

Молекулярная биология, 2020, T. 54, № 3, стр. 497-511
Cys-фланкированные катионные пептиды для доставки гена тимидинкиназы вируса простого герпеса в клетки с целью суицидной генной терапии миомы матки
А. А. Егорова a, С. В. Штыкалова a, b, М. А. Маретина a, А. В. Селютин a, Н. Ю. Швед a, Н. В. Крылова b, А. В. Ильина b, И. А. Пьянков b, С. А. Фрейнд b, С. А. Сельков a, В. С. Баранов a, b, А. В. Киселев a, *
a Институт акушерства, гинекологии и репродуктологии им. Д.О. Отта
199034 Санкт-Петербург, Россия
b Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Россия
* E-mail: ankiselev@yahoo.co.uk
Поступила в редакцию 14.11.2019
После доработки 19.12.2019
Принята к публикации 25.12.2019
Аннотация
Миома матки (ММ) ‒ наиболее часто встречающаяся доброкачественная опухоль у женщин репродуктивного возраста. В качестве подхода к лечению ММ может быть предложена генная терапия с использованием суицидных генов. К ключевым факторам, определяющим успех генной терапии, относится выбор носителя генетической конструкции. Перспективной группой невирусных носителей для доставки экспрессирующихся генных конструкций в клетки считаются катионные фланкированные цистеином (Cys-фланкированные) пептиды, образующие плотные комплексы с ДНК за счет электростатических взаимодействий и наличия межпептидных дисульфидных связей. Проведено сравнительное изучение физико-химических, токсических и трансфекционных свойств ДНК-пептидных комплексов, полученных с помощью матричной полимеризации или окислительной поликонденсации Cys-фланкированных пептидов с использованием ограничителя роста цепи 2-аминоэтантиола. Продемонстрирован генотерапевтический эффект доставки плазмиды pPTK-1, несущей ген тимидинкиназы вируса простого герпеса типа 1 (HSV-1), в клетки культур PANC-1 и HEK-293Т, а также в первичные клетки ММ. Показано, что трансфекция первичных клеток ММ носителями, полученными в результате окислительной поликонденсации, проходит более эффективно, чем с образующимися при матричной полимеризации. Обработка ганцикловиром приводила к гибели почти 40% клеток ММ, трансфицированных плазмидой pPTK-1. Обсуждается перспективность использования носителя polyR6, полученного окислительной поликонденсацией, в качестве основы для создания модульных пептидных носителей в генной терапии ММ.
ВВЕДЕНИЕ
Генная терапия представляет собой лечение с помощью введения в клетку нуклеиновых кислот и их производных с целью исправления/компенсации генных дефектов или направленного воздействия на процессы реализации генетической информации [1]. Генную терапию используют при лечении широкого спектра наследственных и приобретенных заболеваний, в частности, начаты исследования в отношении таких гинекологических заболеваний, как эндометриоз и миома матки [2–5].
Миома матки (ММ) ‒ это доброкачественная гормонозависимая опухоль, развивающаяся в миометрии [6, 7]. На настоящий момент не существует эффективного метода лечения ММ. Хирургический метод (миомэктомия) продолжает оставаться основным для этой категории больных. В случае миомэктомии удаление опухоли в большинстве случаев позволяет сохранить фертильный потенциал женщины, однако увеличивает риск бесплодия в перспективе [8]. Среди консервативных подходов к лечению ММ можно выделить терапию агонистами высвобождающего гонадотропин гормона (GnRH), которые ингибируют синтез стероидов и вызывают химическую менопаузу [9]. Однако применение гормональной терапии ограничено длительностью действия (3–6 мес. после окончания курса) и сопряжено с тяжелыми побочными эффектами, в первую очередь, необратимыми потерями плотности костной ткани [10]. На основании вышесказанного можно сделать вывод о необходимости поиска новых подходов к лечению ММ, не влияющих на овуляцию. Четкая локализованность опухолей ММ и доступность при использовании различных эндоскопических методик позволяет рассматривать их как идеальную мишень для генной терапии с помощью суицидных генов [11].
Актуальной проблемой генной терапии остается проблема эффективной доставки генетических конструкций в клетки и ткани организма. Один из возможных подходов для ее решения ‒ разработка синтетических носителей на основе поли- и олигопептидов. К преимуществам этих соединений относят биодеградируемость и возможность модификации структуры и аминокислотного состава носителя [12, 13]. Например, включение в состав пептидных носителей остатков гистидина позволяет обеспечивать выход комплексов из эндосом [14]. В ряде работ в качестве средств доставки ДНК использованы пептиды, содержащие остатки аргинина [15–20]. Эффективность этих Arg-богатых носителей связывают с наличием в их составе гуанидиновых групп, которые, взаимодействуя с гликозамингликанами клеточной мембраны, повышают эффективность проникновения лекарственного препарата в клетки [21].
Ранее исследовали возможность использования невирусных носителей на основе высокомолекулярных катионных полипептидов в качестве средств доставки нуклеиновых кислот в клетки, но столкнулись с их высокой токсичностью [22]. Что касается низкомолекулярных катионных олигопептидов, то они не способны образовывать стабильные комплексы и эффективно защищать ДНК от деградации [23]. Для решения проблемы стабильности комплексов низкомолекулярных пептидов с ДНК используют метод перекрестных сшивок между молекулами носителя с образованием соединения с большей молекулярной массой. Так, при наличии остатков цистеина, фланкирующих последовательность низкомолекулярных пептидов, возможно образование дисульфидных связей между молекулами пептидов с образованием полимера. Сшивающиеся за счет остатков Cys пептиды способны формировать стабильные комплексы с ДНК [24]. После попадания в клетку происходит деполимеризация носителя за счет разрушения дисульфидных связей глутатионом и высвобождение ДНК из состава комплекса, при этом токсичность соединения снижается до уровня низкомолекулярных пептидов [25, 26]. Сейчас для формирования комплексов Cys-фланкированных пептидов с ДНК используют два подхода. Первый ‒ матричная полимеризация. В этом случае дисульфидные связи между пептидами образуются во время формирования комплекса с матрицей ДНК. Второй ‒ окислительная поликонденсация. Здесь полимеризацию пептидов проводят до компактизации ДНК ‒ путем образования межпептидных дисульфидных связей в присутствии хаотропного агента диметилсульфоксида (DMSO) [17, 27–29].
В работе исследованы комплексы ДНК с Arg-богатыми пептидными носителями, которые получены двумя различными способами полимеризации за счет образования межпептидных цистиновых связей. Изучены физико-химические свойства полученных комплексов (размер, эффективность комплексообразования), их транспорт внутрь клетки, токсичность и трансфекционная эффективность in vitro. Проведено моделирование суицидной генной терапии ММ путем переноса в первичные клетки, полученные из миоматозных узлов, гена тимидинкиназы вируса простого герпеса типа 1 (HSV-1) с последующей оценкой их пролиферативной активности после обработки ганцикловиром.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Получение пептидных носителей. Синтез пептида R6 (аминокислотная последовательность Cys-His-Arg6-His-Cys) проведен в ООО “НПФ Верта” (Санкт-Петербург) методом твердофазного пептидного Вос-синтеза. Степень очистки пептидов, по результатам ВЭЖХ, составляла 90‒95%. В качестве контрольного носителя использовали разветвленный полиэтиленимин (ПЭИ) с молекулярной массой 25 кДа (“Sigma Aldrich”, США). Неполимеризованный пептид R6 растворяли в 0.1%-ной трифторуксусной кислоте для предотвращения образования дисульфидных связей и использовали для последующей полимеризации. Пептидный носитель получали двумя способами: 1) методом матричной полимеризации, где межпептидные цистиновые сшивки образуются в процессе комплексообразования R6 с матрицей ДНК (рН 7.5); и 2) окислительной поликонденсацией, которую проводили в 30%-ном растворе DMSO, содержащем 30 мМ пептид R6 и 33 мол. % 2-аминоэтантиола (АЭТ), в течение 96 ч при доступе кислорода воздуха (полученный продукт далее обозначен как PolyR6) [17, 29].
Плазмидные конструкции. В работе использовали следующие плазмидные ДНК: 1) конструкция pCMV-nls-lacZ, содержащая маркерный ген бактериальной β-галактозидазы Escherichia coli, модифицированной сигналом ядерной транслокации (предоставлена Prof. B. Scholte, Erazmus University Rotterdam, Нидерланды); 2) конструкция pEXPR-IBA5-eGFP, содержащая маркерный ген зеленого флуоресцентного белка GFP (“Iba GmbH”, Германия); 3) конструкция pPTK1, содержащая ген тимидинкиназы HSV-1 (предоставлена к.б.н., с.н.с. С.В. Орловым, Институт экспериментальной медицины, Санкт-Петербург, Россия). Препаративное выделение плазмидных ДНК проводили по стандартной методике щелочного лизиса с последующей двукратной очисткой ДНК в градиенте плотности хлорида цезия [30].
Формирование комплексов ДНК/носитель. К серии проб, содержащих различные количества носителей R6 или polyR6, растворенных в 20 мкл буфера HBM (0.27 М маннитол, 5 мM HEPES, pH 7.5), в соответствии с зарядовыми соотношениями ДНК/носитель (соотношение суммарных зарядов отрицательно заряженных групп ДНК и положительно заряженных групп носителя, которую рассчитывали по концентрации свободных аминогрупп и гуанидиновых групп пептида R6, равной 4.18 мкM), добавляли 1 мкг плазмидной ДНК в 20 мкл буфера HBM и инкубировали 30 мин. Образование комплексов анализировали методом вытеснения бромистого этидия [31]. В качестве контроля использовали комплексы ДНК с ПЭИ (рН 7.5) с зарядовым соотношением 1/8, сформированные в НВМ. Для этого к 40 мкл раствора, содержащего 1.1 мкг ПЭИ, добавляли равный объем раствора, содержащего 1 мкг ДНК, и инкубировали в течение 30 мин.
Анализ образования комплексов ДНК/носитель методом вытеснения бромистого этидия [31]. Анализ проводили в 96-луночных планшетах с плоским дном (ОАО “Медполимер”, Россия). До формирования комплексов в каждую лунку планшета вносили 20 мкл буфера НВМ, содержащего 0.5 мкг ДНК и 400 нг/мл бромистого этидия, после чего добавляли 20 мкл раствора носителя в соответствии с зарядовыми соотношениями ДНК/носитель. Планшет с продуктом окислительной поликонденсации ‒ polyR6 ‒ оставляли в течение 30 мин при комнатной температуре в защищенном от света месте для окончательного формирования комплекса носителя с ДНК. Планшет с R6 оставляли при комнатной температуре в течение 2 ч в защищенном от света месте для образования дисульфидных связей на матрице ДНК. После инкубации вносили в каждую лунку воду до конечного объема 100 мкл. Измерение интенсивности флуоресценции проводили на флуориметре Wallac 1420D (“ThermoFisher Scientific Oy”, Финляндия) при длинах волн 540/590 нм и времени экспозиции 5 с. Для каждого соотношения ДНК/носитель измерение интенсивности флуоресценции (F) проводили в трех повторах. Эффективность вытеснения бромистого этидия рассчитывали по формуле: (F ‒ Ff)/(Fb ‒ Ff), где Ff и Fb – интенсивность флуоресценции бромистого этидия соответственно в отсутствие и в присутствии ДНК.
Определение размера и дзета-потенциала комплексов ДНК/носитель. Для исследования размера и дзета-потенциала комплексов ДНК/носитель с зарядовым соотношением 1/8 брали образцы, содержащие 5 мкг ДНК. Размер комплексов определяли методом динамического рассеяния света, а дзета-потенциал ‒ методом микроэлектрофореза. Измерения проводили на приборе Zetasizer Nano ZS (“Malvern Instruments”, Великобритания).
Клеточные культуры. Эксперименты по трансфекции in vitro проводили на клетках карциномы поджелудочной железы человека PANC-1 и клетках почки эмбриона человека HEK-293T из коллекции клеточных культур Института цитологии РАН (Санкт-Петербург, Россия), а также на первичных клетках ММ, полученных из миоматозных узлов в НИИ АГиР им. Д.О. Отта. Образцы миоматозных узлов были получены при миомэктомии от трех пациенток с ММ. Исследование одобрено этическим комитетом НИИ АГиР им. Д.О. Отта и выполнено с добровольного информированного согласия пациенток.
Образцы узлов помещали в физиологический раствор, измельчали на фрагменты диаметром около 3 мм, обрабатывали коллагеназой IV типа (“Sigma Aldrich”, США) (концентрация 200 ед./мл) в питательной среде Игла MEM (“Биолот”, Россия) и инкубировали в течение 90‒120 мин при 37°С, после чего фрагменты ткани ресуспендировали. К полученной суспензии клеток добавляли питательную среду AmnioMax Basal Medium с 10% сыворотки AmnioMax Supplement (“Sigma Aldrich”) и вносили в культуральные флаконы; после первого пассажа среду меняли на DMEM-F12 с 10% эмбриональной сыворотки крупного рогатого скота (FBS; “Sigma Aldrich”) и продолжали культивирование при 37°С и 5% CO2 в течение 2‒6 недель.
Анализ цитотоксичности комплексов ДНК/носитель. Токсические свойства комплексов носитель/ДНК исследовали с использованием Alamar Blue (“BioSources International”, США). Клетки PANC-1 рассевали на культуральные 96-луночные планшеты (“Nunc”, Дания) из расчета 1.6 × × 104 клеток в лунку, содержащую 175 мкл стандартной культуральной смеси, состоящей из среды DMEM, 10% FBS (“Биолот”) и 0.01 мкг/мл гентамицина, и культивировали в течение 24 ч, после чего промывали средой DMEM и вносили 175 мкл среды и затем суспензию комплексов ДНК/носитель из расчета 0.7 мкг ДНК на лунку. Планшет помещали в инкубатор с температурой 37°C и 5% СО2 на 4 ч, после чего вносили в каждую лунку по 175 мкл стандартной культуральной смеси, содержащей 10% раствора Alamar Blue, и продолжали инкубацию в тех же условиях в течение 16 ч. Интенсивность флуоресценции измеряли на флуориметре Wallac 1420D при длинах волн 530/590 нм. Относительное содержание клеток после добавления комплексов рассчитывали по формуле: [(F ‒ Ff)/(Fb ‒ Ff)] × 100%, где Fb ‒ интенсивность флуоресценции красителя в отсутствие комплексов ДНК/носитель и Ff – интенсивность флуоресценции красителя Alamar Blue в отсутствие клеток.
Проникновение комплексов ДНК/носитель в клетки млекопитающих in vitro. В образец плазмидной ДНК добавляли интеркалирующий краситель YOYO-1 (“Invitrogen”) из расчета 1 молекула красителя на 50 п.н. ДНК и инкубировали смесь 1 ч, после чего вносили раствор носителя для достижения зарядового соотношения ДНК/носитель 1/8. Клетки PANC-1 рассевали на культуральные 48‑луночные планшеты (“Nunc”) из расчета 8.0 × 104 клеток на лунку, содержащую 500 мкл стандартной культуральной смеси, и культивировали в течение 24 ч. Перед трансфекцией среду меняли на не содержащую FBS и добавляли комплексы, содержащие 2 мкг ДНК и растворенные в 80 мкл HBM. Эту процедуру и все последующие проводили в темноте. Планшеты с клетками помещали в термостат с температурой 37°С и 5% СО2 на 2 ч, после чего дважды промывали PBS. Клетки снимали с планшета раствором 0.25%-ного трипсина с 1 мМ EDTA, осаждали центрифугированием при 100 g и ресуспендировали в 100 мкл PBS. В каждую пробирку добавляли по 5 мкл раствора пропидия йодида (PI; 50 мкг/мл PBS, рН 7.4) и инкубировали при 4°C в течение 5 мин. Флуоресценцию 1 × 104 клеток анализировали на проточном цитофлуориметре BD FACS-Canto II (“Becton-Dickinson Biosciences”, США) при длине волны возбуждения 488 нм.
Трансфекция клеток человека комплексами ДНК/носитель. Клетки PANC-1 рассевали на культуральные 48-луночные планшеты (“Nunc”) из расчета 6 × 104 клеток на лунку, содержащую 500 мкл стандартной культуральной смеси, культивировали в течение 24 ч, промывали средой DMEM и вносили 500 мкл среды DMEM без FBS в каждую лунку. Комплексы ДНК/носитель в буфере HBM вносили из расчета 2 мкг ДНК на лунку. Планшет помещали на 4 ч в термостат с температурой 37°С и 5% СО2, промывали средой DMEM, вносили в каждую лунку по 500 мкл стандартной культуральной смеси, инкубировали при температуре 37°С и 5% СО2 в течение 48 ч и анализировали экспрессию репортерного гена.
Анализ экспрессии репортерного гена lacZ. После удаления культуральной смеси планшет дважды промывали PBS и в каждую лунку вносили 80 мкл полного буфера для лизиса, который готовили непосредственно перед использованием (в 10 мл неполного 1× буфера (pH 7.8), содержащего 25 мM глицилглицин, 15 мM MgSO4, 4 мM EGTA (этиленгликольтетрауксусная кислота), 1% Тритон X-100, 1 мМ дитиотреитол (DTT) и 1 мМ фенилметилсульфонилфторид (PMSF)). Лизис вели при ‒70°С в течение не менее 1 ч, после чего из каждой лунки отбирали 50 мкл лизата, вносили в 96-луночный планшет и добавляли 50 мкл буфера, содержащего 200 мM натрий-фосфатный буфер (pH 7.3) и 2 мM MgCl2, с 1 мг/мл 4-метилумбеллиферил-β-D-галактопиранозида (MUG; “Sigma-Aldrich”), используемого в качестве субстрата β-галактозидазы. Реакционную смесь инкубировали в течение 1.5 ч при 37°С и затем останавливали реакцию, внося в каждую лунку 100 мкл стоп-реагента (0.2 M глицин, pH 10.0). Интенсивность флуоресценции измеряли на флуориметре Wallac 1420D при длине волны излучения 355 нм и возбуждения 460 нм и времени экспозиции 0.1 с. В качестве контроля использовали фермент β-галактозидазу с известной активностью, по которой строили калибровочную кривую зависимости интенсивности флуоресценции от активности фермента.
Концентрацию белка в лунке определяли методом Бредфорд с модификациями [32]. Для этого лизат разводили в 100 раз (до конечной концентрации тритона Х-100 0.01%) и переносили 20 мкл разведенного лизата в лунки 96-луночного планшета, добавляли 180 мкл реагента Бредфорд следующего состава (на 1 л водного раствора): 100 мг кумасси G-250, 50 мл 95%-ного этанола, 100 мл 85%-ной фосфорной кислоты. Калибровочную кривую строили по растворам BSA (“Sigma-Aldrich”) с разной концентрацией. В качестве отрицательного контроля использовали 20 мкл 0.01× буфера для лизиса. Измерения проводили на спектрофотометре Multiscan plus P (“Labsystems”, Финляндия). По калибровочной кривой зависимости оптической плотности от концентрации BSA рассчитывали концентрацию белка в анализируемых образцах. Активность β-галактозидазы оценивали в единицах активности (mU) на 1 мг белка в каждой лунке.
Анализ экспрессии репортерного гена gfp. Из лунок планшета удаляли культуральную смесь, после чего промывали лунки PBS. Клетки снимали с планшета путем добавления в каждую лунку по 80 мкл раствора трипсин‒версен (1 : 3) с последующей инкубацией в течение 10 мин при температуре 37°С. Клетки суспендировали в 300 мкл PBS. Количество GFP-положительных клеток определяли методом проточной цитофлуориметрии на приборе FACS Canto II (“Becton Dickinson”, США).
Анализ пролиферативной активности клеток миомы матки после суицидной генной терапии. Клетки, которые были получены из одного миоматозного узла, распределяли по 3 флаконам для одной серии экспериментов. Данные, полученные из каждой серии экспериментов, считали одним повтором (n). В каждом исследовании проведено три‒пять независимых серий экспериментов с клетками из разных миоматозных узлов. Первичные клетки ММ рассевали на культуральные 96-луночные планшеты (“Nunc”) из расчета 1.5 × × 104 клеток на лунку, содержащую 200 мкл стандартной культуральной смеси, и культивировали в течение 24 ч, после чего планшет промывали средой DMEM-F12. В лунки вносили по 200 мкл среды и суспензию комплекса ДНК/носитель из расчета 0.35 мкг ДНК (плазмида pPTK1) на лунку. Планшеты инкубировали в термостате при 37°С и 5% СО2 в течение 2 ч, промывали средой DMEM, вносили в каждую лунку по 200 мкл стандартной культуральной смеси и вновь помещали в термостат на 24 ч. Среду меняли на 200 мкл стандартной культуральной смеси с 50 мкг/мл ганцикловира и оставляли планшет в термостате на 96 ч, после чего среду меняли на 100 мкл стандартной культуральной смеси, содержащей 10% раствора Alamar Blue, и инкубировали планшет еще 2 ч. Интенсивность флуоресценции измеряли на флуориметре Wallac 1420D при длинах волн 530/590 нм. Количество живых клеток после добавления комплексов рассчитывали по формуле (F ‒ Ff)/(Fb ‒ Ff), где Fb – интенсивность флуоресценции красителя в отсутствие комплексов ДНК/носитель, а Ff – интенсивность флуоресценции красителя Alamar Blue в отсутствие клеток. Фотографии клеток получали на микроскопе AxioObserver Z1 (“Carl Zeiss”, Германия) с использованием программы AxioVision при 100-кратном увеличении.
Масс-спектрометрический анализ. Для определения молекулярной массы носителя polyR6 образцы после окислительной поликонденсации анализировали на тандемном времяпролетном масс-спектрометре AB Sciex 5800 TOF/TOF (“AB Sciex”, США) в линейном режиме (linear mode), в качестве вещества матрицы использовали синапиновую кислоту.
Статистические методы. Результаты экспериментов представлены с отображением стандартной ошибки среднего (±SEM) или стандартного отклонения (±SD). Достоверность различий между контролем и опытом, а также между опытами оценивали при помощи t-критерия Стьюдента и U-критерия Манна-Уитни с использованием статистической программы InStat3 (“GraphPad Software”, США).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Анализ конденсации плазмидной ДНК в составе комплексов с носителями
Степень конденсации ДНК в комплексе с Cys-фланкированными Arg-богатыми носителями оценивали методом вытеснения бромистого этидия из комплексов. В каждом образце измеряли флуоресценцию интеркалирующего агента. Свечение бромистого этидия с плазмидной ДНК без носителя принимали за 100%. Выявлено, что с увеличением концентрации носителей происходит гашение флуоресценции в связи с тем, что при повышении плотности формирующихся комплексов снижается доступ молекул красителя к молекулам ДНК. Показано, что при образовании комплекса ДНК с носителями R6 и polyR6 падение флуоресценции происходит при одинаковом зарядовом соотношении ДНК/носитель, равном 1/1.5 (рис. 1). Выход кривых на плато означал, что комплексы достигали плотности, достаточной для полного гашения флуоресценции бромистого этидия.
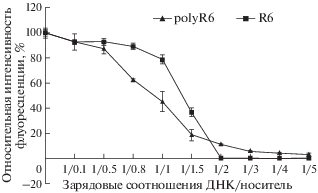
Определение размера и дзета-потенциала комплексов ДНК с носителями
Размер комплексов ДНК/R6 и ДНК/polyR6 с зарядовым соотношением 1/8 определяли методом динамического светорассеяния (табл. 1). Размеры комплексов носителей с ДНК варьировали в диапазоне 88–100 нм и составляли в среднем 98.50 ± 0.89 нм для ДНК/R6 и 89.04 ± 0.35 нм для ДНК/polyR6.
Дзета-потенциал комплексов ДНК/носитель с зарядовым соотношением 1/8 измеряли методом микроэлектрофореза. Как видно из табл. 1, дзета-потенциалы комплексов ДНК/R6 и ДНК/polyR6 отличались: 18.7 ± 0.4 и 32.5 ± 0.6 мВ соответственно.
Определение молекулярной массы носителя, образованного в результате окислительной поликонденсации
Молекулярную массу носителя polyR6, образованного в результате окислительной поликонденсации, определяли методом масс-спектрометрии MALDI-TOF. Обнаружено, что в результате окислительной поликонденсации образуется смесь полимеров с молекулярной массой 2.9‒23.0 кДа (табл. 2), что соответствует полимерам со степенью полимеризации мономеров от 2 до 16, как правило, полностью модифицированных АЭТ. Только в случае димеров и тримеров наблюдали отсутствие или частичную АЭТ-модификацию.
Таблица 2.
Определение молекулярной массы носителя polyR6 масс-спектрометрией MALDI-TOF
| Молекулярная масса, Да | Степень полимеризации | |
|---|---|---|
| теорети-ческая | экспериментальная | |
| 2871.44 | 2865.96 | (R6)2 |
| 2948.59 | 2943.59 | АЭТ-(R6)2 |
| 3025.74 | 3018.79 | АЭТ-(R6)2-АЭТ |
| 4307.16 | 4297.97 | (R6)3 |
| 4384.31 | Не обнаружен | АЭТ-(R6)3 |
| 4461.46 | 4450.28 | АЭТ-(R6)3-АЭТ |
| 5897.18 | 5881.75 | АЭТ-(R6)4-АЭТ |
| 7332.90 | Не обнаружен | АЭТ-(R6)5-АЭТ |
| 8768.62 | 8715.18 | АЭТ-(R6)6-АЭТ |
| 10 204.34 | 10 907.67 | АЭТ-(R6)7-АЭТ |
| 11 640.06 | Не обнаружен | АЭТ-(R6)8-АЭТ |
| 13 075.78 | 13 283.00 | АЭТ-(R6)9-АЭТ |
| 14 511.50 | Не обнаружен | АЭТ-(R6)10-АЭТ |
| 15 947.22 | 15 822.83 | АЭТ-(R6)11-АЭТ |
| 17 382.94 | 16 833.21 | АЭТ-(R6)12-АЭТ |
| 18 818.66 | 18 302.42 | АЭТ-(R6)13-АЭТ |
| 20 254,38 | Не обнаружен | АЭТ-(R6)14-АЭТ |
| 21 690.10 | 21 109.40 | АЭТ-(R6)15-АЭТ |
| 23 125.82 | 22 725.81 | АЭТ-(R6)16-АЭТ |
Анализ цитотоксичности комплексов ДНК/носитель
Цитотоксичность комплексов ДНК/носитель исследовали на клетках PANC-1 с использованием флуоресцентного красителя Alamar Blue (резазурин). Клетки PANC-1 трансфицировали комплексами носителей R6 и polyR6 с плазмидной ДНК, содержащей ген β-галактозидазы. Использовали комплексы ДНК/носитель с зарядовыми соотношениями 1/8 и 1/12. К клеткам добавляли краситель Alamar Blue и измеряли интенсивность флуоресценции. За 100% принимали интенсивность флуоресценции красителя Alamar Blue, добавленного к интактным клеткам. В качестве контроля использовали некомпактизованную в носитель ДНК, а также комплексы ДНК/ПЭИ при зарядовом соотношении 1/8 (рис. 2).
Рис. 2.
Выживаемость клеток PANC-1 после трансфекции плазмидой pCMV-nls-lacZ в комплексе c носителями R6 или polyR6 при зарядовых соотношениях 1/8 и 1/12. Относительное количество живых клеток определяли по флуоресценции реагента Alamar Blue. Результаты представлены как среднее ± SD.
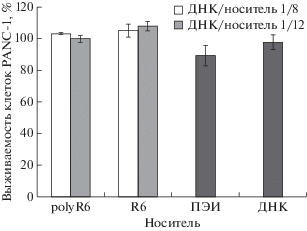
Показано, что комплексы пептидных носителей с ДНК не токсичны для клеток PANC-1 ‒ обработка комплексами не приводила к значимому изменению их количества, как в сравнении с клетками, обработанными некомпактизованной ДНК, так и с клетками без воздействия. Как видно из рис. 2, ПЭИ оказался более токсичным для клеток PANC-1, чем комплексы ДНК с носителями.
Анализ эффективности проникновения комплексов ДНК/носитель в клетки
Способность проникать в клетки исследовали с использованием меченных красителем YOYO-1 комплексов ДНК/носитель с зарядовым соотношением 1/8. Анализ проводили методом проточной цитофлуорометрии на клетках PANC-1 и HEK-293T. Жизнеспособность исходных клеток оценивали по негативной реакции с PI. Эффективность проникновения комплексов с пептидными носителями оценивали по интенсивности флуоресценции в образце, содержащем 5 × 103 клеток. В качестве контроля эффективности проникновения использовали комплексы ДНК с ПЭИ (рис. 3).
Рис. 3.
Эффективность проникновения в клетки комплексов ДНК/носитель. Интенсивность флуоресценции клеток PANC-1 и HEK-293T после трансфекции плазмидной ДНК, меченной интеркалирующим красителем YOYO-1, в комплексе с носителями R6 или polyR6 при зарядовом соотношении ДНК/носитель 1/8. Результаты представлены как среднее ± SEM. **p < 0.01 при сравнении с R6.
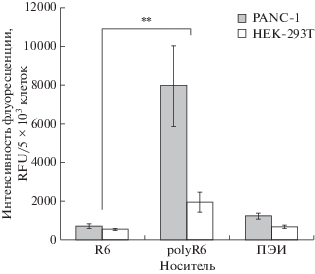
На основании полученных результатов можно предполагать, что эффективность проникновения в клетки комплексов ДНК/носитель зависит от способа полимеризации носителя. Так, в клетки PANC-1 комплексы ДНК/polyR6 проникали на порядок эффективнее, чем ДНК/R6, хотя для клеток HEK-293T эта разница была меньше. Клетки PANC-1 и HEK-293T при трансфекции комплексом ДНК/polyR6 давали сигнал флуоресценции 7 968 ± 2 081 RFU и 1 954 ± 517 RFU соответственно. В случае комплекса ДНК/R6 сигнал был гораздо ниже: 720 ± 125 RFU для клеток PANC-1 и 541 ± 51 RFU для клеток HEK-293T. Кроме того, комплекс ДНК/polyR6 проникал в клетки PANC-1 достоверно эффективнее, чем контрольный комплекс ДНК/ПЭИ: 1247 ± 155 RFU для PANC-1 и 693 ± 84 RFU для HEK-293T (p < 0.05).
Эффективность трансфекции комплексами ДНК/носитель in vitro
Анализ трансфекционной активности проводили на клетках PANC-1 и HEK-293T и комплексах плазмидной ДНК pCMV-nls-lacZ с носителями R6 и polyR6 с двумя зарядовыми соотношениями: 1/8 и 1/12 (рис. 4а, б). В качестве положительного контроля трансфекции использовали плазмидную ДНК, упакованную носителем ПЭИ при зарядовом соотношении 1/8. В качестве отрицательного контроля использовали плазмиду без носителя. В каждом образце измеряли активность фермента β-галактозидазы и нормализовали ее к общему количеству белка в лунке. При трансфекции клеток комплексами ДНК/R6, сформированными в результате матричной полимеризации, эффективность трансфекции была выше при зарядовом соотношении 1/8, чем 1/12: на клетках PANC-1 активность β-галактозидазы составила 325 ± 56 и 63 ± 17 mU/мг белка, а на клетках HEK-293T ‒ 1239 ± 49 и 285 ± 3 mU/мг белка соответственно. Активность β-галактозидазы после трансфекции клеток PANC-1 комплексами ДНК/polyR6 при соотношении 1/8 была на уровне 2405 ± 377 mU/мг белка, тогда как в клетках HEK-293T она была ниже: 1 499 ± ± 82 mU/мг белка. Самая высокая ферментативная активность после проведения трансфекции зарегистрирована для комплексов ДНК/polyR6 при зарядовом соотношении 1/12 и составила 3517 ± 614 mU/мг белка в клетках PANC-1 и 2407 ± ± 327 mU/мг белка в клетках HEK-293T. Стоит отметить, что эффективность трансфекции обеих линий клеток комплексами, сформированными с ДНК полимерами, полученными окислительной поликонденсацией, была сравнима или выше таковой для комплексов ДНК с ПЭИ (2034 ± 146 mU/мг белка в клетках PANC-1 и 2033 ± 334 mU/мг белка в клетках HEK-293T). На основании полученных результатов можно утверждать, что трансфекционная активность комплексов ДНК/polyR6 выше, чем комплексов, образованных в результате матричной полимеризации (p < 0.001).
Рис. 4.
Трансфекционная активность комплексов ДНК/носитель. Активность β-галактозидазы в клетках PANC-1 (а) и HEK293T (б) после трансфекции плазмидой pCMV-nls-lacZ в комплексе с R6 и polyR6 при зарядовых соотношениях 1/8 и 1/12. Активность β-галактозидазы выражена в условных единицах активности на 1 мг общего белка (mU/мг белка). в ‒ Относительное количество GFP-положительных клеток PANC-1 (%) после трансфекции плазмидой pEXPR-IBA5-eGFP в комплексе c R6 и polyR6 при зарядовых соотношениях 1/8. Результаты представлены как среднее ± SEM. *p < 0.05, ***p < 0.001 по сравнению с (а, б) активностью β-галактозидазы и (в) относительным количеством GFP-положительных клеток после трансфекции комплексами ДНК/R6.
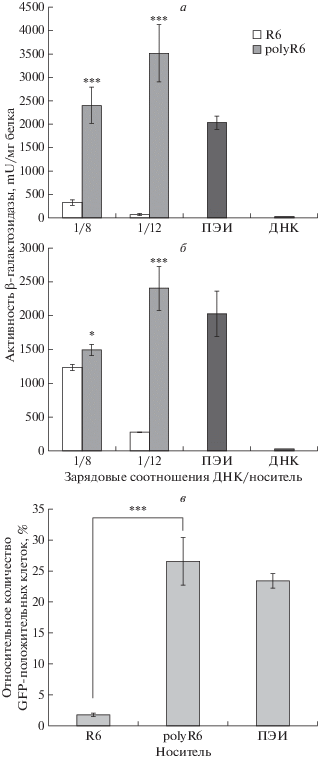
Далее был проведен ряд экспериментов по трансфекции клеток линии PANC-1 комплексами плазмиды pEXPR-IBA5-eGFP, содержащей ген белка GFP, с носителями R6 и polyR6 при зарядовом соотношении 1/8 (рис. 4в). В результате показано, что комплексы ДНК/R6 трансфицировали 1.8% клеток PANC-1, а ДНК/polyR6 ‒ 28%. Для комплексов ДНК/ПЭИ зарегистрировано около 23% GFP-положительных клеток. Таким образом, результаты, полученные с использованием плазмиды pEXPR-IBA5-eGFP и цитологического метода анализа, согласуются с данными, полученными биохимическим методом с использованием плазмиды pCMV-nls-lacZ; и на этом основании можно говорить о высокой эффективности комплексов ДНК с носителем polyR6, сформированным в результате окислительной поликонденсации.
Пролиферативная активность клеток миомы матки после проведения суицидной генной терапии
Для проведения суицидной генной терапии была получена первичная культура клеток из миоматозных узлов пациенток с ММ после миомэктомии. Для экспериментов использовали культуры клеток 2 и 3 пассажей. Для трансфекции использована плазмидная ДНК рРТК1, содержащая ген тимидинкиназы HSV-1. Комплексы ДНК/R6 и ДНК/polyR6 были сформированы при зарядовых соотношениях 1/8 и 1/12. В качестве контрольных использовали комплексы с плазмидной ДНК, содержащей только репортерный ген lacZ. После трансфекции клеток ММ и последующей обработки ганцикловиром оценивали их пролиферативную активность с помощью красителя Alamar Blue. Количество необработанных клеток принимали за 100% (рис. 5). Показано, что обработка клеток только ганцикловиром (контроль) не приводит к значимому снижению их количества ‒ сохраняется на уровне 93%. Трансфекция клеток ММ комплексами плазмиды pCMV-nls-lacZ с носителями приводила к небольшому снижению их пролиферации относительно необработанных клеток, что может быть обусловлено слабым токсическим действием комплексов на эти клетки. В то же время токсичность изучаемых комплексов (23‒34%) не превышала таковую для комплексов с коммерческим носителем ПЭИ (38%).
Рис. 5.
Выживаемость первичных клеток миомы матки после трансфекции плазмидой pPTK1 (tk) или pCMV-nls-lacZ (lacZ) в комплексе c носителями R6 или polyR6 при зарядовых соотношениях 1/8 и 1/12 и последующей обработки ганцикловиром. Относительное количество клеток определяли с помощью реагента Alamar Blue. Результаты представлены как среднее ± SD. *p < 0.05, **p < 0.01 при сравнении с комплексами, несущими плазмиду pCMV-nls-lacZ. Контроль ‒ клетки, обработанные только ганцикловиром.
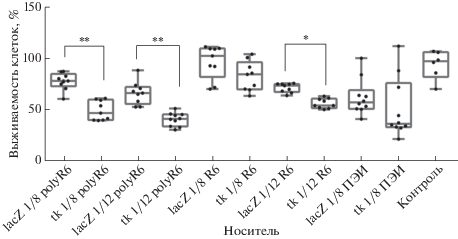
При использовании плазмиды рРТК1, содержащей в своем составе ген тимидинкиназы HSV-1, наблюдали достоверное снижение количества живых клеток в отличие от использования плазмиды pCMV-nls-lacZ, несущей репортерный ген lacZ. Доставка комплексов рРТК1 с носителем R6 при зарядовых соотношениях ДНК/носитель 1/8 и 1/12 приводила к снижению количества клеток соответственно на 15 и 21%, а с носителем polyR6 ‒ на 39 и 40%. Стоит отметить, что при использовании контрольного носителя ПЭИ доставка терапевтической плазмиды приводила к снижению количества живых клеток только на 19% по сравнению с контрольными комплексами с геном lacZ. Таким образом, после доставки в первичные клетки ММ плазмиды рРТК1, содержащей ген тимидинкиназы HSV-1, в составе комплекса с носителем polyR6 наблюдали выраженный эффект суицидной генной терапии in vitro.
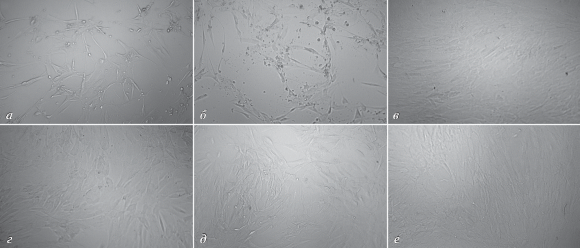
На рис. 6 представлены микрофотографии клеток ММ, обработанных ганцикловиром после трансфекции контрольными и терапевтическими плазмидами в комплексе с носителем polyR6 ‒ как более эффективным, чем R6. Из представленных фотографий видно, что трансфекция терапевтическими комплексами приводила к значительному снижению количества клеток ММ по сравнению с нетрансфицированными клетками и трансфицированными контрольными комплексами.
Рис. 6.
Микрофотографии первичных клеток миомы матки, обработанных ганцикловиром после трансфекции терапевтическими и контрольными комплексами: а ‒ pPTK1/polyR6 (1/8); б ‒ pPTK1/polyR6 (1/12); в – интактные клетки (без обработки ганцикловиром); г – pCMV-nls-lacZ/polyR6 (1/8); д – pCMV-nls-lacZ/polyR6 (1/12); е – клетки, обработанные только ганцикловиром. Увеличение 100×.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Проблема эффективной доставки генетических конструкций в клетки и ткани организма не потеряла актуальности в технологии генной терапии. Одним из путей ее решения может быть создание Cys-фланкированных Arg-богатых пептидных носителей, способных обеспечить стабильность ДНК-комплексов за счет образования межпептидных дисульфидных связей. Первоначально для образования перекрестных сшивок между молекулами носителя использовали глутаральдегид, который повышал стабильность комплексов ДНК с пептидами как in vitro, так и in vivo [33]. Основным недостатком такой модификации была низкая эффективность высвобождения ДНК из состава комплексов, что в свою очередь отрицательно влияло на уровень экспрессии целевого гена.
Ранее нами разработан Cys-фланкированный пептидный носитель R6. На основании результатов собственных исследований, а также литературных данных в его состав был включен остаток His для обеспечения эндосомолитических свойств [17, 34, 35], а также остатки Arg, что было продиктовано данными о роли этой аминокислоты в природных пептидах-пенетратинах, в частности в ТАТ-пептидах [36], а также о повышенной способности олигоаргининов, по сравнению с олиголизинами, проникать в клетки [37, 38]. Дополнительным преимуществом разработанного пептидного носителя была возможность модификации его состава. Это может быть присоединение лигандов для направленной доставки, сигналов ядерной локализации и др. [18, 19]. Кроме того, показана перспективность использования пептидного носителя R6 для доставки малых интерферирующих РНК in vitro и in vivo [3, 39, 40].
Ранее было высказано предположение, что различные типы полимеризации Cys-фланкированных пептидов оказывают влияние на их способность проникать в клетку и способствовать трансфекции ДНК [27]. К настоящему моменту уже исследованы ДНК-пептидные и РНК-пептидные комплексы, сформированные в результате матричной полимеризации [25, 39, 41–43] или после окислительной поликонденсации Cys-фланкированных пептидов [26, 29, 44, 45]. В ряде работ проведено сравнение биологических свойств носителей, образованных этими двумя способами полимеризации [17, 27, 46, 47].
В проведенном нами исследовании для матричной полимеризации пептид R6 растворяли в трифторуксусной кислоте ‒ с целью сохранения свободных тиольных групп Cys до их “использования” при полимеризации R6 (за счет образования межпептидных дисульфидных связей) на матрице ДНК. Синтез носителя polyR6 проводили до образования комплексов с ДНК: окислительную поликонденсацию пептида R6 вели в присутствии прерывателя роста цепи АЭТ. Использование АЭТ позволяет контролировать реакцию поликонденсации и, следовательно, молекулярную массу полимеров [27, 29], а также во время полимеризации присоединять к концам полимера тиолсодержащие молекулы лигандов, флуоресцентных меток и т.д. По результатам масс-спектрометрического анализа продуктов реакции окислительной поликонденсации использование АЭТ позволило ограничить молекулярную массу полимеров 23 кДа (табл. 2). Parker c соавт. [24] сообщали о получении полимерных носителей сходной молекулярной массы в результате окислительной поликонденсации пептида Cys-Lys16-Cys. Следует отметить, что в отсутствие прерывателя окислительной поликонденсации молекулярная масса полимеров может превышать 180 кДа, что негативно отражается на токсических свойствах Cys-фланкированных пептидных носителей [29].
В результате исследования ДНК-компактизующих свойств носителей R6 и polyR6 показано, что эти соединения обеспечивают конденсацию плазмиды при зарядовом соотношении ДНК/носитель 1/2 (рис. 1). Таким образом, способ полимеризации пептида не влияет на ДНК-конденсирующие свойства получаемых носителей.
Одним из ключевых параметров, влияющих на трансфекционную активность комплексов ДНК с невирусными носителями, считается их размер. Этот параметр в значительной степени влияет на механизм проникновения комплексов в клетку. Согласно литературным данным, размер частиц, способных проникать в клетки по пути клатринзависимого эндоцитоза, составляет 100‒200 нм, в то время как более крупные частицы проникают в клетку путем макропиноцитоза [48]. Если размер комплексов катионных полимеров с ДНК не превышает 100 нм, комплексы могут входить в клетку с помощью кавеолинопосредованного эндоцитоза (потоцитоза) [49]. Ранее, при исследовании комплексов ДНК с невирусными носителями, показали, что наибольшей продолжительностью циркуляции в кровотоке и наибольшей эффективностью проникновения в клетки опухоли обладают частицы размером 70‒200 нм [50]. Размеры полученных нами комплексов ДНК с носителями R6 и polyR6 варьировали в диапазоне 88–100 нм (табл. 1), то есть имели оптимальный размер для проникновения в клетки путем эндоцитоза.
Дзета-потенциал (заряд) комплексов ДНК/носитель также оказывает влияние на их трансфекционную активность. Так, комплексы с положительным дзета-потенциалом трансфицируют клетки эффективнее, чем отрицательно заряженные аналоги [51]. Дзета-потенциал комплексов ДНК с изученными носителями зависел от способа полимеризации пептида. Несмотря на то, что оба типа комплексов были положительно заряженными, дзета-потенциал комплексов ДНК с носителем polyR6 был выше (табл. 1). Известно, что при избытке положительного заряда комплексов увеличивается сила отталкивания между ними, а способность к образованию агрегатов снижается [52]. Таким образом, отсутствие агрегации может быть одной из причин высокой трансфекционной активности комплексов ДНК с носителем polyR6.
К важным критериям эффективности носителя относится его способность доставлять ДНК в клетки, не оказывая на них негативного действия. Известно 3 основных механизма токсического действия поликатионов на клетки: дестабилизация плазматической мембраны, дестабилизиция внутриклеточных мембран (мембраны лизосом, ядерная мембрана), взаимодействие с клеточными белками и нуклеиновыми кислотами [27]. Цитотоксичность комплексов ДНК с носителями определяется такими параметрами, как плотность заряда, молекулярная масса и др. Мы оценивали токсичность комплексов ДНК с самосшивающимися Cys-богатыми пептидами с использованием красителя Alamar Blue (рис. 2). Alamar Blue представляет собой нефлуоресцирующий резазурин, который при проникновении в живые клетки под действием восстановительного потенциала превращается во флуоресцирующий резоруфин. Таким образом, Alamar Blue окрашивает только пролиферирующие клетки. Токсичность исследованных комплексов сравнивали с токсичностью комплексов ДНК/ПЭИ, которая составляла около 10% от общего числа живых клеток. Обнаружено, что относительное количество живых клеток после обработки комплексами ДНК/R6 и ДНК/polyR6 было сравнимо с таковым после внесения в лунку некомпактизованной ДНК. Возможной причиной низкой токсичности исследованных комплексов может быть редукция дисульфидных связей в полимере R6 после проникновения комплексов в клетку. Эта гипотеза согласуется с данными о более низкой токсичности аналогичных носителей по сравнению с поликатионами, не способными к деполимеризации [17, 26, 27].
Эффективность проникновения комплексов ДНК/носитель в клетку изучена нами на клетках культуры PANC-1 (рис. 3). Выявлено, что эффективность проникновения напрямую зависит от способа полимеризации пептидов. Так, комплексы, сформированные после окислительной поликонденсации, проникают в клетки в 10 раз более эффективно, чем комплексы, образованные в результате матричной полимеризации. Следует отметить, что у комплексов ДНК/polyR6 положительный заряд выше, чем у ДНК/R6, поэтому комплексы, образованные в результате окислительной поликонденсации, могут более эффективно взаимодействовать с отрицательно заряженной мембраной клетки. Manickam и соавт. [27], изучая эффективность проникновения в клетку Arg-богатых пептидных носителей, полученных разными способами полимеризации, пришли к выводу, что комплексы с полимерами, образованными поликонденсацией, более эффективны, что может быть обусловлено более сильным взаимодействием Arg-богатых мотивов с поверхностью клетки по сравнению с комплексами, образованными в результате матричной полимеризации. Повышенная проникающая способность комплексов c Arg-содержащими носителями, сформированными в результате окислительной поликонденсации, недавно продемонстрирована и в других работах [46, 53, 54].
На клетках культуры PANC-1 мы сравнили трансфекционную активность комплексов с носителями, полученными разными способами полимеризации (рис. 4). Комплексы с носителями, образованными поликонденсацией, трансфицировали клетки в 7‒50 раз эффективнее, чем с носителями, полученными матричной полимеризацией. Ранее Manickam и др. [27], используя ТАТ-пептид, терминированный остатками Cys, обнаружили, что в большинстве случаев матричная полимеризация и окислительная поликонденвация дают сравнимые по эффективности трансфекции комплексы, хотя в некоторых экспериментах сформированные после поликонденсации комплексы проявляют повышенную трансфекционную активность.
На клетках ММ, полученных от пациенток после миомэктомии, мы смоделировали суицидную генотерапию in vitro. Этот подход активно применяется в генной терапии опухолей [55]. В опухолевые клетки больного доставляют “суицидный” ген, экспрессия которого вызывает гибель клеток (чаще всего это ген тимидинкиназы HSV-1) и обрабатывают клетки ацикловиром или ганцикловиром (аналоги гуанозина). Тимидинкиназа HSV-1 эффективно фосфорилирует эти аналоги нуклеотидов, это приводит к их включению в реплицирующуюся ДНК и, следовательно, к преждевременной терминации репликации, что ведет к гибели клеток. В отличие от тимидинкиназы HSV-1 аналогичный фермент млекопитающих не имеет сродства к таким аналогам гуанозина, как ганцикловир, и поэтому не фосфорилирует их [56]. Таким образом, обработка ацикловиром или ганцикловиром опухолевых клеток, трансфицированных плазмидой с геном тимидинкиназы HSV-1, индуцирует их гибель. Нами показано, что использование носителя polyR6 для доставки плазмиды pPTK-1, содержащей ген тимидинкиназы HSV-1, в клетки ММ приводило к снижению их количества в 1.7 раз по сравнению с контролем (рис. 5). Использование комплексов pPTK-1 с polyR6 было менее эффективным. Следует отметить тенденцию к снижению выживаемости клеток ММ при доставке плазмид, как с геном tk, так и lacZ, с помощью ПЭИ, но достоверной разницы между плазмидами не выявлено. По-видимому, это связано с токсичностью собственно ПЭИ.
Достигнутая в данной работе эффективность трансфекции ДНК с использованием носителя polyR6 сравнима либо превосходит известный коммерческий носитель ПЭИ, который считают “золотым стандартом” невирусных средств доставки [53]. Таким образом, polyR6 можно рассматривать в качестве перспективного невирусного носителя ДНК и основы для создания более совершенных модульных систем доставки. Полученные в ходе работы результаты могут быть использованы в разработках по генной терапии опухолей.
Авторы выражают благодарность к.б.н., с.н.с. А.Г. Миттенбергу (Институт цитологии РАН) за помощь в проведении масс-спектрометрического анализа и аналитику О.В. Лигай (ООО “КД Системы и Оборудование”, Санкт-Петербург) за помощь в определении размеров и дзета-потенциала наночастиц.
Создание клеточной модели миомы матки проведено в соответствии с планами научно-исследовательской работы НИИАГиР им. Д.О. Отта (тема № АААА-А19-119021290033-1). Синтез и исследование пептидных носителей проведено при поддержке гранта РНФ № 19-15-00108. Эксперименты по суицидной генотерапии проведены при поддержке гранта РФФИ № 17-04-01463. Марианна Маретина ‒ стипендиат Совета по грантам Президента РФ (СП-822.2018.4).
Все процедуры, выполненные в данной работе, соответствуют этическим стандартам институционального комитета по исследовательской этике и Хельсинкской декларации 1964 года и ее последующим изменениям или сопоставимым нормам этики.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Егорова А.А., Богачева М.С., Киселев А.В. (2012) Поли- и олигопептидные носители для доставки нуклеиновых кислот в клетки. Мед. генетика. 11(5), 3–14.
Shubina A., Egorova A., Baranov V., Kiselev A. (2013) Recent advances in gene therapy of endometriosis. Recent Pat. DNA Gene Seq. 7, 169–178. https://doi.org/10.2174/18722156113079990021
Egorova A., Petrosyan M., Maretina M., Balashova N., Polyanskih L., Baranov V., Kiselev A. (2018) Anti-angiogenic treatment of endometriosis via anti-VEGFA siRNA delivery by means of peptide-based carrier in a rat subcutaneous model. Gene Ther. 25, 548–555. https://doi.org/10.1038/s41434-018-0042-7
Abdelaziz M., Sherif L., Elkhiary M., Nair S., Shalaby S., Mohamed S., Eziba N., El-Lakany M., Curiel D., Ismail N., Diamond M. P., Al-Hendy A. (2016) Targeted adenoviral vector demonstrates enhanced efficacy for in vivo gene therapy of uterine leiomyoma. Reprod. Sci. 23, 464–474. https://doi.org/10.1177/1933719116630413
Шубина А.Н., Киселев А.В., Егорова А.А., Баранов В.С. (2012) На пути к генной терапии эндометриоза. Журнал акушерства и женских болезней. 61, 148–158.
De Vos S., Wilczynski S.P., Fleischhacker M., Koeffler P. (1994) p53 alterations in uterine leiomyosarcomas versus leiomyomas. Gynecol. Oncol. 54, 205–208. https://doi.org/10.1006/gyno.1994.1194
Bodner K., Bodner-Adler B., Kimberger O., Czerwenka K., Mayerhofer K. (2004) Estrogen and progeste-rone receptor expression in patients with uterine smooth muscle tumors. Fertil. Steril. 81, 1062–1066. https://doi.org/10.1016/j.fertnstert.2003.08.043
Surrey E.S., Lietz A.K., Schoolcraft W.B. (2001) Impact of intramural leiomyomata in patients with a normal endometrial cavity on in vitro fertilization-embryo transfer cycle outcome. Fertil. Steril. 75, 405–410. https://doi.org/10.1016/S0015-0282(00)01714-3
Vercellini P., Trespidi L., Zaina B., Vicentini S., Stellato G., Crosignani P.G. (2003) Gonadotropin-releasing hormone agonist treatment before abdominal myomectomy: a controlled trial. Fertil. Steril. 79, 1390–1395.
Andreyko J.L., Marshall L.A., Dumesic D.A., Jaffe R.B. (1987) Therapeutic uses of gonadotropin-releasing hormone analogs. Obstet. Gynecol. Surv. 42, 1–21. https://doi.org/10.1097/00006254-198701000-00001
Al-Hendy A., Salama S. (2006) Gene therapy and ute-rine leiomyoma: a review. Hum. Reprod. Update. 12, 385–400. https://doi.org/10.1093/humupd/dml015
Gardlík R., Pálffy R., Hodosy J., Lukács J., Turna J., Celec P. (2005) Vectors and delivery systems in gene therapy. Med. Sci. Monit. 11, RA110–RA121.
Egorova A.A., Kiselev A.V. (2016) Peptide modules for overcoming barriers of nucleic acids transport to cells. Curr. Top. Med. Chem. 16, 330–342. https://doi.org/10.2174/1568026615666150812120755
Medina-Kauwe L.K., Xie J., Hamm-Alvarez S. (2005) Intracellular trafficking of nonviral vectors. Gene Ther. 12, 1734–1751. https://doi.org/10.1038/sj.gt.3302592
Hyun H., Won Y., Kim K., Lee J., Lee M., Kim Y. (2010) Therapeutic effects of a reducible poly (oligo-D-arginine) carrier with the heme oxygenase-1 gene in the treatment of hypoxic-ischemic brain injury. Biomaterials. 31, 9128–9134. https://doi.org/10.1016/j.biomaterials.2010.08.038
Woo J., Bae S.-H., Kim B., Park J. S., Jung S., Lee M., Kim Y.-H., Choi D. (2015) Cardiac usage of reducible poly (oligo-D-arginine) as a gene carrier for vascular endothelial growth factor expression. PLoS One. 10, e0144491. https://doi.org/10.1371/journal.pone.0144491
Kiselev A., Egorova A., Laukkanen A., Baranov V., Urtti A. (2013) Characterization of reducible peptide oligomers as carriers for gene delivery. Int. J. Pharm. 441, 736–747. https://doi.org/10.1016/j.ijpharm.2012.10.020
Egorova A., Bogacheva M., Shubina A., Baranov V., Kiselev A. (2014) Development of a receptor-targeted gene delivery system using CXCR4 ligand-conjugated cross-linking peptides. J. Gene Med. 16, 336–351. https://doi.org/10.1002/jgm.2811
Bogacheva M., Egorova A., Slita A., Maretina M., Baranov V., Kiselev A. (2017) Arginine-rich cross-linking peptides with different SV40 nuclear localization signal content as vectors for intranuclear DNA delivery. Bioorg. Med. Chem. Lett. 27, 4781–4785. https://doi.org/10.1016/j.bmcl.2017.10.001
Zhang Y., Buhrman J.S., Liu Y., Rayahin J. E., Gemeinhart R.A. (2016) Reducible micelleplexes are stable systems for anti-mirna delivery in cerebrospinal fluid. Mol. Pharm. 13, 1791–1799. https://doi.org/10.1021/acs.molpharmaceut.5b00933
Futaki S. (2005) Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv. Drug Deliv. Rev. 57, 547–558. https://doi.org/10.1016/j.addr.2004.10.009
Brown M.D., Schätzlein A.G., Uchegbu I.F. (2001) Gene delivery with synthetic (non viral) carriers. Int. J. Pharm. 229, 1–21.
Ward C.M., Read M.L., Seymour L.W. (2001) Syste-mic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: differential circulation in mice and rats and the implications for human gene therapy. Blood. 97, 2221–2229. https://doi.org/10.1182/blood.V97.8.2221
Parker A.L., Eckley L., Singh S., Preece J. A., Collins L., Fabre J.W. (2007) (LYS)16-based reducible polycations provide stable polyplexes with anionic fusogenic peptides and efficient gene delivery to post mitotic cells. Biochim. Biophys. Acta. 1770, 1331–1337. https://doi.org/10.1016/j.bbagen.2007.06.009
McKenzie D.L., Kwok K.Y., Rice K.G. (2000) A potent new class of reductively activated peptide gene delivery agents. J. Biol. Chem. 275, 9970–9977. https://doi.org/10.1074/jbc.275.14.9970
Read M.L., Bremner K.H., Oupický D., Green N.K., Searle P.F., Seymour L.W. (2003) Vectors based on reducible polycations facilitate intracellular release of nucleic acids. J. Gene Med. 5, 232–245. https://doi.org/10.1002/jgm.331
Manickam D.S., Bisht H.S., Wan L., Mao G., Oupicky D. (2005) Influence of TAT-peptide polymerization on properties and transfection activity of TAT/DNA polyplexes. J. Control. Release. 102, 293–306. https://doi.org/10.1016/j.jconrel.2004.09.018
McKenzie D.L., Smiley E., Kwok K.Y., Rice K.G. (2000) Low molecular weight disulfide cross-linking peptides as nonviral gene delivery carriers. Bioconjug. Chem. 11, 901–909. https://doi.org/10.1021/bc000056i
Oupický D., Parker A.L., Seymour L.W. (2002) Laterally stabilized complexes of DNA with linear reducible polycations: strategy for triggered intracellular activation of DNA delivery vectors. J. Am. Chem. Soc. 124, 8–9. https://doi.org/10.1021/ja016440n
Sambrook J., Fritsch E.F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual. 2nd ed., Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press. https://doi.org/10.1016/0092-8674(90)90210-6
Киселев А.В., Ильина П.Л., Егорова А.А., Баранов А.Н., Гурьянов И.А., Баянова Н.В., Тарасенко И.И., Лесина Е.А., Власов Г.П., Баранов В.С. (2007) Изучение лизиновых дендримеров как векторов доставки генных конструкций в клетки эукариот. Генетика. 43, 725–733.
Bradford M.M. (1976) A rapid and sensitive method for the quantitation microgram quantities of protein utili-zing the principle of protein-dye binding. Anal. Biochem. 72, 248–254.
Adami R.C., Rice K.G. (1999) Metabolic stability of glutaraldehyde cross-linked peptide DNA condensates. J. Pharm. Sci. 88, 739–746. https://doi.org/10.1021/js990042p
Midoux P., Monsigny M. (1999) Efficient gene transfer by histidylated polylysine/pDNA complexes. Bioconjug. Chem. 10, 406–411.
Егорова А.А., Киселев А.В., Тарасенко И.И., Ильина П.Л., Панкова Г.А., Ильина И.Е., Баранов В.С., Власов Г.П. (2009) Гиперразветвленные полилизины, модифицированные гистидином и аргинином: оптимизация ДНК-компактизирующих и эндосомолитических свойств. Биоорг. химия. 35, 483–492.
Ignatovich I.A., Dizhe E.B., Pavlotskaya A.V., Akifiev B.N., Burov S.V., Orlov S.V., Perevozchikov A.P. (2003) Complexes of plasmid DNA with basic domain 47‒57 of the HIV-1 Tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J. Biol. Chem. 278, 42625–42636. https://doi.org/10.1074/jbc.M301431200
Mitchell D.J., Steinman L., Kim D.T., Fathman C.G., Rothbard J.B. (2000) Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 56, 318–325. https://doi.org/10.1034/j.1399-3011.2000.00723.x
Mann A., Thakur G., Shukla V., Singh A.K., Khanduri R., Naik R., Jiang Y., Kalra N., Dwarakanath B.S., Langel U., Ganguli M. (2011) Differences in DNA condensation and release by lysine and arginine homopeptides govern their DNA delivery efficiencies. Mol. Pharm. 8, 1729–1741. https://doi.org/10.1021/mp2000814
Egorova A., Shubina A., Sokolov D., Selkov S., Bara-nov V., Kiselev A. (2016) CXCR4-targeted modular peptide carriers for efficient anti-VEGF siRNA delivery. Int. J. Pharm. 515, 431–440. https://doi.org/10.1016/j.ijpharm.2016.10.049
Egorova A.A., Maretina M.A., Kiselev A.V. (2019) VEGFA gene silencing in CXCR4-expressing cells via siRNA delivery by means of targeted peptide carrier. Methods Mol. Biol. 1974, 57–68. https://doi.org/10.1007/978-1-4939-9220-1_5
Subia B., Reinisalo M., Dey N., Tavakoli S., Subrizi A., Ganguli M., Ruponen M. (2019) Nucleic acid delivery to differentiated retinal pigment epithelial cells using cell-penetrating peptide as a carrier. Eur. J. Pharm. Biopharm. 140, 91–99. https://doi.org/10.1016/j.ejpb.2019.05.003
Tanaka K., Kanazawa T., Horiuchi S., Ando T., Sugawara K., Takashima Y., Seta Y., Okada H. (2013) Cytoplasm-responsive nanocarriers conjugated with a functional cell-penetrating peptide for systemic siRNA delivery. Int. J. Pharm. 455, 40–47. https://doi.org/10.1016/j.ijpharm.2013.07.069
Anita M., Vasundhara S., Richa K., Spoorti D., Harpal S., Munia G. (2014) Linear short histidine and cysteine modified arginine peptides constitute a potential class of DNA delivery agents. Mol. Pharm. 11, 689–696.
Stevenson M., Ramos-perez V., Singh S., Soliman M., Preece J.A., Briggs S.S., Read M.L., Seymour L.W. (2008) Delivery of siRNA mediated by histidine-containing reducible polycations. J. Control. Release. 130, 46–56. https://doi.org/10.1016/j.jconrel.2008.05.014
Hu C., Gu F., Tai Z., Yao C., Gong C. (2016) Synergistic effect of reduced polypeptide micelle for co-delivery of doxorubicin and TRAIL against drug-resistance in breast cancer. Oncotarget. 7, 61832–61844.
Tai Z., Wang X., Tian J., Gao Y., Zhang L., Yao C., Wu X., Zhang W., Zhu Q., Gao S. (2015) A biodegradable stearylated peptide with internal disulfide bonds for efficient delivery of siRNA in vitro and in vivo. Biomacromolecules. 16, 1119–1130. https://doi.org/10.1021/bm501777a
Yao C., Tai Z., Wang X., Liu J., Zhu Q., Wu X., Zhang L., Zhang W., Tian J., Gao Y., Gao S. (2015) Reduction-responsive cross-linked stearyl peptide for effective delivery of plasmid DNA. Int. J. Nanomedicine. 10, 3403–3416. https://doi.org/10.2147/IJN.S82413
Simões S., Pires P., Düzgünes N., de Lima Pedrosa M.C. (1999) Cationic liposomes as gene transfer vectors: barriers to successful application in gene therapy. Curr. Opin. Mol. Ther. 1, 147–157.
Grosse S., Aron Y., Thévenot G., François D., Monsigny M., Fajac I. (2005) Potocytosis and cellular exit of complexes as cellular pathways for gene delivery by polycations. J. Gene Med. 7, 1275–1286. https://doi.org/10.1002/jgm.772
Litzinger D.C., Buiting A.M.J., van Rooijen N., Huang L. (1994) Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. Biochim. Biophys. Acta. 1190(1), 99–107. https://doi.org/10.1016/0005-2736(94)90038-8
Jones N.A., Hill I.R.C., Stolnik S., Bignotti F., Davis S.S., Garnett M.C. (2000) Polymer chemical structure is a key determinant of physicochemical and colloidal properties of polymer–DNA complexes for gene deli-very. Biochim. Biophys. Acta. 1517(1), 1–18. https://doi.org/10.1016/S0167-4781(00)00220-7
Bloomfield V.A. (1997) DNA condensation by multivalent cations. Biopolymers. 44, 269–282. https://doi.org/10.1002/(SICI)1097-0282(1997)44:3<269::AID-BIP6>3.0.CO;2-T
Yao C., Tai Z., Wang X., Liu J., Zhu Q., Wu X., Zhang L., Zhang W., Tian J., Gao Y., Gao S. (2015) Reduction-responsive cross-linked stearyl peptide for effective delivery of plasmid DNA. Int. J. Nanomedicine. 10, 3403–3416. https://doi.org/10.2147/IJN.S82413
Lu Y., Jiang W., Wu X., Huang S., Huang Z., Shi Y., Dai Q., Chen J., Ren F., Gao S. (2018) Peptide T7-modified polypeptide with disulfide bonds for targeted delivery of plasmid DNA for gene therapy of prostate cancer. Int. J. Nanomedicine. 13, 6913–6927. https://doi.org/10.2147/IJN.S180957
Navarro A., Diaz T., Martinez A., Gaya A., Pons A., Gel B., Codony C., Ferrer G., Martinez C., Montserrat E., Monzo M. (2009) Regulation of JAK2 by miR-135a: prognostic impact in classic Hodgkin lymphoma. Blood. 114, 2945–2951. https://doi.org/10.1182/blood-2009-02-204842
Frank K.B., Chiou J.F., Cheng Y.C. (1984) Interaction of herpes simplex virus-induced DNA polymerase with 9-(1,3-dihydroxy-2-propoxymethyl)guanine triphosphate. J. Biol. Chem. 259, 1566–1569.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология