипичный паркинсонизм могут сочетаться с любым из этих вариантов. Кроме того, в диагностических критериях ППА выделяют логопенический вариант, однако на сегодняшний день этот фентип считается ассоциированным с БА.
В патогенезе ЛВД большую роль играет генетический фактор – до 40% пациентов имеют отягощенный семейный анамнез в виде диагноза деменция по крайней мере у одного члена семьи, при этом аутосомно-доминантный характер наследования может быть прослежен примерно в 13.4% всех случаев [2]. В настоящее время идентифицировано более 20 генов, мутации в которых могут быть ассоциированы с ЛВД (табл. 1). Чаще всего патогенные мутации встречаются в трех генах – MAPT, C9orf72, GRN. Мутации в остальных генах встречаютя с меньшей частотой. Однако в большинстве случаев ЛВД это спорадическое заболевание, возникающее в результате взаимодействия большого количества генетических и средовых факторов [3].
Таблица 1.
Основные гены, патогенетически связанные с лобно-височной деменцией
| Ген | Функция белка |
|---|---|
| MAPT | Сборка и стабилизация микротрубочек, организация цитоскелета |
| GRN | Фактор роста (регуляция процессов раннего эмбриогенеза, восстановления тканей и процессов воспаления во взрослом возрасте) |
| C9orf72 | Ядерно-цитоплазматический транспорт, аутофагия, межклеточный транспорт |
| CHMP2B | Аутофагия, участие в транспорте и деградации белков |
| CP-1 | Аутофагия, участие в транспорте и деградации белков |
| SQSTM1 | Аутофагия, деградация белков |
| CHCHD10 | Митохондриальный белок |
| TBK1 | Аутофагия, участие в транспорте и деградации белков |
| TARDBP | Фактор транскрипции |
| FUS | Фактор транскрипции |
| UBQLN2 | Аутофагия, участие в транспорте и деградации белков |
| TUBA4A | Организация цитоскелета |
Для гистологической картины ЛВД характерно присутствие в нейрональных и глиальных клетках аномальных агрегатов трех типов белков. В большинстве случаев обнаруживают патологическое накопление белков тау (до 45%) или TDP-43 (50%), в небольшом числе случаев (около 5‒10%) видны включения белка FUS [4, 5]. Аномальная агрегация тау описана как у пациентов со спорадическими случаями пвЛВД, КБС, авППА и ПНП, так и в отягощенных семьях, где заболевание обсуловлено мутацией в гене MAPT [6‒9]. Однако агрегация тау, как правило, отсутствует при свППА. Включения белка TDP-43 встречаются у большинства тау-негативных пациентов. Патология белка ТDP-43 описана при свППА, ЛВД-БАС и пвЛВД, а также при мутациях в генах C9orf72, GRN, VCP и др. Интересно, что такая гистологическая картина редко встречается при мутациях в гене TARDBP, кодирующем белок TDP-43 [7‒9]. FUS-патология, как правило, ассоциирована с более ранним началом ЛВД, доминирующими нервнопсихическими симптомами и большей скоростью течения заболевания. FUS-патология встречается преимущественно у пациентов со спорадической формой ЛВД, однако она возможна и при мутациях в гене FUS [10].
Таким образом, к настоящему времени не найдены четкие корреляции между клиническими фенотипами, гистологической картиной и генетическими вариантами ЛВД, что значительно затрудняет диагностику даже при семейных случаях заболевания. Кроме того, отсутствие корреляций затрудняет анализ течения патогенетического процесса при семейных и спорадических случаях ЛВД, что препятствует разработке болезнь-модифицирующей терапии. В представленном обзоре рассмотрены основные гены, мутации в которых могут служить важным генетическим фактором развития ЛВД.
ГЕН MAPT
В 1994 году впервые было показано, что аутосомно-доминантные случаи ЛВД с паркинсонизмом могут быть ассоциированы с локусом 17q21.2 [11, 12], впоследствии названным FTDP-17. Локализованный в этой области ген обнаружили на несколько лет позже и назвали MAPT (Microtubule Associated Protein Tau; OMIM *157140) [13]. Ген MAPT состоит из 16 экзонов и кодирует тау-белок, который участвует в процессах сборки и стабилизации микротрубочек, в организации цитоскелета нейронов [13]. мРНК МАРТ подвергается альтернативному сплайсингу, в результате чего образуется шесть изоформ белка, каждая из которых участвует в поддержании структуры микротрубочек. Избыток тау приводит к формированию белковых агрегатов, которые заполняют клетку и вызывают токсический эффект. В С-концевой области тау локализованы четыре повторяющихся домена, которые опосредуют взаимодействие с микротрубочками. Эти домены кодируются экзонами 9‒12, в которых обнаружено большинство патогенных мутаций. Кроме того, в результате альтернативного сплайсинга экзона 10 возникают изоформы, содержащие три (3R) или четыре (4R) повтора из 31 аминокислотного остатка [13] (рис. 1).
Рис. 1.
Ген MAPT и изоформы белка тау, образующиеся в результате альтернативного сплайсинга в головном мозге человека. Ген MAPT состоит из 16 экзонов (верхняя часть рисунка). Экзоны E1, E4, E5, E7, E9, E11, E12 и E13 ‒ конститутивные (светло-серые квадраты), остальные подвергаются альтернативному сплайсингу. Экзоны E0 и E1 кодируют 5'-нетранслируемую область мРНК MAPT, а экзон E14 кодирует часть 3'-нетранслируемой области. Экзон E0 входит в состав промоторной области, он транскрибируется, но не транслируется. Инициаторный кодон ATG локализован в экзоне E1. Экзоны E4a, E6 и E8 транскрибируются только в периферических тканях. Шесть изоформ белка тау головного мозга человека образуются в результате альтернативного сплайсинга экзонов E2 (N1, синий), E3 (N2, желтый) и E10 (R2, красный). Эти изоформы отличаются друг от друга присутствием 0, 1 или 2 около-N-концевых инсерций (0N, 1N, 2N соответственно), а также повтора R2 (красный), который приводит к возникновению трех и четырех С-концевых повторяющихся доменов (3R или 4R соответственно) в разных подтипах тау.
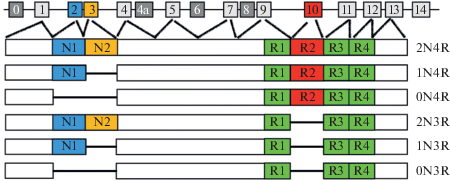
Патоморфологическая картина при всех мутациях в гене MAPT характеризуется отложением нерастворимых гиперфосфорилированных агрегатов белка тау в нейронах и клетках глии в коре головного мозга и других его отделах [14]. Описано более 40 патогенных мутаций в гене MAPT, каждый из которых классифицирован согласно положению, влиянию на транскрипцию и типу таупатии. Частота мутации в гене MAPT при ЛВД составляет 6‒11%. ЛВД с мутациями в этом гене, которые характеризуются высокой пенетрантностью, наследуется по аутосомно-доминантному типу и развивается быстрее, чем при мутациях в других генах [15, 16].
Патологический эффект каждой мутации зависит от типа и локализации генетического дефекта и проявляется нарушением нормального функционирования белка тау, например, стабилизации микротрубочек посредством их связывания с тубулином. Некоторые мутации приводят к повышению количества свободного белка в цитоплазме, поддерживающего агрегацию тау, в то время как другие мутации изменяют фосфорилирование тау, что нарушает стабилизацию микротрубочек [17]. Показано, что мутации в донорном сайте сплайсинга, локализованном за экзоном 10, повышают вероятность включения экзона 10 гена MAPT в мРНК путем дестабилизации шпилечной структуры, которая маскирует сайт сплайсинга. Это приводит к увеличению продукции изоформы 4R белка тау. Мутации в акцепторном сайте сплайсинга, следующим за экзоном 10, также усиливают включение этого экзона в мРНК [18]. Другие мутации, влияющие на процесс альтернативного сплайсинга, приводят к сдвигу соотношения изоформ белка тау (3R/4R). Большинство миссенс-вариантов, таких как p.P301L, ухудшают способность белка тау связывать микротрубочки и поддерживать их сборку [19]. Более того, в исследованиях in vitro показано, что некоторые мутации, локализованные в белоккодирующих участках гена, ускоряют агрегацию белка тау [20]. В 2009 году у одного из пациентов с ЛВД была обнаружена гетерозиготная делеция размером 17.3 т.п.н., ответственная за исключение экзонов 6‒9 из мРНК гена MAPT [21]. Эта делеция привела к потере первого домена, ответственного за связь с микротрубочками, и уменьшению способности тау связываться с микротрубочками. Описана также дупликация участка, включающего гены CRHR1, MAPT и STH, у одного из пациентов с клинической картиной поведенческих и мнестических расстройств [22].
При аутопсии у носителей мутаций в гене MAPT выявлена атрофия лобно-височных долей и базальных ядер, присутствие тау-позитивных включений [23]. Клиническая картина у носителей мутаций в гене MAPT чаще всего представлена сочетанием ЛВД с синдромом паркинсонизма со средним возрастом начала заболевания около 50 лет. Поведенческие нарушения у носителей мутации в гене MAPT наиболее часто включают растормаживание, стереотипии и обсессивные расстройства. Примечательно, что для данных пациентов апатия менее характерна, чем для носителей мутаций в генах GRN и C9orf72. Семантические нарушения развиваются, как правило, на более поздних стадиях заболевания. Несмотря на вариабельную клиническую картину, практически у всех носителей мутации в гене MAPT наблюдается выраженная атрофия височных долей головного мозга, преимущественно справа.
ГЕН GRN
Обнаружение мутаций в гене MAPT не объяснило все семейные случаи ЛВД с аутосомно-доминантным наследованием (в сочетании с паркинсонизмом или без него); более того, у некоторых пациентов без мутаций в гене MAPT выявлена генетическая ассоциация с тем же хромосомным регионом (chr17q21) [24, 25]. Патоморфологическая картина в этих случаях была совершенно другой, представленной тау-негативным и убиквинтин-позитивным окрашиванием. В результате в 2006 году был идентифицирован новый ген GRN (Granulin precursor; OMIM *138945), локализованный на расстоянии 6.2 млн.п.н. от локуса MAPT. Первой мутацией, обнаруженной в гене GRN, стала инсерция 4 п.н. CTGC между нуклеотидами 90 и 71, приводящая к сдвигу рамки считывания и преждевременной терминации трансляции програнулина (C31LfsX34) [23]. В параллельном исследовании нашли мутацию в области интрона, следующего за первым некодирующим экзоном GRN (IVS1+5G>C). Эта мутация приводит к вырезанию интрона 0 при сплайсинге, задержке мРНК в ядре и ее последующей деградации [25].
В дальнейшем мутации в гене GRN обнаружили в 5‒20% семейных случаев c аутосомно-доминантным типом наследования и в 1‒5% спорадических случаев ЛВД [3]. GRN кодирует белок програнулин ‒ гликопротеин размером 88 кДа, который экспрессируется в нейронах и клетках микроглии [26]. В раннем возрасте GRN экспрессируется на низком уровне, а по мере взросления экспрессия возрастает. Програнулин это секретируемый фактор роста из 593 аминокислотных остатков, состоящий из 7.5 обогащенных цистеином доменов, расположенных в следующем порядке: P–G–F–B–A–C–D–E, где A–G ‒ полные повторы, а P – половинный мотив, известный как парагранулин [27]. Считается, что некоторые протеазы могут расщеплять програнулин на субъединицы размером 6 кДа, так называемые гранулины. Гранулины принадлежат к семейству белков, имеющих множество биологических функций (развитие, заживление ран, воспаление), которые реализуются путем активации сигнального каскада, контролирующего клеточный цикл и подвижность клеток [28].
К настоящему времени описано более 70 мутаций в гене GRN при ЛВД. Наиболее известные патогенные мутации (сплайсинговые, нонсенс-мутации) приводят к преждевременному образованию стоп-кодона. В результате мРНК с нарушенной структурой подвергается деградации посредством нонсенс-опосредованного механизма, что приводит к гаплонедостаточности [28]. Описаны более редкие частичные и полные делеции всего гена [29]. Пенетрантность мутаций GRN зависит от возраста: к 60 годам только у половины носителей мутаций проявляются симптомы заболевания, в 70 лет этот показатель вырастает уже до 90%. Возраст манифестации заболевания значительно варьирует даже среди членов одной семьи (от 47 до 79 лет) [30].
При патоморфологическом исследовании ЛВД, ассоциированной с мутациями в гене GRN, выявлены убиквитин-позитивные, тау-негативные включения, отличные от тау-позитивных включений, обнаруживаемых при мутациях в гене MAPT. Главным компонентом убиквитинсодержащих включений являются укороченные и гиперфосфорилированные изоформы ДНК-связывающего белка TDP-43 (transactive response (TAR) DNA-binding рrotein of 43 kDa). Эти изоформы описаны при семейных случаях ЛВД, ассоциированных с мутациями в гене GRN, при спорадических случаях ЛВД и в некоторых случаях БАС [31]. Интересно, что мутации GRN в гомозиготном состоянии ассоциированы с другим заболеванием – взрослой формой нейронального цероидного липофусциноза [32, 33], в то время как гетерозиготные мутации приводят к появлению характерных для ЛВД нейрональных и глиальных включений, иммунопозитивных по TDP-43 [32].
Клинически мутации в гене GRN ассоциированы с разнообразными фенотипами, однако наиболее часто встречается ППА [34]. Около 40% пациентов имеют в клинической картине паркинсонизм; часто наблюдаются трудности с эпизодической памятью, что в некоторых случаях приводит к ошибочному диагнозу БА [35]. Кроме того, в редких случаях показана ассоциация генетических вариантов ЛВД с психическими расстройствами. Например, описаны два случая ЛВД с мутацией Thr272fs в гене GRN с преморбидным биполярным расстройством в анамнезе [36].
Свой вклад в постановку корректного диагноза вносит определение уровня програнулина в плазме крови, который снижается у носителей мутаций GRN даже в отсутствие симптомов болезни [37, 38].
ГЕН C9orf72
Около 10 лет назад в семьях, отягощенных одновременно ЛВД и БАС, обнаружили ассоциацию этих заболеваний с локусом 9q21-22. Первые данные о локализации гена в этом локусе получены при изучении ЛВД-БАС-ассоциированных семей с аутосомно-доминантным типом наследования [39]. В 2011 году двумя международными группами в локусе 9q21-22 был идентифицирован ген C9orf72 (Chromosome 9 open reading frame 72; OMIM *614260) [40, 41]. Мутация представляет собой экспансию гексануклеотидных повторов (GGGGCC) в первом интроне, локализованным между экзонами 1а и 1b гена C9orf72. Более того, экспансия повтора может располагаться либо в интроне, либо в промоторной области в зависимости от того, какой из альтернативных сайтов начала транскрипции используется [40]. В норме количество повторов может варьировать от 2 до 20. У пациентов с ЛВД и БАС размер экспансии обычно составляет от 100 до нескольких тысяч копий повтора. Минимальный размер экспансии, который считается фактором риска развития заболевания, еще не установлен, возможно, из-за соматического мозаицизма. Известно, что размер экспансии варьирует в разных тканях даже одного человека, что затрудняет точное выявление корреляций ген-фенотип [42].
Экспансия повторов в гене C9orf72 считается самой частой причиной ЛВД (вне зависимости от ассоциации с БАС) в мире. Она особенно высока в популяции Финляндии, что, возможно, обусловлено эффектом основателя, тогда как в азиатской когорте пациентов распространенность экспансии, напротив, более низкая [43]. Клинический фенотип данной формы молекулярной нейродегенерации очень разнообразен, так же как возраст манифестации и длительность заболевания. По различным данным возраст манифестации варьирует между 27 и 83 годами, а длительность заболевания – от 1 до 22 лет. Наиболее частые клинические фенотипы заболевания – ЛВД, БАС или сочетание этих синдромов. Как уже упоминалось, в отягощенных семьях с клиническим фенотипом ЛВД-БАС экспансия в гене C9orf72 встречается весьма часто – более 50% случаев [44]. Клинический вариант ЛВД представлен в основном поведенческими нарушениями, в то время как речевые расстройства встречаются реже. Кроме классических поведенческих нарушений, включающих апатию, расторможенность, асоциальное поведение и потерю эмпатии, для носителей экспансии повторов в гене C9orf72 характерна высокая частота галлюцинаций, психозов и иллюзий [45], что может приводить к первичному диагнозу шизофрении и биполярного расстройства [46, 47]. В некоторых случаях описаны трудности в сфере эпизодической памяти в начале заболевания, что может приводить к первичному диагнозу БА [44, 48]. У носителей экспансии гексануклеотидных повторов в гене C9orf72 наблюдается раннее развитие синдрома паркинсонизма, очень редкого у носителей мутаций в генах MAPT и GRN [45].
При патоморфологическом исследовании в различных участках ткани головного мозга выявлены включения, содержащие белок TDP-43. Кроме того, описаны убиквитин- и p62-позитивные нейрональные включения в гранулярном слое коры мозжечка, пирамидных нейронах гиппокампа и в других анатомических областях. Эти включения образованы DPR-дипептидами (dipeptide repeat protein), которые транслируются с области GGGGCC-копий путем ассоциированной с повторами не-ATG-трансляции (repeat associated non-ATG translation – RAN). Выделяют пять разновидностей DPR-дипептидов, которые транслируются со смысловой РНК – поли-Gly-Pro (GP), поли-Gly-Аla (GA) и поли-Glu-Аrg (GR), и с антисмысловой РНК – поли-Pro-Аla (РА), поли-Pro-Аrg (PR) и дополнительный поли-GP с разных рамок считывания (рис. 2) [49, 50].
Рис. 2.
Разновидности DPR-дипептидов. Не-ATG-трансляция РНК-повторов приводит к образованию пяти различных дипептидов DPR в зависимости от рамки считывания, три из которых синтезируются со смысловой цепи: поли-GR, поли-GA, поли-GP. Три других образуются с антисмысловой цепи: поли-PR, поли-PA и снова поли-GP. Различия в эффектах перечисленных дипептидов показаны на разных модельных системах.
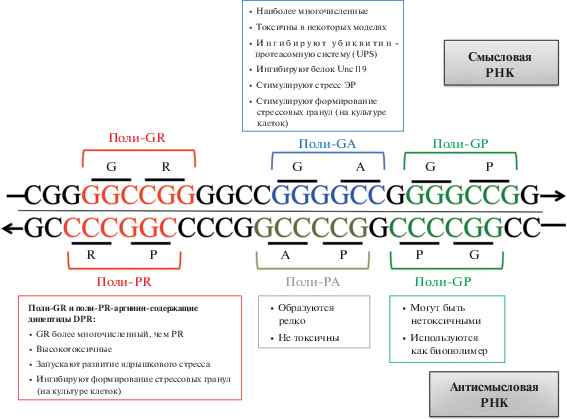
На трансгенных Drosophila показана высокая токсичность содержащих аргинин дипептидов (поли-GR и поли-PR), которые также могут вызывать формирование ядерных и цитоплазматических включений в нейронах [51, 52]. На культуре клеток и первичных нейронов показано, что сверхэкспрессия поли-GA приводит к образованию p62-позитивных включений и нейротоксичности, характерной для нарушений убиквитин-протеасомной системы [53]. Однако до сих пор не получен ответ на вопрос о клинической значимости дипептидов, а также об их изменениях в процессе прогрессирования заболевания. С целью поиска пациентов с экспансией гексануклеотидных повторов в гене C9orf72 в 2017 году Lehmer и соавт. разработали иммунологический метод определения дипептида поли-GP в цереброспинальной жидкости [54]. Уровень поли-GP-дипептида, обнаруженный в ликворе бессимптомных носителей экспансии (в отличие от здоровых доноров), сходен с его уровнем у носителей с симптомами, что потенциально можно использовать в качестве диагностического биомаркера в дополнение к генетическому скринингу [54].
В норме функции C9orf72 связаны с процессами ядерно-цитоплазматического транспорта, аутофагии, межклеточного транспорта и агрегации белка TDP-43 [55]. В 2013 году показали, что РНК, состоящие из повторяющихся гексануклеотидов (GGGGCC)n, формируют чрезвычайно стабильные G-квадруплексные структуры, которые теоретически могут влиять на активность промотора, генетическую стабильность, процессы сплайсинга, трансляции и локализацию РНК в аксоне [56]. В нескольких исследованиях, выполненных на полученных от пациентов клетках и тканях, показано, что эти структуры способны секвестрировать РНК-связывающие белки, нарушая систему ядерного транспорта [51]. Однако механизм, связывающий образование фокусов РНК и секвестрированных белков с нейродегенеративным процессом, изучен не полностью. Помимо формирования фокусов РНК и образования DPR-дипептидов, предложен еще один патологический механизм подавления экспрессии гена C9orf72 посредством метилирования [57].
ГЕН CHMP2B
В нескольких семьях с отягощенным семейным анамнезом по ЛВД описаны мутации в гене CHMP2B (charged multivesicular body protein 2B, OMIM *609512), который кодирует один из компонентов гетеромерного комплекса ESCRT III (Endosomal Sorting Complexes Required for Transport), вовлеченного в эндосомный транспорт и деградацию [58]. Белковый продукт гена CHMP2B участвует в сортировке и транспорте поверхностных рецепторов или белков в интралюминальные везикулы для деградации в лизосомах, а также связывает белок Vps4 (Vacuolar рrotein sorting), ответственный за диссоциацию компонентов ESCRT [59].
Первая мутация в гене CHMP2B была идентифицирована в большой семье из Дании [60]. Все описанные мутации (миссенс-мутации и укорачивающие мутации) имеют общий механизм действия: делецию С-концевой области белка с потерей связывающего Vps4 домена [59]. Это приводит к аккумуляции мутированного CHMP2B на мембране эндосом и препятствует вовлечению других белков, необходимых для слияния эндосомы с лизосомой. Такой феномен приводит к нарушению позднего эндосомного транспорта и способствует развитию нейродегенеративного процесса при ЛВД [60]. В ткани мозга таких пациентов наблюдаются увеличенные аномальные эндосомные структуры [61]. Гистологическое исследование выявляет у пациентов с мутацией в гене CHMP2B убиквитин- и р62-позитивные и TDP-43-негативные нейрональные цитоплазматические включения [62]. К основным особенностям клинической картины таких пациентов относятся поведенческие и когнитивные нарушения, ассоциированные с экстрапирамидными и пирамидными симптомами.
ГЕНЫ VCP-1 И SQSTM1
Мутации в гене VCP (valosin containing protein; OMIM *601023) впервые были описаны у пациентов с аутосомно-доминантным типом наследования и клинической триадой, включающей миопатию с тельцами включения, болезнь Педжета и ЛВД (IBMPFD – inclusion body myopathy with Paget’s disease of the bone and frontotemporal dementia) [63]. Миопатия – наиболее частый клинический симптом, характерный для 90% больных, тогда как ЛВД наблюдается примерно у 33% и обычно развивается много лет спустя после манифестации симптомов поражения мышц. Таким образом, с учетом своеобразия клинической картины отнесение VCP-1 к генам ЛВД является небесспорным, однако возможная связь данного гена с ЛВД [64, 65] находит свое патоморфологическое подтверждение. Так, у носителей мутации в гене VCP в ядрах нейронов головного мозга обнаружены TDP-43 и p62-позитивные включения [66].
VCP-1 кодирует белок из 806 аминокислотных остатков. VCP-1 регулирует большое количество процессов, таких как убиквитинзависимый контроль качества белков и создание меток для последующей деградации и координация процессов удаления белковых агрегатов путем формирования мультивезикулярных телец [67].
SQSTM1 (sequestosome 1 gene; OMIM *601530) ‒ еще один ген, вовлеченный в деградацию белков и патогенез ЛВД. Этот ген кодирует белок p62, который служит связующим звеном между убиквитинированными белками и рецептором аутофагии или протеасомными путями деградации [68]. Мутации в гене SQSTM1, впервые описанные при болезни Педжета, являются причиной примерно 30% семейных случаев этого заболевания [69].
ГЕН CHCHD10
Ген CHCHD10 (coiled-coil-helix-coiled-coil-helix domain containing 10; OMIM *615903) кодирует митохондриальный белок, который поддерживает структуру крист в межмембранном пространстве. Появление методов массового параллельного секвенирования позволило идентифицировать первую патогенную мутацию p.S59L в этом гене в семье с поздней манифестацией болезни двигательного нейрона, ЛВД, мозжечковой атаксией и митохондриальной миопатией [70]. В последующих генетических исследованиях у пациентов с ЛВД и БАС идентифицированы другие потенциально патогенные мутации с частотой 1–3% [71]. Относительно недавно у пациента с атипичной клинической картиной ЛВД и патоморфологически подтвержденной болезнью Паркинсона описана новая нонсенс-мутация (p.Gln108*) [72].
ГЕН TBK1
В 2015 году в когорте пациентов со спорадическим БАС в исследовании методом случай–контроль с использованием полноэкзомного секвенирования обнаружены мутации в гене TBK1 (TANK binding kinase 1; OMIM *604834) [73]. В дальнейшем мутации в гене TBK1, приводящие к потере его функции, выявили в семьях, отягощенных ЛВД-БАС, и при изолированных вариантах ЛВД [73]. Большинство идентифицированных мутаций приводят к потере функции гена в результате снижения его экспрессии до 50%. Миссенс-мутации нарушают связывание TBK1 с оптинейрином (OPTN). Как VCP и p62, TBK1 участвует в процессах деградации белков и аутофагии. Он фосфорилирует р62 или OPTN – дополнительные участники процессов аутофагии. В 2015 году в группе C9orf72- и GRN-негативных пациентов с ЛВД и патоморфологически подтвержденной агрегацией белка TDP-43 (самый частый патоморфологический вариант среди всех случаев ЛВД – до 50%) выявили пять случаев (4.8%) с вариантами нуклеотидной последовательности в генах OPTN и TBK1, которые расценили как высокопатогенные. Эти данные подтверждают, что оба гена вовлечены в патогенез рассмотренного варианта ЛВД [74].
ГЕН TARDBP
Ген TARDBP (TAR DNA-binding protein; OMIM *605078) кодирует ядерный белок TDP-43, способный формировать гетерогенные ядерные рибонуклеопротеидные комплексы (гяРНП), выполняющие различные функции, связанные с регуляцией РНК, такие как контроль сплайсинга, стабильности и транспорта мРНК. Предположение о связи между развитием ЛВД и БАС и нарушениями в белке TDP-43 основано на том, что TDP-43 регулирует рост аксонов в моделях in vivo и in vitro, т.е. изменение функционирования TDP-43 влияет на способность нейронов образовывать и поддерживать правильную структуру аксонов [75].
ГЕН FUS
Ген FUS (Fused in sarcoma; OMIM *137070) кодирует высококонсервативный белок, экспрессируемый в различных тканях. Белок FUS ‒ один из компонентов гяРНП, участвующий в транспорте и сплайсинге РНК, в метаболизме ДНК/РНК [67]. Мутации в гене FUS идентифицированы в 2009 году в 3% семейных случаев БАС. Большинство мутаций локализованы в участке, кодирующем С-концевую область белка FUS, особенно в участке ядерной локализации, что приводит к нарушению опосредуемого транспортином ядерного транспорта FUS [76]. Мутации в этом гене связаны с семейной формой БАС, которая может быть как ассоциированной, так и не ассоциированной с ЛВД. У носителей мутации в гене FUS находят аномальные цитоплазматические FUS-позитивные нейрональные и глиальные включения. Однако в нескольких случаях БАС с FUS-позитивными включениями мутации в гене FUS не найдены. Немногочисленные случаи ЛВД с мутацией в этом гене характеризовались атрофией лобных и височных долей, а также области стриатума.
ГЕН UBQLN2
Ген UBQLN2 (ubiquilin 2; OMIM *300264) связан с развитием редкой X-сцепленной семейной формы БАС и ЛВД-БАС [77]. Мутации в белке UBQLN2 расположены, как правило, в обогащенном остатками пролина высококонсервативном домене, содержащем повторы PXXP, участвующем в убиквитин-протеасомной деградации неправильно свернутых белков и аутофагии.
ГЕН TUBA4A
Ген TUBA4A (Tubulin, alpha-4a; OMIM *191110) кодирует один из восьми α-тубулинов человека, который полимеризуется с β-тубулином и формирует цитоскелет нейронов. Мутации в гене TUBA4A ассоциированы в первую очередь с БАС, хотя у некоторых пациентов описаны когнитивные нарушения, степень выраженности которых варьирует от умеренных до ЛВД. К настоящему времени при семейных и спорадических случаях БАС, часть которых ассоциирована с ЛВД, описано 10 миссенс- и одна нонсенс-мутация, а также одна мутация в донорном сайте сплайсинга [72].
ГЕНЫ-МОДИФИКАТОРЫ
Кроме описанных генов, мутации в которых встречаются преимущественно при семейных случаях с аутосомно-доминантным наследованием, обнаружены и другие гены, мутации которых могут быть факторами риска развития заболевания. Наиболее важный из них – ген TMEM106B (transmembrane protein 106b; *613413). В 2010 году Van Deerlin и соавт. опубликовали результаты полногеномного поиска ассоциаций у 515 пациентов с ЛВД и TDP-43-патологией, выявленными при патоморфологическом исследовании. Идентифицирован возможный локус хромосомы 7p21, содержащий ген TMEM106B [78]. Обнаружены три однонуклеотидных полиморфизма (SNP) ‒ rs102004, rs6966915 и rs1990622, ассоциированных со снижением экспрессии TMEM106B. Установлено, что у носителей мутации в гене GRN С-аллель SNP rs1990622 может действовать как протективный в отношении ЛВД [79]. Обнаружен также протективный эффект вариантов гена TMEM106B у носителей экспансии гексануклеотидных повторов в гене C9orf72 [80, 81]. Ген TMEM106B кодирует гликозилированный мембранный белок типа 2, локализованный в поздних эндосомах и лизосомах, где он, вероятно, выполняет важные функции. Сверхэкспрессия белка TMEM106b в культурах клеток вызывает нарушения формирования вакуолей и процессов эндолизосомного пути [82].
Изучена также роль различных SNP в основных генах, мутации в которых приводят к ЛВД, в качестве факторов риска этого заболевания, например, SNP rs5848 в предполагаемом сайте связывания микроРНК в 3'-нетранслируемой области (3'UTR) гена GRN. Роль этого полиморфизма до сих пор остается неясной, так как значимая его ассоциация показана в первой изученной когорте пациентов с ЛВД-TDP-43 и не найдена в последующих исследованиях [83]. На 11-й международной конференции по ЛВД в Сиднее в 2018 году группа исследователей из клиники Мейо (США) представила данные о том, что носительство минорного Т-аллеля в гомозиготном состоянии может приводить к снижению экспрессии гранулина (в меньшей степени, чем при патогенной мутации). Эти данные могут говорить о том, что снижение уровня програнулина при ЛВД возможно не только у носителей мутации в гене GRN.
Проведение полногеномного поиска ассоциаций позволило идентифицировать два других локуса: локус HLA (human leukocyte antigen; лейкоцитарные антигены человека, или антигены тканевой совместимости человека) на хромосоме 6p21.3 и локус на хромосоме 11q14, содержащий гены RAB8 (RAS-associated protein; *606281) и CTSC (cathepsin C; *602365) [84]. В последних двух генах выявлена связь между некоторыми SNP локуса RAB8/CTSC и 50%-ным снижением уровня RAB8 в крови пациентов. Это может служить подтверждением того факта, что утрата функции RAB8, участвующего в регуляции лизосом и транспорте белков, может играть роль в развитии ЛВД. Ассоциация с локусом HLA может указывать на связь патогенеза ЛВД с иммунной системой [84].
ЗАКЛЮЧЕНИЕ
До 40% пациентов с ЛВД имеют положительный семейный анамнез, при этом мутации в трех основных генах (GRN, MAPT, C9orf72) выявляются почти в 60% случаев ЛВД. Проведение ДНК-диагностики следует обсуждать со всеми пациентами, имеющими отягощенный семейный анамнез по таким неврологическим заболеваниям, как ЛВД, БА, паркинсонизм, БАС, миопатия с включениями или психозы во взрослом возрасте с доминантным типом наследования. Несмотря на преобладание спорадических форм, детальное изучение генетических вариантов способствует лучшему пониманию молекулярных и клеточных механизмов ЛВД. Дальнейшее изучение патогенеза ЛВД открывает новые потенциальные терапевтические мишени для разработки эффективных препаратов, модифицирующих течение заболевания. На сегодняшний день таких препаратов не существует, и лечение направлено на симптоматическую коррекцию и использование некоторых препаратов, предложеных ранее для БА. Однако разрабатываются и проходят клинические испытания лекарственные средства, модифицирующие течение заболевания, ассоциированного с мутациями в трех основных генах ЛВД [85]. Таким образом, определение генетической причины ЛВД важно не только для медико-генетического консультирования здоровых членов отягощенных семей, но и потенциально может быть необходимо для персонифицированного подбора препаратов, модифицирующих течение заболевания как в манифестной, так и в доклинической стадиях.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (грант № 19-015-00533).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Bang J., Spina S., Miller B.L. (2015) Frontotemporal dementia. Lancet. 386, 1672–1682.
Goldman J.S., Farmer J.M., Wood E.M., Johnson J.K., Boxer A., Neuhaus J., Lomen-Hoerth C., Wilhelmsen K.C., Lee V.M.Y., Grossman M., Miller B.L. (2005) Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. 65, 1817–1819.
Rademakers R., Neumann M., Mackenzie I.R. (2012) Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 8, 423–434.
Shi J., Shaw C.L., Plessis D.D., Richardson A.M.T., Bailey L.K., Julien C., Stopford C., Thompson J., Varma A., Craufurd D, Tian J., Pickering-Brown S., Neary D., Snowden J.S., Mann D.M.A. (2005) Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 110, 501‒512.
Mackenzie I.R.A., Neumann M., Baborie A., Sampathu D.M., Plessis D.D., Jaros E., Perry R.H., Trojanowski J.Q., Mann D.M.A., Lee V.M.Y. (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111‒113.
Forman M.S., Farmer J., Johnson J.K., Clark C.M., Arnold S.E., Coslett H.B., Chatterjee A., Hurtig H.I., Karlawish J.H., Rosen H.J., Van Deerlin V., Lee V.M.Y., Miller B.L., Trojanowski J.Q., Grossman M. (2006) Frontotemporal dementia: clinicopathological correlations. Ann. Neurol. 59, 952‒962.
Josephs K.A., Hodges J.R., Snowden J.S., Mackenzie I.R., Neumann M., Mann D.M., Dickson D.W. (2011) Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 122, 137‒153.
Rohrer J.D., Lashley T., Schott J.M., Warren J.E., Mead S., Isaacs A.M., Beck J., Hardy J., de Silva R., Warrington E., Troakes C., Al-Sarraj S., King A., Borroni B., Clarkson M.J., Ourselin S., Holton J.L., Fox N.C., Revesz T., Rossor M.N., Warren J.D. (2011) Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. 134, 2565‒2581.
Snowden J.S., Thompson J.C., Stopford C.L., Richar-dson A.M.T., Gerhard A., Neary D., Mann D.M.A. (2011) Clinical diagnosis of early-onset dementias: diagnostic accuracy and clinico-pathological relationships. Brain. 134, 2478‒2492.
Snowden J.S., Hu Q., Rollinson S., Halliwell N., Robinson A., Davidson Y.S., Momeni P., Baborie A., Griffiths T.D., Jaros E., Perry R.H., Richardson A., Pickering-Brown S.M., Neary D., Mann D.M.A (2011) The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol. 122, 99‒110.
Wilhelmsen K.C., Lynch T., Pavlou E., Higgins M., Nygaard T.G. (1994) Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21-22. Am. J. Hum.Genet. 55, 1159–1165.
Lynch T., Sano M., Marder K.S., Bell K.L., Foster N.L., Defending R.F., Sima A.A.F., Keohane C., Nygaard T.G., Fahn S., Mayeux R., Rowland L.P., Wilhelmsen K.C. (1994) Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology. 44, 1878–1884.
Hutton M., Lendon C.L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., Petersen R.C., Stevens M., de Graaff E., Wauters E., van Baren J., Hillebrand M., Joosse M., Kwon J.M., Nowotny P., Che L.K., Norton J., Morris J.C., Reed L.A., Trojanowski J., Basun H., Lannfelt L., Neystat M., Fahn S., Dark F., Tannenberg T., Dodd P.R., Hayward N., Kwok J.B., Schofield P.R., Andreadis A., Snowden J., Craufurd D., Neary D., Owen F., Oostra B.A., Hardy J., Goate A., van Swieten J., Mann D., Lynch T., Heutink P. (1998) Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 393, 702–705.
Wang Y., Mandelkow E. (2016) Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21.
Boeve B., Hutton M. (2008) Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch. Neurol. 65, 460–464.
Pickering-Brown S.M., Rollinson S., Plessis D., Morrison K.E., Varma A., Richardson A.M.T., Neary D., Snowden J.S., Mann D.M.A. (2008) Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort : comparison with patients with MAPT and no known mutations. Brain. 131, 721–731.
Rademakers R., Cruts M., Van Broeckhoven C. (2004) The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 24, 277–295.
Malkani R., D’Souza I., Gwinn-Hardy K., Schellenberg G.D., Hardy J., Momeni P. (2006) A MAPT mutation in a regulatory element upstream of exon 10 causes frontotemporal dementia. Neurobiol. Dis. 22, 401–403.
Hong M., Zhukareva V., Vogelsberg-Ragaglia V., Wszolek Z., Reed L., Miller B.I., Geschwind D.H., Bird T.D., McKeel D., Goate A., Morris J.C., Wilhelmsen K.C., Schellenberg G.D., Trojanowski J.Q., Lee V.M.Y. (1998) Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 282, 1914–1917.
Goedert M., Jakes R., Crowther R.A. (1999) Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 450, 306–311.
Rovelet-Lecrux A., Lecourtois M., Thomas-Anterion C., Le Ber I., Brice A., Frebourg T., Hannequin D., Campion D. (2009) Partial deletion of the MAPT gene: A novel mechanism of FTDP-17. Hum. Mutat. 30, E591‒E602.
Rovelet-Lecrux A., Hannequin D., Guillin O., Legallic S., Jurici S.б Wallon D., Frebourg T., Campion D. (2010) Frontotemporal dementia phenotype associated with MAPT gene duplication. J. Alzheimer’s Dis. 21, 897–902.
Mann D.M.A., Snowden J.S. (2017) Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 27, 723–736.
Baker M., Mackenzie I.R., Pickering-Brown S.M., Gass J., Rademakers R., Lindholm C., Snowden J., Adamson J., Sadovnick A.D., Rollinson S., Cannon A., Dwosh E., Neary D., Melquist S., Richardson A., Dickson D., Berger Z., Eriksen J., Robinson T., Zehr C., Dickey C.A., Crook R., McGowan E., Mann D., Boeve B., Feldman H., Hutton M. (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 442, 916–919.
Cruts M., Gijselink I., Van Der Zee J., Engelborghs S., Wils H., Pirici D., Rademakers R., Vandenberghe R., Dermaut B., Martin J.J., van Duijn C., Peeters K., Sciot R., Santens P., De Pooter T., Mattheijssens M., Van den Broeck M., Cuijt I., Vennekens K., De Deyn P.P., Kumar-Singh S., Van Broeckhoven C. (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 442, 920–924.
Petkau T.L., Leavitt B.R. (2014) Progranulin in neurodegenerative disease. Trends Neurosci. 37, 388–398.
Hrabal R., Chen Z., James S., Bennett H.P., Ni F. (1996) The hairpin stack fold, a novel protein architecture for a new family of protein growth factors. Nat. Struct. Biol. 3, 747–752.
Gass J., Cannon A., Mackenzie I.R., Boeve B., Baker M., Adamson J., Josephs K. (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 15, 2988–3001.
Yu C.E., Bird T.D., Bekris L.M., Montine T.J., Leverenz J.B., Steinbart E., Wood E.M. (2010) The spectrum of mutations in progranulin: A collaborative study screening 545 cases of neurodegeneration. Arch. Neurol. 67, 161–170.
Pietroboni A.M., Fumagalli G.G., Ghezzi L., Fenoglio C., Cortini F., Serpente M., Bassi M. (2011) Phenotypic heterogeneity of the GRN Asp22fs mutation in a large italian kindred. J. Alzheimer’s Dis. 24, 253–259.
Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., McCluskey L.F. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314, 130–133.
Smith K.R., Damiano J., Franceschetti S., Carpenter S., Canafoglia L., Morbin M., Sims, K.B. (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 90, 1102–1107.
Mole S.E., Cotman S.L. (2015) Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta (BBA) – Mol. Basis Dis. 1852, 2237–2241.
Benussi A., Padovani A., Borroni B. (2015) Phenotypic heterogeneity of monogenic frontotemporal dementia. Front. Aging Neurosci. 7, 171.
Le Ber I., Camuzat A., Hannequin D., Pasquier F., Guedj E., Rovelet-Lecrux A., Puel M. (2008) Phenotype variability in progranulin mutation carriers: A clinical, neuropsychological, imaging and genetic study. Brain. 131, 732–746.
Cerami C., Marcone A., Galimberti D., Villa C., Scarpini E., Cappa S.F. (2011) From genotype to phenotype: Two cases of genetic frontotemporal lobar degeneration with premorbid bipolar disorder. J. Alzheimer’s Dis. 27, 791–797.
Ghidoni R., Benussi L., Glionna M., Franzoni M., Binetti G. (2008) Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 71, 1235–1239.
Carecchio M., Fenoglio C., De Riz M., Guidi I., Comi C., Cortini F., Monaco F. (2009) Progranulin plasma levels as potential biomarker for the identification of GRN deletion carriers. A case with atypical onset as clinical amnestic mild cognitive impairment converted to Alzheimer’s disease. J. Neurol. Sci. 287, 291–293.
Hosler B., Siddique T., Sapp P.C., Sailor W., Huang M.C., Hossain A., Hung W.Y. (2000) Linkage of familial amyotrophic lateral to chromosome 9q21-q22. J. Am. Med. Assoc. 284, 1664–1669.
DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Kouri N. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 72, 245–256.
Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Kalimo H. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 72, 257–268.
van Blitterswijk M., DeJesus-Hernandez M., Niemantsverdriet E., Murray M.E., Heckman M.G., Diehl N.N., Serrano G. (2013) Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 12, 978–988.
Ishiura H., Tsuji S. (2015) Epidemiology and molecular mechanism of frontotemporal lobar degeneration/amyotrophic lateral sclerosis with repeat expansion mutation in C9orf72. J. Neurogenet. 29, 85–94.
Cooper-Knock J., Kirby J., Highley R., Shaw P.J. (2015) The spectrum of C9orf72-mediated neurodegeneration and amyotrophic lateral sclerosis. Neurotherapeutics. 12, 326–339.
Galimberti D., Fenoglio C., Serpente M., Villa C., Bonsi R., Arighi A., Clodomiro A. (2013) Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: late-onset psychotic clinical presentation. 2013. Biol. Psychiatry. 74, 384–391.
Galimberti D., Reif A., Dell’Osso B., Palazzo C., Villa C., Fenoglio C., Paoli R.A. (2014) C9ORF72 hexanucleotide repeat expansion as a rare cause of bipolar disorder. Bipolar Disorders. 16, 448–449.
Galimberti D., Reif A., Dell’Osso B., Kittel-Schneider S., Leonhard C., Herr A., Cioffi S.M. (2014) C9ORF72 hexanucleotide repeat expansion is a rare cause of schizophrenia. Neurobiol. Aging. 35, 1214.e7‒1214.e10.
Majounie E., Abramzon Y., Renton A.E., Perry R., Bassett S.S., Pletnikova O., Traynor B.J. (2012) Repeat expansion in C9ORF72 in Alzheimer’s disease. N. Engl. J. Med. 366, 283–284.
Gendron T.F., Bieniek K.F., Zhang Y.J., Jansen-West K., Ash P.E., Caulfield T., Cosio, D.M. (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844.
Mori K., Arzberger T., Grässer F.A., Gijselinck I., May S., Rentzsch K., Van Broeckhoven C. (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893.
Mizielinska S., Isaacs A.M. (2014) C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: gain or loss of function? Curr. Opin. Neurol. 27, 515–523.
Freibaum B.D., Lu Y., Lopez-Gonzalez R., Kim N.C., Almeida S., Lee K.H., Petrucelli L. (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 525, 129–133.
May S., Hornburg D., Schludi M.H., Arzberger T., Rentzsch K., Schwenk B.M., Mann M. (2014) C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 128, 485–503.
Lehmer C., Oeckl P., Weishaupt J.H., Volk A.E., Diehl-Schmid J., Schroeter M.L., Landwehrmeyer B. (2017) Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol. Med. 9, 859–868.
Haeusler A.R., Donnelly C.J., Rothstein J.D. (2016) The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 17, 383–395.
Reddy K., Zamiri B., Stanley S.Y., Macgregor R.B., Pearson C.E. (2013) The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem. 288, 9860–9866.
Gijselinck I., Van Mossevelde S., van der Zee J., Sieben A., Engelborghs S., De Bleecker J., Heeman B. (2016) The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry. 21, 1112–1124.
Skibinski G., Parkinson N.J., Brown J.M., Chakrabarti L., Lloyd S.L., Hummerich H., Brandner S. (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, P. 806.
Urwin H., Ghazi-Noori S., Collinge J., Isaacs A. (2009) The role of CHMP2B in frontotemporal dementia. Biochem. Soc. Transactions. 37, 208–212.
Lindquist S.G., Brændgaard H., Svenstrup K., Isaacs A.M., Nielsen J.E., FReJA Consortium. (2008) Frontotemporal dementia linked to chromosome 3 (FTD-3)-current concepts and the detection of a previously unknown branch of the Danish FTD-3 family. Eur. J. Neurol. 15, 667–670.
Urwin H., Authier A., Nielsen J.E., Metcalf D., Powell C., Froud K., Fisher E.M. (2010) Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet. 19, 2228–2238.
M. Isaacs A., Johannsen P., Holm I., E. Nielsen J. (2011) Frontotemporal dementia caused by CHMP2B mutations. Curr. Alzheimer Res. 8, 246–251.
Watts G.D.J., Wymer J., Kovach M.J., Mehta S.G., Mumm S., Darvish D., Kimonis V.E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381.
Kimonis V.E., Fulchiero E., Vesa J., Watts G. (2008) VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1782, 744‒748.
Mehta S.G., Khare M., Ramani R., Watts G.D.J., Simon M., Osann K.E., Donkervoort S., Dec E., Nalbandian A., Platt J., Pasquali M., Wang A., Mozaffar T., Smith C.D., Kimonis V.E. (2013) Genotype–phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin. Genet. 83, 422‒431.
Spina S., Van Laar A.D., Murrell J.R., Hamilton R.L., Kofler J.K., Epperson F., Ghetti B. (2013) Phenotypic variability in three families with valosin-containing protein mutation. Eur. J. Neurol. 20, 251–258.
Ju J.S., Weihl C.C. (2010) Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: A disorder of autophagy. Hum. Mol. Genet. 19, 38–45.
Ng A.S.L., Rademakers R., Miller B.L. (2015) Frontotemporal dementia: A bridge between dementia and neuromuscular disease. Ann. N.Y. Acad. Sci. 1338, 71–93.
Rea S.L., Majcher V., Searle M.S., Layfield R. (2014) SQSTM1 mutations ‒ Bridging Paget disease of bone and ALS/FTLD. Exp. Cell Res. 325, 27–37.
Bannwarth S., Ait-El-Mkadem S., Chaussenot A., Genin E.C., Lacas-Gervais S., Fragaki K., Verschueren A. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 137, 2329–2345.
Zhang M., Xi Z., Zinman L., Bruni A.C., Maletta R.G., Curcio S.A., Sorbi S. (2015) Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain. 138, e380.
Perrone F., Nguyen, H.P., Van Mossevelde S., Moisse M., Sieben A., Santens P., Cras P. (2017) Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol. Aging. 51, 177.e9‒177.e16.
Cirulli E.T., Lasseigne B.N., Petrovski S., Sapp P.C., Dion P.A., Leblond C.S., Ren Z. (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 347, 1436–1441.
Pottier C., Bieniek K.F., Finch N., van de Vorst M., Baker M., Perkersen R., DeTure M. (2016) Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar dementia without motor neuron disease. Acta Neuropathol. 130, 77–92.
Rainero I., Rubino E., Michelerio A., D’Agata F., Gentile S., Pinessi L. (2017) Recent advances in the molecular genetics of frontotemporal lobar degeneration. Funct. Neurol. 32, 7‒16.
Neumann M., Valori C.F., Ansorge O., Kretzschmar H.A., Munoz D.G., Kusaka H., Mackenzie I.R. (2012) Transportin 1 accumulates specifically with FET proteins but no other transportin cargos in FTLD-FUS and is absent in FUS inclusions in ALS with FUS mutations. Acta Neuropathol. 124, 705–716.
Dillen L., Van Langenhove T., Engelborghs S., Vandenbulcke M., Sarafov S., Tournev I., Jordanova A. (2013) Explorative genetic study of UBQLN2 and PFN1 in an extended Flanders-Belgian cohort of frontotemporal lobar degeneration patients. Neurobiol. Aging. 34, 1711.e1‒1711.e5.
Deerlin V.M., Sleiman P.M., Martinez-Lage M., Chen-Plotkin A., Wang L.S., Graff-Radford N.R., Arnold S.E., Mann D.M.A., Pickering-Brown S.M., Seelaar H., Heutink P., van Swieten J.C., Murrell J.R., Ghetti B., Spina S., Grafman J., Hodges J., Spillantini M.G., Gilman S., Lieberman A.P., Kaye J.A., Woltjer R.L., Bigio E.H., Mesulam M., al-Sarraj S., Troa-kes C., Rosenberg R.N., White III C.L., Ferrer I., Lladó F., Neumann M., Kretzschmar H.A., Hulette C.M., Welsh-Bohmer K.A., Miller B.L., Alzualde A., de Munain A.L., McKee A.C., Gearing M., Levey A.I., Lah J.J., Hardy J., Rohrer J.D., Lashley T., Mackenzie I.R.A., Feldman H.H., Hamilton R.L., Dekosky S.T., van der Zee J., Kumar-Singh S., Van Broeckhoven C., Mayeux R., Vonsattel J.P.G., Troncoso J.C., Kril J.J., Kwok J.B.J., Halliday G.M., Bird T.D., Ince P.G., Shaw P.J., Cairns N.J., Morris J.C., McLean C.A., DeCarli C., Ellis W.G., Freeman S.H., Frosch M.P., Growdon J.H., Perl D.P., Sano M., Bennett D.A., Schneider J.A., Beach T.G., Reiman E.M., Woodruff B.K., Cummings J., Vinters H.V., Miller C.A., Chui H.C., Alafuzoff I., Hartikainen P., Seilhean D., Galasko D., Masliah E., Cotman C.W., Tuñón M.T., Martínez M.C.C., Munoz D.G., Carroll S.L., Marson D., Riederer P.F., Bogdanovic N., Schellenberg G.D., Hakonarson H., Trojanowski J.Q., Lee V.M.Y. (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP43 inclusions. Nat. Genet. 42, 234–239.
Cruchaga C., Graff C., Chiang H.H., Wang J., Hinrichs A.L., Spiegel N., Goate A. (2011) Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch. Neurol. 68, 581–586.
Gallagher M.D., Suh E., Grossman M., Elman L., McCluskey L., Van Swieten, J.C., Rohrer J.D. (2014) TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 127, 407–418.
Van Blitterswijk M., Mullen B., Nicholson A.M., Bieniek, K.F., Heckman M.G., Baker M.C., Hsiung G.Y.R. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 127, 397–406.
Brady O.A., Zheng Y., Murphy K., Huang M., Hu F. (2013) The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum. Mol. Genet. 22, 685–695.
Rollinson S., Rohrer J.D., van der Zee J., Sleegers K., Mead S., Engelborghs S., Pickering-Brown S.M. (2011) No association of PGRN 3’UTR rs5848 in frontotemporal lobar degeneration. Neurobiol. Aging. 32, 754–755.
Ferrari R., Hernandez D.G., Nalls M.A., Rohrer J.D., Ramasamy A., Kwok J.R. (2014) Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 13, 686–699.
Tsai R.M., Boxer A.L. (2016) Therapy and clinical trials in frontotemporal dementia: past, present, and future. J. Neurochem. 20, 211–221.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология