

Молекулярная биология, 2020, T. 54, № 2, стр. 252-261
PARP10 влияет на пролиферацию клеток колоректального рака: предварительное исследование
C. F. Wu a, M. Xiao a, Y. L. Wang a, *, M. D. Threadgill b, M. Li a, Y. Tang a, X. Lin a, L. Yang a, Q. S. Li a, X. Li a
a Department of Pathology, Molecular Medicine and Cancer Research Center,
Chongqing Medical University
400016 Chongqing, China
b Department of Pharmacy and Pharmacology, University of Bath
BA2 7AY Bath, United Kingdom
* E-mail: wangyalan@cqmu.edu.cn
Поступила в редакцию 02.03.2019
После доработки 28.03.2019
Принята к публикации 29.05.2019
Аннотация
Внутриклеточная моно(АDP)-рибозилтрансфераза PARP10 регулирует пролиферацию некоторых типов клеток. Однако влияние этого фермента на пролиферацию клеток колоректального рака до сих пор систематически не изучали. Предварительное исследование механизмов, лежащих в основе влияния PARP10 на пролиферацию клеток колоректального рака, проведено нами на примере клеток LoVo и CT26. Ингибирование ферментативной активности PARP10 приводит к значительному снижению пролиферативной способности клеток LoVo и CT26 in vitro и подавлению роста опухолей CT26 в подмышечной области мышей BALB/с in vivo, а также к задержке клеточного цикла. Отмечены изменения экспрессии β-катенина и его транслокации в ядро, а также связанных с ним сигнальных белков аксина-2 и c-Myb – экспрессия аксина-2 увеличилась, а с-Myb уменьшалась. Показано, что PARP10 способствует пролиферации тех клеток колоректального рака, которые продуцируют значительные количества PARP10. Этот эффект подавляется при ингибировании ферментативной активности. По всей видимости, β-катенин можно рассматривать как медиатор антипролиферативного эффекта.
Поли(АDP)-рибоза-полимераза-10 (PARP10/ ARTD10) входит в семейство PARP и обладает моно(АDP)-рибозилтрансферазной (MAR) активностью. Первоначально PARP10 был идентифицирован как белок, взаимодействующий с MYC и способный моно(АDP)-рибозилировать и себя, и коровые гистоны [1]. Впоследствии установили, что PARP10 действует на различные клеточные процессы [2], в том числе контролирует апоптоз и пролиферацию опухолевых клеток [3, 4], метастазирование опухолей [5], модулирует функции митохондрий [6]. PARP10 присутствует как в ядре, так и в цитоплазме [7]. Сообщается, что локализованный в ядре PARP10 способствует пролиферации опухолевых клеток [4], участвуя в репарации повреждений ДНК [8] и изменении клеточного цикла [9]. С другой стороны, PARP10 может моно(АDP)-рибозилировать один из сигнальных белков, который регулирует пролиферацию клеток посредством транслокации β-катенина в ядро [10, 11].
В представленной работе изучено влияние PARP10 на пролиферацию клеток колоректального рака в условиях in vitro и in vivo. С этой целью использованы линии клеток со значительной экспрессией PARP10 и селективный ингибитор PARP10. Исследованы также некоторые сигнальные пути, участвующие в этих процессах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Клеточные линии колоректального рака и PARP10-селективный ингибитор. Клетки LoVo и СВ480 любезно предоставлены Wei Xue Tang (Chongqing Medical University, КНР). Клетки CT26 получены от Yu Quan Wei (Sichuan University, КНР). Клетки LoVo культивировали в среде DMEM (“Hyclone” США) с добавлением 10% фетальной сыворотки крупного рогатого скота (“Hyclone”) и 1% пенициллина/стрептомицина (“Beyotime”, КНР). Клетки CT26 и SW480 культивировали в среде RPMI-1640 (“Hyclone”), содержащей 10% фетальной сыворотки крупного рогатого скота и 1% пенициллина/стрептомицина, инкубировали в условиях 5% CO2 при 37°C. PARP10-селективный ингибитор ‒ 3-(Z-4-метиламинометокси-4-оксобутеноиламино)-бензамид ‒ любезно предоставлен Mikael Elofsson (Umeå University, Швеция). Этот ингибитор выбран из 26 аналогов, разработанных в качестве ингибиторов PARP10, ARTD8 и ARTD7 (pис. 1) [12]. Ингибитор растворяли в DMSO (“Sigma”, США), добавляли RPMI-1640 до концентрации от 0.5 до 4 мкМ и хранили при 4°С в темноте.
Определение экспрессии PARP10 в клеточных линиях колоректального рака методом иммунофлуоресценции. Экспрессию PARP10 в клетках CT26, LoVo и SW480 определяли с помощью иммунофлуоресцентного анализа. Клетки высевали на предметные стекла в 24-луночные планшеты, инкубировали в течение ночи при 37°C, фиксировали в течение 15 мин 4%-ным параформальдегидом, пропитывали в течение 20 мин 0.5%-ным тритоном и блокировали в течение 30 мин козьей сывороткой при комнатной температуре. Антитела против PARP10 (“Santa Cruz”, США 1 : 20) добавляли в каждую лунку и культивировали при 4°C в течение ночи в темноте. После инкубации с вторичными антителами кролика против IgG козы, меченными флуоресцеинизотиоцианатом (FITC, “ZSGB-Bio”, Китай), в течение 1 ч при 37°С и DAPI в течение 5 мин для окрашивания ядер уровень экспрессии и распределение PARP10 оценивали с использованием лазерного сканирующего конфокального микроскопа (Leica TCS SP2, “Leica Microsystems GmbH”, Германия). Параметры мощность лазера (laser power) и фотоумножитель (PMT) поддерживали на одном уровне в каждой линии клеток и считывали интенсивность флуоресценции 10 клеток в каждой клеточной линии. Среднее значение определяли как среднюю интенсивность флуоресценции в каждой клеточной линии. Эксперименты проводили в трех параллельных сериях.
Изучение пролиферации клеток CT26, LoVo и SW480 in vitro с использованием анализа CCK-8. Пролиферацию клеток анализировали с помощью набора CCK-8 (“Beyotime”). Клетки CT26 (5 × 103), LoVo (5 × 103) и SW480 (5 × 103) в логарифмической фазе роста высевали в 96-луночные планшеты и инкубировали при 37°C в 5% CO2 в течение 24 ч. В лунки добавляли ингибитор PARP10 в концентрации 0.1, 0.2, 0.5, 2.0 и 4.0 мкМ. Клетки без ингибитора PARP10 использовали как контрольную группу, а культуральную среду ‒ в качестве пустой пробы. Для определения зависимости ингибирования пролиферации клеток от времени клетки CT26 и LoVo обрабатывали ингибитором PARP10 (0.5 мкМ) в течение 24, 48, 72, 96 и 120 ч. Реагент ССК-8 (10 мкл) добавляли в каждую лунку и инкубировали при 37°C в течение 1 ч. Жизнеспособность клеток определяли по оптической плотности (OD) при 450 нм с помощью микропланшетного ридера (“Bio-Tek”, США). В каждом опыте заполняли по пять дублирующих лунок, и испытания повторяли трижды.
Рис. 1.
Структура PARP10-селективного ингибитора 3-(Z-4-метиламинометокси-4-оксобутенамино)-бензамида.
Изучение влияния PARP10 на клеточный цикл клеток CT26 методом проточной цитометрии. Клетки CT26 (1 × 104) собирали, дважды промывали PBS, суспендировали в культуральной среде и центрифугировали при 1000 об./мин в течение 5 мин. Затем клетки промывали 70%-ным этанолом, центрифугировали при 1000 об./мин в течение 5 мин и анализировали методом проточной цитометрии (FACS Vantage SE, “Becton Dickinson”, США). Индекс пролиферации (PI) измеряли по формуле: PI = (G2 + S)/(G1 + G2 + S). Измерения повторяли в трех повторностях.
Исследования на животных: обнаружение роста опухолей из клеток CT26, пересаженных мышам BALB/с. Все исследования на мышах соответствовали требованиям комитета по этике Chongqing Medical University, получены все необходимые разрешения. Восемь 6‒8-недельных мышей-самцов линии BALB/с (Центр лабораторных животных Chongqing Medical University) весом 22‒25 г, получавших стандартный корм и воду ad libitum, разделили произвольным образом на две группы ‒ группу, получавшую PARP10, и контрольную группу. Клетки CT26 (2 × 106) прививали мышам в правую подмышечную область. С восьмого дня после этого мышам опытной группы 1 раз в сутки внутрибрюшинно вводили селективный ингибитор PARP10 (2.0 мг/кг, в 200 мкл физиологического раствора), мышам контрольной группы вводили только физиологический раствор (200 мкл). Мышей забивали на 14-й день. Опухоль вырезали, определяли ее вес и объем. Объем опухоли рассчитывали по формуле V = ab2/2 (a ‒ наибольший диаметр; b ‒ наименьший диаметр).
Обнаружение потенциальных ассоциированных сигнальных белков в клетках CT26 in vitro и in vivo с помощью Вестерн-блотинга. Клетки CT26 культивировали в течение 72 ч с селективным ингибитором PARP10 (0.5 мкМ) или без него. Затем клетки дважды промывали ледяным PBS и собирали. Суммарный клеточный белок и ядерные белки экстрагировали реагентами для выделения ядерного и цитоплазматического материала (“Pierce”, США). Концентрацию белка определяли с помощью набора BCA protein assay kit (“Byotime”). Белки (20 мкг) разделяли в помощью электрофореза в 8%-ном полиакриламидном геле и переносили на поливинилиденфторидную мембрану HybondTM (“AmershamTM”, “GE Healthcare Life Sciences”, США). Затем мембраны в течение 2 ч блокировали 5%-ным обезжиренным молоком при комнатной температуре и инкубировали с β‑катенином (“Cell Signaling”, 1 : 1000), аксином-2 (“Proteintech”, CША, 1 : 200), c-Myb (“Proteintech”, 1 : 500), ламином B1 (“Proteintech”, 1 : 1000) и β‑актином (“Ding Guo”, Китай, 1 : 1000) в течение ночи при 4°C. Затем мембраны дважды промывали TBST и инкубировали с соответствующими вторичными антителами (конъюгированные с пероксидазой хрена антикроличьи, 1 : 1000, или иммуноглобулин G мыши, 1 : 1000) в течение 2 ч при комнатной температуре. После трехкратного промывания TBST мембраны визуализировали с помощью BeyoECL Plus (“Beyotime”) и переносили в форму для формирования гелей (“Bio-Rad”, США). Блоты анализировали с помощью программного обеспечения Quantity One (“Bio-Rad”). Все эксперименты повторяли трижды.
Статистический анализ. Все количественные данные представлены как среднее ± стандартное отклонение (SD). Статистический анализ проводили с помощью одностороннего теста ANOVA или t-критерия Стьюдента с использованием пакета программ SPSS 19.0. Р < 0.05 считали статистически значимыми.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
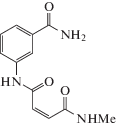
PARP10 экспрессируется в двух клеточных линиях колоректального рака
Нами выявлены различия в экспрессии PARP10 в трех клеточных линиях ‒ от очень слабой, практически отсутствующей в клетках SW480, до сильно выраженной в клетках CT26 и LoVo, со статистически значимыми различиями при средней интенсивности флуоресценции (Р < 0.001). Большая часть положительного сигнала находилась в цитоплазме, при этом несколько ядер были окрашены положительно (рис. 2).
Рис. 2.
Экспрессия PARP10 в клеточных линиях колоректального рака. а ‒ Экспрессия PARP10 в клетках SW480 слабая или почти отсутствует, но в клетках CT26 и LoVo выражена достаточно сильно. В клетках CT26 и LoVo окрашивание выявлено в основном в цитоплазме. Некоторые ядра также окрашены. б ‒ Средняя интенсивность флуоресценции в клетках CT26 и LoVo значительно выше, чем в клетках SW480 (*Р < 0.001). Использовали одинаковые параметры “мощность лазера” и “фотоумножитель” (PMT).
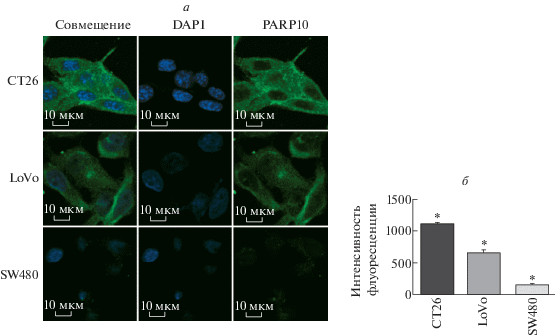
Ингибирование ферментативной активности PARP10 подавляет пролиферацию клеток CT26 и LoVo in vitro
Явное ингибирование пролиферации наблюдали при культивировании клеток CT26 с различными концентрациями ингибитора PARP10 в течение 72 ч. Выявлены статистически значимые различия между клетками, получавшимми ингибитор PARP10 и контрольной группой (P < 0.05), за исключением концентрации ингибитора 0.1 мкМ (P > 0.05). Интенсивность подавления пролиферации клеток становится более выраженной при увеличении концентрации ингибитора, что указывает на значительную дозовую зависимость (P < 0.05) (рис. 3а). Кроме того, временная зависимость эффекта была существенной, когда клетки CT26 культивировали с 0.5 мкМ PARP10-селективного ингибитора (P < 0.05) (рис. 3б). Результаты, полученные на клетках LoVo, соответствовали результатам на клетках CT26 (P < 0.05) (рис. 3в, г). Напротив, никакие концентрации PARP10-селективного ингибитора не смогли значимо подавить пролиферацию клеток SW480; эта клеточная линия отличается низким уровнем PARP10. Это подтверждает селективность используемого ингибитора (P > 0.05) (рис. 3д). В свете этих данных в дальнейшем клетки обрабатывали ингибитором в концентрации 0.5 мкМ в течение 72 ч.
Рис. 3.
Влияние PARP10 на пролиферацию клеток CT26. Жизнеспособность клеток определяли с помощью CCK-8-анализа. а ‒ Клетки CT26 инкубировали с PARP10-селективным ингибитором (0.1, 0.2, 0.5, 2.0, 4.0 мкМ) в течение 72 ч. Видно, что влияние ингибитора на жизнеспособность клеток СТ26 зависит от его концентрации (0.5 мкМ и более). Результаты выражены как среднее значение ± SD (*P < 0.05). б ‒ Клетки CT26 обрабатывали ингибитором PARP10 в течение 24, 48, 72, 96 и 120 ч. Ингибирующий эффект значительно зависел от времени. Результаты выражены в виде среднего значения ± SD (*P < 0.05, **P < 0.01). в ‒ Клетки LoVo инкубировали с PARP10-селективным ингибитором (0.1, 0.2, 0.5, 2.0, 4.0 мкМ) в течение 72 ч. Показано, что жизнеспособность клеток зависит от дозы ингибитора (эффекты выражены при 0.5 мкМ и выше). Приведены средние значения ± SD (*P < 0.05). г ‒ Клетки LoVo обрабатывали PARP10-селективным ингибитором в течение 24, 48, 72, 96 и 120 ч. Эффект ингибитора существенно зависел от времени инкубации. Данные выражены как: среднее значение ± SD (*P < 0.05, **P < 0.01). д ‒ Клетки SW480 инкубировали с PARP10-селективным ингибитором в разных концентрациях (0.1, 0.2, 0.5, 2.0, 4.0 мкМ) в течение 72 ч. Не выявлено статистически значимого эффекта ингибитора (P > 0.05).
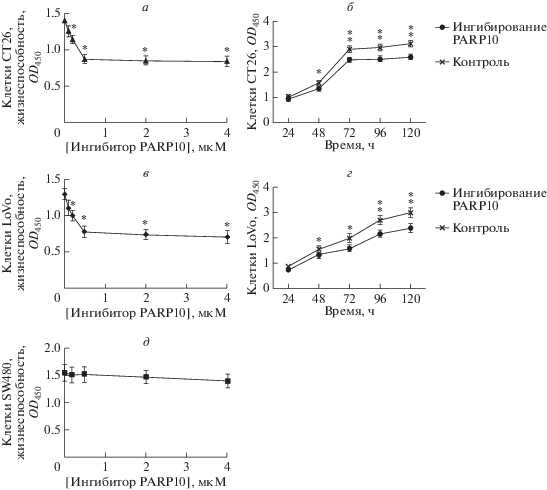
Ингибирование ферментативной активности PARP10 блокирует клеточный цикл клеток CT26 и LoVo с остановкой в фазе G1
Ингибирование PARP10 приводило к увеличению содержания клеток CT26 в фазе G1 с 58.1 ± 1.1 до 67.4 ± 0.2% (P < 0.001). При этом величина индекса пролиферации (PI) в группе с ингибитором PARP10 снижалась более существенно, чем в контрольной группе (Р < 0.001), что свидетельствует о снижении скорости пролиферации клеток CT26 (рис. 4).
Рис. 4.
Проточная цитометрия клеток СТС26, обработанных ингибитором PARP10 (а), и клеток контрольной группы (б). Красный пик слева ‒ число клеток в фазе G1, заштрихованный участок ‒ число клеток в S-фазе, красный участок справа ‒ число клеток в фазе G2. Определение индекса пролиферации (PI) в группе, получавшей ингибитор PARP10, и в контроле (в). Видно, что в группе, обработанной ингибитором PARP10, величина PI статистически значимо ниже, чем в контроле (*Р < 0.001). Доля клеток CT26 в фазе G1 значительно выше в группе, обработанной ингибитором PARP10 (67.4 ± 02%), чем в контрольной группе ‒ 58.1 ± 1.1% (*P < 0.001) (г).
Ингибирование ферментативной активности PARP10 подавляет рост опухолей из трансплантированных клеток CT26 у мышей BALB/c
Объем и масса опухолей из клеток CT26, подкожно трансплантированных мышам BALB/c, получавшим ингибитор PARP10, была значительно меньше, чем в контрольной группе (P < 0.001) (рис. 5 и табл. 1), т.е. ингибирование PARP10 оказывало постоянный эффект на пролиферацию клеток CT26 in vitro и in vivo.
Рис. 5.
Влияние PARP10 на рост опухолей из клеток CT26 в подмышечной области мышей BALB/c. Видно, что объем опухолей в группе с ингибитором меньше, чем в контрольной группе.
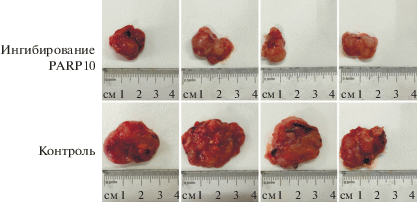
Ингибирование активности PARP10 снижает экспрессию и перенос β-катенина в клеточное ядро
Нами обнаружена экспрессия β-катенина в клетках CT26. Методом Вестерн-блотинга показано, что ингибирование PARP10 приводило к статистически значимому снижению экспрессии β-катенина по сравнению с контрольной группой, причем как в целых клетках, так и в клеточных ядрах (Р < 0.05) (рис. 6).
Рис. 6.
Экспрессия β-катенина в клетках CT26, обработанных PARP10-селективным ингибитором в течение 72 ч. Вестерн-блот-анализ; β-актин и ламин B1 использовали в качестве контроля суммарного и ядерного белка соответственно. а и б ‒ Ингибирование PARP10 снижает общее содержание β-катенина в клетках CT26. в и г ‒ Ингибирование PARP10 снижает количество β-катенина в ядре. Данные представлены как: среднее ± SD (*P < 0.05).
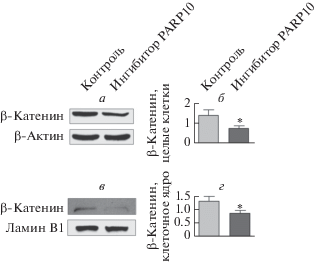
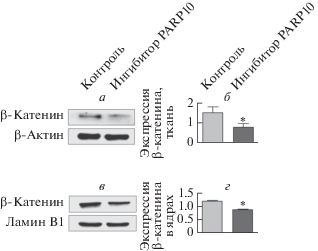
Показано, что ингибирование PARP10 уменьшает как содержание внутриклеточного β-катенина, так и перенос β-катенина в ядро. Чтобы сопоставить этот эффект с функционированием PARP10 in vivo, мы оценили экспрессию β-катенина в опухолях, которые образовались из клеток CT26, пересаженных мышам BALB/с подкожно. Результаты этих опытов соответствуют данным, полученным in vitro: при подавлении активности PARP10 экспрессия β-катенина как в целых опухолевых клетках, так и в их ядрах была существенно ниже, чем в контроле (P < 0.05) (рис. 7).
Рис. 7.
Экспрессия β-катенина в опухолях CT26 in vivo. Мышам BALB/c вводили селективный ингибитор PARP10 (2.0 мг/кг) или 0.9% нормального физиологического раствора внутрибрюшинно в течение 1 недели. После этого содержание β-катенина в пересаженной опухоли CT26 анализировали методом вестерн-блотинга. При определении содержания суммарного и ядерного белка в качестве контроля использовали β-актин и ламин B1. а и б ‒ Ингибирование PARP10 снижает суммарное содержание β-катенина в клетках. в и г ‒ Ингибирование PARP10 снижает количество β-катенина в ядре. Данные выражены как: среднее значение ± SD (*P < 0.05, **Р < 0.01).
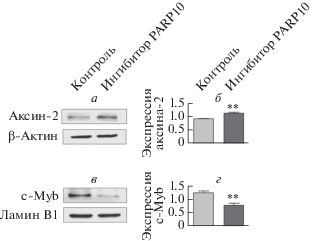
Ингибирование ферментативной активности PARP10 изменяет экспрессию аксина-2 и белка c-Myb
Аксин-2 и c-Myb ‒ белки сигнальной системы, функционально тесно связанные с β-катенином, которые участвуют в регуляции пролиферации клеток. При ингибировании PARP10 экспрессия аксина-2 значительно повышалась, а экспрессия ядерного c-Myb заметно снижалась (P < 0.05) (рис. 8).
Рис. 8.
Экспрессия аксина-2 и белка c-Myb в клетках CT26. После обработки ингибитором PARP10 (0.5 мкМ, 72 ч) с помощью вестерн-блотинга измеряли уровень аксина-2 в клетках СТ26 и белка c-Myb в клеточном ядре. Содержание суммарного клеточного белка и ядерного белка определяли с использованием в качестве контроля β-актина и ламина B1 соответственно. а и б ‒ Ингибирование PARP10 повышает уровень аксина-2. в и г ‒ Ингибирование PARP10 снижает экспрессию белка c-Myb по сравнению с контрольной группой. Данные представлены как среднее значение ± SD (**Р < 0.01).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Функции недавно обнаруженной моно(АDP)-рибозилтрансферазы PARP10 включают влияние на репликацию вируса [13, 14], регуляцию пролиферации эукариотических клеток, апоптоз, прохождение клеточного цикла, а также на метастазирование [2]. Сообщается, что нокдаун PARP10 в клетках HeLa приводит к подавлению роста опухолей и замедлению онкогенеза. Повышенная экспрессия PARP10 в нетрансформированных клетках RPE-1 понижает чувствительность клеток к репликационному стрессу, способствует запуску остановленной репликации и после ксенотрансплантации приводит к образованию опухолей у мышей [4]. Сайленсинг PARP10 снижает скорость пролиферации клеток MCF7, в то время как увеличение экспрессии PARP10 может способствовать пролиферации [6]. Однако влияние PARP10 на пролиферацию различных клеток колоректального рака не изучено.
Нами изучено влияние активности PARP10 на пролиферацию трех линий клеток колоректального рака: LoVo, CT26 и SW480. PARP10 экспрессируется как в ядрах, так и в цитоплазме клеток LoVo и CT26, но практически не экспрессируется в клетках SW480, что согласуется с данными [7]. Ингибирование ферментативной активности PARP10 приводит к статистически значимому снижению скорости пролиферации в клетках LoVo и CT26, причем это снижение зависит как от концентрации ингибитора, так и от времени инкубации. Это дает основания полагать, что клетки колоректального рака, экспрессирующие PARP10, могут быть чувствительными к подавлению ферментативной активности PARP10. Для определения эффекта PARP10 в опухоли in vivo мы пересадили клетки CT26 в подмышечные области мышей BALB/с и получили таким образом опухолевую модель. Рост этих опухолей был существенно подавлен в группе мышей, которым вводили ингибитор PARP10, что указывает на способность PARP10 стимулировать пролиферацию клеток колоректального рака in vivo. Кроме того, при ингибировании PARP10 наблюдалось значительное увеличение доли клеток CT26 в фазе G1 и снижение PI, что позволяет рационально объяснить выявленное ранее снижение пролиферации клеток CT26 при подавлении активности PARP10.
β-Катенин ‒ это вторичный посредник, который действует как фактор транскрипции, вовлеченный в стимуляцию пролиферации клеток [15, 16], а также как один из сигнальных белков, связанных с одним или несколькими целевыми белками, регулируемыми PARP10 [11]. Для поиска ядерных факторов транскрипции, регулирующих пролиферацию клеток, на которые может влиять PARP10, мы использовали β-катенин. Когда мы избирательно ингибировали PARP10, экспрессия β-катенина снижалась статистически значимо и в клетках CT26, и в клетках перевиваемых CT26-опухолей у мышей BALB/с, указывая на снижение содержания β-катенина и в клетках CT26, и в ядрах этих клеток in vitro и in vivo, что позволяет говорить о существенной роли PARP10 в регуляции экспрессии β-катенина.
Киназа 3 гликоген-синтазы (GSK3β) ‒ сигнальный белок, локализованный выше β-катенина в каскаде Wnt-сигнального пути, который участвует в образовании комплекса деградации β‑катенина и деградации β-катенина посредством фосфорилирования. При отсутствии сигнала Wnt β-катенин образует комплекс с аксином, GSK3β, APC и Fzd и фосфорилируется GSK3β или гидролизуется лизосомами или убиквитин-протеасомной системой (UPS) [11]. Когда Wnt-путь активирован, комплекс GSK3β/аксин/APC распадается и β-катенин невозможно спасти от деградации. С использованием белковых микрочипов GSK3β недавно была идентифицирована в качестве субстрата катализируемого PARP10 моно(АDP)-рибозилирования, причем киназная активность этого фермента ингибируется при его моно(АDP)-рибозилировании под действием PARP10 [7, 17]. Таким образом, мы предполагаем, что PARP10 может подавлять деградацию β-катенина путем ингибирования киназной активности GSK3β в результате моно(АDP)-рибозилирования. При ингибировании PARP10 киназная активность GSK3β восстанавливается, β-катенин может распадаться, и его содержание в клетках уменьшается. Как именно осуществляется регуляция β-катенина в PARP10-положительных клетках колоректального рака с помощью PARP10 и GSK3β еще предстоит выяснить.
β-Катенин транслоцируется в ядро с помощью нуклеопорина 62 (NUP62), замещая CoR, связывается с такими факторами транскрипции, как TCF/LEF, HIF-1α и TBX5, и активирует транскрипцию генов, кодирующих сигнальные белки, связанные с пролиферацией, включая аксин-2 и c-Myb [15, 16]. Чтобы определить, как влияет изменение уровня транслокации β-катенина в ядро на экспрессию белков нисходящего сигнального пути, измеряли экспрессию c-Myb – одного из белков, регулируемых β-катенином. Белок c-Myb взаимодействует с большим количеством потенциальных регуляторов, включая протеинкиназы, регуляторы клеточного цикла и факторы транскрипции. Он действует как онкогенный белок, активно экспрессируется в опухолях, в том числе в клетках колоректального рака, и способствует пролиферации опухолевых клеток путем активации генов, ответственных за переходы G/S и G/M [18‒20]. Показано, что количество белка c-Myb в ядре снижается в тех клетках CT26, в которых подавлена ферментативная активность PARP10. Это уменьшение указывает на то, что экспрессия и функциональные изменения β-катенина вызывают последующие специфические эффекты. Стоит отметить, что наше предположение о влиянии изменений уровня белка c-Myb на прохождение клеточного цикла клетками CT26 с ингибированным PARP10, подтверждается остановкой клеток в фазе G1. Влияет ли сниженная экспрессия β-катенина и его транслокация в ядро на пролиферацию клеток CT26 с ингибированным PARP10, через c-Myb-опосредованный механизм? Для ответа на этот вопрос необходима дополнительная проверка.
В то же время в опухолях с нарушением функций β-катенина обнаружена аномально высокая экспрессия аксина-2 [21]. Аксин-2 способствует деградации β-катенина путем сборки мультибелкового комплекса GSK3β/аксин/APC/Fzd, который разрушает β-катенин [22, 23]. В настоящем исследовании выявлено повышение экспрессии аксина-2 в группе с ингибированием PARP10 по сравнению с контролем. Таким образом, мы делаем вывод, что в клетках CT26, обработанных ингибитором PARP10, снижение уровня ядерной транслокации β-катенина приводит к регулируемому повышению уровней аксина-2. Затем аксин-2 действует как репрессор по типу обратной связи, способствуя усилению эффекта снижения β-катенина и дальнейшему снижению экспрессии и функции β-катенина. В настоящее время предприняты попытки увеличить экспрессию аксинов-1/2, а следовательно, снизить конститутивную стабильность β-катенина, с целью возможного применения в терапии колоректального рака [24, 25]. Так, β-катенин с высокой вероятностью регулирует экспрессию аксина-2, а также увеличивает эффект снижения уровня аксина-2 по механизму отрицательной обратной связи в клетках колоректального рака с положительной экспрессией PARP10.
На субклеточном уровне PARP10 моно(АDP)-рибозилирует GSK3β в цитоплазме. Нами показано, что PARP10 в основном присутствует в цитоплазме клеток LoVo и CT26. Это дает нам основание полагать, что в цитоплазме PARP10 регулирует пролиферацию клеток колоректального рака, воздействуя на экспрессию и функции β-катенина [7, 10, 17].
Сообщалось также, что ядерная PARP10 принимает прямое и эффективное участие в пролиферации клеток. PARP10 служит партнером PCNA (ядерный антиген пролиферирующих клеток), способствуя стабильности генома, а также участвует в трансляции и синтезе ДНК [26, 27]. Пониженная экспрессия PARP10 замедляет прохождение фазы G1 и приводит к гибели клеток HeLa [9]. Среди клеток колоректального рака с блокированным PARP10 увеличивается доля клеток в фазе G1 и снижается величина PI. Эта тенденция согласуется с сообщениями, безусловно поддерживающими этот механизм регуляции пролиферации клеток с участием ядерного PARP10. Мы полагаем, что PARP10, предположительно рекрутируемый PTEN в ядра клеток, в комплексе с PTEN присоединяется к поврежденным участкам ДНК, стимулирует репликацию и репарацию, регулирует клеточный цикл, а также влияет на пролиферацию клеток колоректального рака [4, 26].
Следует отметить, что влияние PARP10 на поведение опухолевых клеток никогда не является простым и односторонним [3, 5, 8, 28]. Более детальное изучение механизма регуляции пролиферации клеток колоректального рака белком PARP10 позволит выяснить роль PARP10. Кроме того, очень важно понять, можно ли распространить представления о способности PARP10 регулировать пролиферацию клеток колоректального рака на другие типы опухолевых клеток.
ВЫВОДЫ
Обнаружено, что PARP10 способствует пролиферации тех клеток колоректального рака, которые содержат значительное количество PARP10. И наоборот, ингибирование ферментативной активности PARP10 практически нивелирует эту способность. Показано также, что в осуществлении этой функции участвует транслоцированный в ядро β-катенин.
Список литературы
Kleine H., Poreba E., Lesniewicz K., Hassa P.O., Hottiger M.O., Litchfield D.W., Shilton B.H., Lüscher B. (2008) Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol. Cell. 32, 57–69.
Bütepage M., Eckei L., Verheugd P., Lüscher B. (2015) Intracellular mono-ADP- ribosylation in signaling and disease. Cells. 4, 569‒595.
Herzog N., Hartkamp J.D., Verheugd P., Treude F., Forst A.H., Feijs K.L.H., Lippok B.E., Kremmer E., Kleine H., Lüscher B. (2013) Caspase-dependent cleavage of the mono-ADP-ribosyltransferase ARTD10 interferes with its pro-apoptotic function. FEBS J. 280, 1330‒1343.
Schleicher E.M., Galvan A.M., Imamura-Kawasawa. Y., Moldovan G.-L., Nicolae C.M. (2018) PARP10 promotes cellular proliferation and tumorigenesis by alleviating replication stress. Nucl. Acids Res. 46, 8908‒8916.
Zhao Y., Hu X., Wei L., Song D., Wang J., You L., Saiyin H., Li Z., Yu W., Yu L., Ding J., Wu J. (2018) PARP10 suppresses tumor metastasis through regulation of Aurora A activity. Oncogene. 37, 2921‒2935.
Márton J., Fodor T., Nagy L., Vida A., Kis G., Brunyán-szki A., Antal M., Lüscher B., Bai P. (2018) PARP10 (ARTD10) modulates mitochondrial function. PLoS One. 13, e0187789.
Kleine H., Herrmann A., Lamark T., Forst A.H., Verheugd P., Lüscher-Firzlaff. J., Lippok B., Feijs K.L., Herzog N., Kremmer E., Johansen T., Muller-Newen G., Lüscher B. (2012) Dynamic subcellular localization of the mono-ADP-ribosyltransferase ARTD10 and interaction with the ubiquitin receptor p62. Cell. Commun. Signal. 10, 10‒28.
Kaufmann M., Feijs K.L., Lüscher B. (2015) Function and regulation of the mono-ADP-ribosyltransferase ARTD10. Curr. Topics Microbiol. Immunol. 384, 167‒188.
Chou H.Y., Chou H.T., Lee S.C. (2006) CDK-dependent activation of poly(ADP-ribose) polymerase member 10 (PARP10). J. Biol. Chem. 281, 15201‒15207.
Feijs K.L., Kleine H., Braczynski A., Forst A.H., Herzog N., Verheugd P., Linzen U., Kremmer E., Lüscher B. (2013) ARTD10 substrate identification on protein microarrays: regulation of GSK3beta by mono-ADP-ribosylation. Cell. Commun. Signal. 11, 11‒15.
Wu D., Pan W. (2010) GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 35, 161‒168.
Ekblad T., Lindgren A.E.G., Andersson C.D., Caraballo R., Thorsell A.G., Karlberg T., Spjut S., Linusson A., Schüler H., Elofsson M. (2015) Towards small molecule inhibitors of mono-ADP-ribosyltransferases. Eur. J. Med. Chem. 95, 546‒551.
Atasheva S., Frolova E.I., Frolov I. (2014) Interferon-stimulated poly(ADP-ribose) polymerases are potent inhibitors of cellular translation and virus replication. J. Virol. 88, 2116‒2130.
Yu M., Zhang C., Yang Y., Yang Z., Zhao L., Xu L., Wang R., Zhou X., Huang P. (2011) The interaction between the PARP10 protein and the NS1 protein of H5N1 AIV and its effect on virus replication. Virol. J. 8, 546.
Majidinia M.A.J., Jahanban-Esfahlani R., Yousefi B. (2018) The roles of Wnt/β-catenin pathway in tissue development and regenerative medicine. J. Cell. Physiol. 233, 5598‒5612.
Yang X., Gu Q., Lin L., Li S., Zhong S., Li Q., Cui Z. (2015) Nucleoporin 62-like protein activates canonical Wnt signaling through facilitating the nuclear import of β-catenin in zebrafish. Mol. Cell. Biol. 35, 1110‒1124.
Yu M., Schreek S., Cerni C., Schamberger C., Lesniewicz K., Poreba E., Vervoorts J., Walsemann G., Grotzinger J., Kremmer E., Mehraein Y., Mertsching J., Kraft R., Austen M., Lüscher-Firzlaff J., Lüscher B. (2005) PARP-10, a novel Myc-interacting protein with poly(ADP-ribose) polymerase activity, inhibits transformation. Oncogene. 24, 1982‒1993.
Biroccio A., Benassi B., D’Agnano I., D’Angelo C., Buglioni S., Mottolese M., Ricciotti A., Citro G., Cosimelli M., Ramsay R.G., Calabretta B., Zupi G. (2001) c-Myb and Bcl-x overexpression predicts poor prognosis in colorectal cancer: clinical and experimental findings. Am. J. Pathol. 158, 1289‒1299.
Ramsay R.G., Gonda T.J. (2008) MYB function in normal and cancer cells. Nat. Rev. Cancer. 8, 523‒534.
McCubrey J.A., Steelman L.S., Bertrand F.E., Davis N.M., Abrams S.L., Montalto G., D’Assoro A.B., Libra M., Nicoletti F., Maestro R., Basecke J., Cocco L., Cervello M., Martelli A.M. (2014) Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia. 28, 15‒33.
Leung J.Y., Kolligs F.T., Wu R., Zhai Y., Kuick R., Hanash S., Cho K.R., Fearon E.R. (2002) Activation of AXIN2 expression by β-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J. Biol. Chem. 277, 21657‒21665.
Herbst A., Jurinovic J., Krebs S., Thieme S.E., Blum H., Göke B., Kolligs F. T. (2014) Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signaling. BMC Genomics. 15, 74.
Wu Z.Q., Brabletz T., Fearon E., Willis A.L., Hu C.Y., Li X.Y., Weiss S.J. (2012) Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc. Natl. Acad. Sci. USA. 109, 11312‒11317.
Nathubhai A., Haikarainen T., Koivunen J., Murthy S., Koumanov F., Lloyd M.D., Holman G.D., Pihlajaniemi T., Tosh D., Lehtiö L., Threadgill M.D. (2017) Highly potent and isoform selective dual site binding tankyrase/Wnt signaling inhibitors that increase cellular glucose uptake and have antiproliferative activity. J. Med. Chem. 60, 814‒820.
Jeong W.J., Ro E.J., Choi K.Y. (2018) Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. N.P.J. Precis Oncol. 2, 5.
Nicolae C.M., Aho E.R., Vlahos A.H., Choe K.N., De S., Karras G.I., Moldovan G.L. (2014) The ADP-ribosyltransferase PARP10/ARTD10 interacts with proliferating cell nuclear antigen (PCNA) and is required for DNA damage tolerance. J. Biol. Chem. 289, 13627‒13637.
Shahrour M.A., Nicolae C.M., Edvardson S., Ashhab M., Galvan A.M., Constantin D., Abu-Libdeh B., Moldovan G.L., Elpeleg O. (2016) PARP10 deficiency manifests by severe developmental delay and DNA repair defect. Neurogenetics. 17, 227‒232.
Verheugd P., Forst A.H., Milke L., Herzog N., Feijs K.L., Kremmer E., Kleine H., Luscher B. (2013) Regulation of NF-κB signalling by the mono-ADP-ribosyltransferase ARTD10. Nat. Commun. 4, 1683.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология