

Молекулярная биология, 2020, T. 54, № 4, стр. 580-595
Регуляция экспрессии генов раково-тестикулярных антигенов у больных колоректальным раком
Д. С. Кутилин *
Национальный медицинский научно-исследовательский центр онкологии
344037 Ростов-на-Дону, Россия
* E-mail: k.denees@yandex.ru
Поступила в редакцию 30.10.2019
После доработки 27.01.2020
Принята к публикации 02.03.2020
Аннотация
Транскрипционная активность генов, кодирующих раково-тестикулярные антигены (CTA), и ее регуляция при колоректальном раке (КРР) изучены недостаточно. В исследовании проанализирована экспрессия CTA-кодирующих генов (СT-генов) и возможные механизмы ее регуляции, в том числе экспрессия и копийность генов ДНК-метилтрансфераз, копийность CT-генов, экспрессия микроРНК и метилирование LINE-1 при КРР. Методом количественной ПЦР в режиме реального времени определяли уровни относительной экспрессии и копийность 19 генов: MAGE-A1, -A2, -A3, -A4, -B1, -B2, GAGE-1, -3, -4, MAGEC1, BAGE, XAGE3, NY-ESO1, SSX2, SCP1, PRAME1, DNMT1, DNMT3A, DNMT3B. Количественное метилирование CpG-сайтов LINE-1 оценивали методом пиросеквенирования, а для определения уровня экспрессии микроРНК использовали метод множественного параллельного секвенирования. Обнаружено, что в опухолевой ткани толстой кишки наблюдается разнонаправленная дестабилизация транскрипционной активности DNMT3A и DNMT3B, ассоциированная с их копийностью и изменением экспрессии CT-генов BAGE, SSX2 и PRAME1. Выявлена сильная положительная корреляция между копийностью и экспрессией генов BAGE, SSX2 и PRAME1. В результате множественного параллельного секвенирования обнаружено 6 дифференциально экспрессирующихся микроРНК: hsa-miR-143-3p, hsa-miR-26a-5p, hsa-miR-25-3p, hsa-miR-92a-3p, hsa-miR-21-5p и hsa-let-7i-5p, ‒ таргетирующих CT-гены GAGE1, SSX2, PRAME и SCP1, а также ген ДНК-метилтрансферазы 3A (DNMT3A). Данные о механизмах регуляции транскрипционной активности CT-генов в злокачественных опухолях толстой кишки имеют важное значение для разработки СTA-зависимых иммунотерапевтических подходов к лечению опухолей этого типа.
ВВЕДЕНИЕ
Колоректальный рак (КРР) ‒ одно из наиболее распространенных онкологических заболеваний в мире. По данным ВОЗ каждый год регистрируется около 1 млн новых случаев [1]. По числу диагностированных случаев и числу умерших пациентов эта патология уступает лишь раку легкого, желудка и молочной железы [2]. В настоящее время, несмотря на достигнутые успехи в диагностике этих опухолей, их часто выявляют на поздних стадиях. Даже применение новых комбинаторных схем химиотерапии не позволяет достичь существенного улучшения по такому показателю, как выживание пациентов. Возможно, ситуацию могут изменить такие методы лечения, как иммунотерапия, нацеленная на раково-тестикулярные антигены (CТА). CТА имеют особые характеристики, которые позволяют рассматривать их как потенциальные мишени для иммунотерапии, а также как биомаркеры для раннего выявления злокачественных опухолей [3, 4].
В конце ХХ века было установлено, что в опухолях экспрессируются высокоспецифичные антигены, позже названные “cancer-testis antigens”, так как они экспрессировались в опухолях различного гистологического происхождения и, как считалось на тот момент, практически не экспрессировались в нормальных тканях (за исключением семенников и плаценты) [5]. Экспрессия CТА и их генов носит яркий тканеспецифический характер, по-видимому, благодаря наличию сложных эпигенетических механизмов регуляции [6].
Метилирование ДНК, изменение экспрессии микроРНК и копийности генов (CNV) ‒ на сегодняшний день наиболее полно охарактеризованные факторы, контролирующие экспрессию CТ-генов [1]. Однако, несмотря на многочисленные исследования экспрессии CТ-генов в опухолях различных локализаций, их транскрипционная активность и ее регуляция при КРР остаются недостаточно изученными [1]. Цель исследования заключалась в анализе экспрессии CТ-генов и механизмов ее регуляции (экспрессии и копийности генов ДНК-метилтрансфераз, копийности CТ-генов, экспрессии микроРНК и метилирования LINE-1) при КРР.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пациенты. В исследование был включен 61 пациент с умеренно (G2) и низкодифференцированным (G3) раком толстой кишки в возрасте от 33 до 80 лет. Пациенты проходили стационарное лечение в ФГБУ РНИОИ в 2015‒2018 гг. (табл. 1). Выборку гистологически подтвержденных КРР-опухолей составили аденокарциномы. Диагноз у всех пациентов установлен на основании клинико-инструментального обследования и подтвержден морфологически. Каждый пациент подписал добровольное информированное согласие на участие в проведении исследования. На проведение исследования получено разрешение этического комитета РНИОИ (протокол № 11).
Таблица 1.
Клиническая характеристика пациентов
| Пол | Число пациентов | Возраст, медиана | Стадия | Степень дифференциации опухоли |
|---|---|---|---|---|
| Женский | 22 | 51 | ||
| 11 | 45 | 2 | G2 | |
| 8 | 51 | 3 | G2‒3 | |
| 3 | 56 | 4 | G3 | |
| Мужской | 39 | 59 | ||
| 19 | 65 | 2 | G2 | |
| 14 | 58 | 3 | G2 | |
| 6 | 59 | 4 | G3 |
В исследовании использовали операционные биоптаты нормальных и опухолевых тканей толстой кишки. Образцы тканей для доставки в лабораторию мгновенно (не более 20 с после удаления) замораживали в жидком азоте.
На основании анализа баз данных CT Database (LNCC, www.cta.lncc.br), TCGA (The Cancer Genome Atlas, https://portal.gdc.cancer.gov/), BIOGPS (http://biogps.org/) и DisGeNET (https://www.disgenet.org), а также данных литературы [1] были отобраны 16 СТ-генов для исследования их экспрессии и копийности в образцах опухолей толстой кишки (основными критериями выбора генов были следующие: ассоциация с КРР, экспрессия в тканях толстой кишки, как в нормальной, так и в опухолевой).
Анализ экспрессии генов. Фрагменты ткани гомогенизировали в лизирующем буфере, содержащем 4 М тиоцианат гуанидина, 25 мМ цитрат натрия, 0.5% саркозил и 0.1 М β-меркаптоэтанол. Выделение суммарной РНК из лизата тканей проводили методом фенол-хлороформной экстракции. Для очистки от примесей ДНК образцы обрабатывали препаратами ДНКазы I. Концентрацию полученных препаратов РНК измеряли на флуориметре Qubit 2.0® с использованием набора Qubit RNA HS Assay Kit (“Invitrogen”, США). Для оценки качества полученного препарата РНК проводили электрофорез в 2%-ном агарозном геле (интенсивность полос 18S и 28S рРНК в соотношении 1 : 1 свидетельствовала о приемлемом для дальнейшей работы качестве РНК) (рис. 1) [7].
Рис. 1.
Электрофореграмма РНК, выделенной из свежезамороженных тканей опухолей толстой кишки. Для визуализации использована система GelDoc XR PLUS (“BioRad”, США). L (ladder RNA) ‒ маркер длин РНК, T ‒ образец из опухолевой ткани, N ‒ образец из нормальной ткани.
Синтез кДНК проводили с использованием коммерческих наборов Reverta-L (“Интерлабсервис”, Россия) по инструкции производителя.
Методом количественной ПЦР в режиме реального времени определяли величины относительной экспрессии 19 генов: MAGE-A1, -A2, -A3, -A4, -B1, -B2, GAGE-1, -3, -4, MAGEC1, BAGE, XAGE3, NY-ESO1, SSX2, SCP1, PRAME1, DNMT1, DNMT3A, DNMT3B. При выборе референсного гена использовали алгоритм, описанный в работе Водолажского и др. [8], по нему рассчитывали величину M-value и Stability (Stability = ln(1/M-value). Гены с M-value менее 0.5 считали идеальными. В качестве референсных выбрали два гена: GAPDH (M-value = 0.063, Stability = 2.762) и GUSB (M-value = 0.184, Stability = 1.695). В исследовании использовали синтетические олигонуклеотидные праймеры (табл. 2), разработанные Водолажским и соавт. [9].
Таблица 2.
Панель праймеров для определения относительной экспрессии генов
| № | Ген | Последовательность праймера 5' → 3' | |
|---|---|---|---|
| Прямой | Обратный | ||
| 1 | GAPDH | GTCAAGGCTGAGAACGGGAA | TCGCCCCACTTGATTTTGGA |
| 2 | GUSB | CAGGACCTGCGCACAAGAC | CTAGCGTGTCGACCCCATTG |
| 3 | MAGEA1 | GAAGGAACCTGATCCAGGC | AGGGAATTCTGTCCTCTGGG |
| 4 | MAGEA2 | CGAAGGCTCCGTGAGGA | CTGTATTGACCTGAGTCACC |
| 5 | MAGEA3 | TGAGCAAAGAGCGACGG | TCAGACTGTCCCCTCAGAA |
| 6 | MAGEB1 | TTCAGTGTGGTGTCCAGCAA | CGAGTTGTACTCCTGGATGATCA |
| 7 | MAGEB2 | AGCCAGGGGTGAATTCTCTG | GGCACGGAGCTTACTCTCCT |
| 8 | GAGE1 | CTGATGGGCACGAGATGGAC | CCAGTCTCGGCAACATAGTGA |
| 9 | GAGE3 | TCACACAGCTGAGTTGGCGA | CTGTGTGAAATATGAGTTGGCGC |
| 10 | GAGE4 | GAGGAGGTGAAAACGCCTGG | GCATCATTTCAACGTGCCTTCG |
| 11 | MAGEC1 | ACGAGGATCGTCTCAGGTCC | CCAGGTCTTCAACTCCTGCC |
| 12 | MAGEA4 | CTGACCAGCAGCTTGGGATC | TCCAGGGAATCCTGTCCTCCT |
| 13 | BAGE | GCCGGCTCCTTTCAGGATTT | ACATCTTTCAGGAGCTTGGTCAC |
| 14 | XAGE3 | ACTTGCCCTGAGACTTAGTT | ACTTGCCCTGAGACTTAGTTT |
| 15 | NY-ESO1 | GAGTTCACTGTGTCCGGCAC | TGGAGACAGGAGCTGATGGA |
| 16 | SSX2 | TACGGTTGGTGCTCAAATACC | CCGAGGCTTTCATCTTTTCCT |
| 17 | SCP1 | AGGTGAAACCTCAGACCCT | AGTCTTTGCAAATGGAAACTCAAT |
| 18 | PRAME1 | GCTGAGCCATTGTCTCGTTACT | AGGTCTCAGTCACTTGTTGCC |
| 19 | DNMT1 | AGAGTTATGAGGCGCTTCCC | GACACAGGTGACCGTGCTTA |
| 20 | DNMT3A | GTGCTGGATGCGGGGAC | TGTTGAGCCCTCTGGTGAAC |
| 21 | DNMT3B | CTCACAGGGCCCGATACTTC | TGGACACGTCTGTGTAGTGC |
ПЦР в реальном времени (в трех технических повторах для каждого образца) проводили на термоциклере Bio-Rad CFX96 (“Bio-Rad”, США) в 25 мкл реакционной смеси, содержащей 12 нг кДНК, 0.25 мМ каждого из dNTPs, 2.5 мМ MgCl2, ПЦР-буфер и 1 ед. ДНК-полимеразы SynTaq (с ингибирующими активность фермента антителами) (“Синтол”, Россия), краситель EVA-Green и по 400 нМ прямого и обратного праймеров для референсных генов или гена-мишени, по следующей программе: первичная денатурация при 95°С в течение 3 мин; 40 циклов: 10 с при 95°С, 30 с при 58°С (отжиг праймеров ‒ температура подобрана экспериментальным путем с использованием температурного градиента), 30 с при 72°С. Специфичность ПЦР контролировали с использованием анализа кривых плавления (melting curve analysis). Относительную экспрессию (RЕ) рассчитывали по формуле RЕ = Е–ΔΔCt, где Е ‒ эффективность амплификации, рассчитанная по формуле E = 10–1/k , где k ‒ коэффициент из уравнения прямой C(T) = = klg P0 + b, полученного путем линейной аппроксимации экспериментальных данных (Еср = 1.954). Нормализацию результатов проводили по двум референсным генам: GAPDH и GUSB ‒ и уровню экспрессии соответствующих генов-мишеней в образцах нормальной ткани последовательно, по приведенной ниже схеме:
1. Нормализация по среднему референсных генов: ΔC(t) = C(t)target ‒ C(t)g_reference, где C(t)g_reference ‒ среднее геометрическое C(t) референсных генов (изменялось в диапазоне 19.4‒21.1), C(t)target ‒ среднее геометрическое C(t) генов-мишеней (изменялось в диапазоне 24.2‒32.8 в зависимости от гена и типа ткани – норма или опухоль).
2. Расчет E–ΔC(t) по каждому гену для условно нормальной и опухолевой ткани каждого пациента.
3. Расчет медианы E–ΔC(t) по каждому гену для условно нормальной и опухолевой ткани пациентов.
4. Нормализация по условно нормальной ткани и окончательный результат как кратность изменения (fold change, FC) [9]: FC = = E–ΔC(t)медиана опухолевой ткани/E–ΔC(t)медиана нормальной ткани (что тождественно RЕ = E–ΔΔC(t), или RЕ = E–(ΔC(t)медиана опухолевой ткани – ΔC(t)медиана нормальной ткани).
Выделение и бисульфитное конвертирование ДНК. Из опухолевых и условно нормальных образцов тканей методом фенол-хлороформной экстракции выделяли ДНК. Для дальнейшей процедуры отбирали препараты, содержащие 0.5‒2.0 мкг тотальной ДНК, и проводили реакцию бисульфитной конверсии, используя набор EpiJET Bisulfite Conversion Kit (“Thermo Fisher Scientific”), согласно инструкции производителя.
Пиросеквенирование. Количественное определение уровня метилирования CpG-сайтов LINE-1 проводили методом пиросеквенирования с использованием системы генетического анализа PyroMarkQ24 (“Qiagen”, Германия). Для пиросеквенирования использовали праймеры, указанные в табл. 3.
Постановку пиросеквенирования каждого образца проводили в двух повторах. Полученные пирограммы анализировали с помощью программного обеспечения PyroMark Q24 Software (“Qiagen”, Германия). Значение метилирования отдельного CpG-сайта, рассчитанного как отношение содержания нуклеотидов С/Т, использовали для вычисления усредненного метилирования.
Оценка копийности CТ-генов и генов ДНК-метилтрансфераз. Анализ проводили методом количественной ПЦР на термоциклере Bio-Rad CFX96 (“Bio-Rad”), используя ДНК, выделенную фенол-хлороформным методом, и высокоспецифичные праймеры, представленные в табл. 4.
Таблица 4.
Последовательности праймеров для определения копийности генов
| № | Ген | Последовательности праймеров, 5' → 3' | |
|---|---|---|---|
| Прямой | Обратный | ||
| 1 | GAPDH | GCTGAACGGGAAGCTCACT | GCAGGTTTTTCTAGACGGCAG |
| 2 | ACTB | CACCCTGAAGTACCCCATCG | TGTAGAAGGTGTGGTGCCAG |
| 3 | B2M | TGAGTGCTGTCTCCATGTTTGA | ATTCTCTGCTCCCCACCTCT |
| 4 | DNMT1 | TCCGTTCCATCCTTCTGCAC | GCGTCTACCACCCCAGTC |
| 5 | DNMT3A | GCTGGATGCGGGGACC | TGTTGAGCCCTCTGGTGAAC |
| 6 | DNMT3B | TGTTTCTGTGTGGAGTGCCT | ACATGTAACAGCTCCAGGGC |
| 7 | MAGEA1 | TTTCCATTCTGAGGGACGGC | TCCAGAGCTTGGGGAGAAGA |
| 8 | MAGEA2 | TGACTCAGGTCAACACAGGG | TCCTACCTCAGGCTCTCCAC |
| 9 | MAGEA3 | TGCCCTCTCACTTCCTCCTT | CAGTGCTGACTCCTCTGCTC |
| 10 | MAGEA4 | TGGGGGTTAGAGAGAAGCGA | CTCCTCAGCGTCTCACCTTG |
| 11 | MAGEB1 | TAGGAAGCCTTGGCGGAGAT | CCCAATTAGACGGAACCAGTCA |
| 12 | MAGEB2 | CCACTAAGAGCCCAAGCGAA | TGAAGGTGTAAGTGTGGCCG |
| 13 | GAGE1 | GCCGAAGCCTGAAGCTGATA | TCAGGCGTTTTCACCTCCTC |
| 14 | GAGE3 | TTCACACAGATGAGTTGGCGA | GGCCCAATCACTTCAGGAGG |
| 15 | GAGE4 | TGGAACCAGCAACACCTGAA | GCAGATGCTCCCTCATCCTC |
| 16 | MAGEC1 | TATGACCTCCTCCTTCTCCTCT | AGCACTTTGAGCACTCTCAGGA |
| 17 | BAGE | CCTTGAGTCCGGGAAGAGGA | CAGCAGGTTGTGGAGGTGG |
| 18 | XAGE3 | GGAAAGGATCTCAAACATTTGCT | CATTATATCCATGAAGCTGCAAACT |
| 19 | NY-ESO1 (CTAG1B) | GGCCCTGACCTTCTCTCTGA | CGCATCTGCAGCATCCATTC |
| 20 | SSX2 | GGGAGGCCCAAGAAAAGGAA | TTGGGGGTACCATGAACTGC |
| 21 | SYCP1 | GCTGCTCATTCGTTTGTGGTTA | ACATGAGACCTGACTACAGTTG |
| 22 | PRAME1 | TTCCTTCCAGCAACTTCGC | ACGCACGTCTGAGAGTAATAAT |
Относительную копийность (rC) вычисляли по формуле:
Множественное параллельное секвенирование микроРНК. Для множественного параллельного секвенирования микроРНК было выбрано 5 пациентов с КРР, от которых были получены парные биоптаты тканей (опухолевых и нормальных, всего 10 образцов).
Для выделения фракции микроРНК использовали набор mirVana miRNA Isolation Kit (“Ambion, Life Science Technologies”, США). Создание и последующую очистку библиотеки транскриптома микроРНК проводили с помощью набора TruSeq Small RNASample Preparation Kit (“Illumina”, США). К фракции микроРНК последовательно пришивали 3'- и 5'-адаптеры, проводили обратную транскрипцию и амплификацию созданной конструкции микроРНК согласно прилагаемому протоколу. Копии кДНК очищали электрофорезом в 6%-ном ПААГ. кДНК из геля экстрагировали водой и осаждали 95%-ным этанолом (в качестве соосадителя использовали гликоген). Содержание кДНК определяли на флуориметре Qubit 2.0® с использованием набора Qubit dsDNA HS Assay Kit (“Invitrogen”). Множественное параллельное секвенирование нуклеотидных последовательностей библиотек кДНК проводили на приборе MiSeq (“Illumina”). Методом параллельного множественного секвенирования идентифицировали нуклеотидную последовательность в случайной выборке созданных кДНК-копий.
Определение экспрессии микроРНК проводили путем сравнения нуклеотидной последовательности секвенированных молекул в каждом образце с известными нуклеотидными последовательностями микроРНК, представленными в базах данных miRBase и mirGeneDB. Число проанализированных молекул кДНК менялось от 143 000 до 1 млн на образец, из которых от 0.1 до 2% приходилось на зрелую микроРНК. Примененный подход основан на хорошо зарекомендовавшем себя алгоритме, реализованном в miRanalyzer [11].
Статистический анализ выполняли с использованием прикладных пакетов программ Microsoft Excel 2013, STATISTICA 8.0, IBM SPSS Statistics v.23.0. Для проведения кластерного анализа (Hierarchical Clustering, Euclidean distance) и построения тепловых карт использовали приложение Morpheus (Broad Institute, https://software.broadinstitute.org/morpheus). Оценку различий проводили с использованием критерия Манна‒Уитни для порогового уровня статистической значимости р < 0.05, корреляционный анализ проводили с использованием коэффициента ранговой корреляции Спирмена (r). При анализе дифференциальной экспрессии микроРНК использовали алгоритм DESeq2, реализованный в среде R [12]. DESeq2 предоставляет методы для проверки дифференциальной экспрессии с использованием отрицательных биномиальных обобщенных линейных моделей; оценки дисперсии и изменения логарифмической кратности. В анализ были включены микроРНК, представленные в выборках не менее чем 10 копиями. Для учета влияния интегральной экспрессии микроРНК на экспрессию сравниваемых микроРНК проводили нормализацию сравниваемых библиотек методом RLE (Relative Log Expression). При оценке статической значимости различий использовали тест Вальда. Для учета множественности сравнений использовали метод оценки вероятности появления ложнопозитивных результатов (false discovery rate, FDR).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
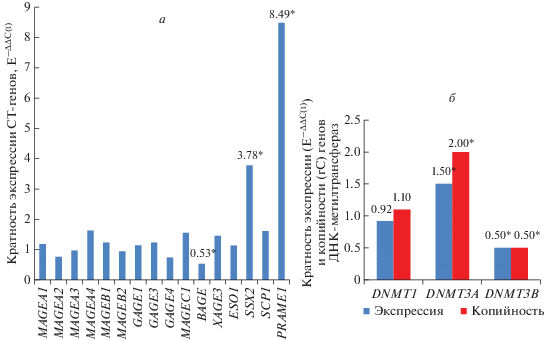
В результате проведенного исследования обнаружено статистически значимое (р < 0.05) увеличение экспрессии двух CТ-генов: SSX2 и PRAME1 в 3.8 и 8.5 раз соответственно ‒ в опухолевой ткани толстой кишки относительно нормальной ткани и снижение экспрессии одного CТ-гена, BAGE, в 1.9 раза в опухолевой ткани относительно нормальной (рис. 2а). Экспрессия генов MAGEA1, MAGEA2, MAGEA3, MAGEB1, MAGEB2, GAGE1, GAGE3, GAGE4, MAGEC1, MAGEA4, XAGE3, NY-ESO1 и SCP1 в ткани опухоли статистически значимо не отличалась от уровня экспрессии в нормальной ткани толстой кишки (р > 0.05). Также обнаружено статистически значимое (р < 0.05) изменение экспрессии и копийности генов ДНК-метилтрансфераз в опухолевой ткани толстой кишки относительно нормальной: увеличение экспрессии и копийности гена DNMT3A в 1.5 и 2.0 раза соответственно и снижение экспрессии и копийности гена DNMT3B в 2.0 раза. Однако ни копийность, ни экспрессия гена DNMT1 не изменялись. Следует отметить, что выявлена сильная положительная корреляция (r = 0.997, р < 0.05) между экспрессией и копийностью генов ДНК-метилтрансфераз (рис. 2б).
Рис. 2.
Дифференциальная экспрессия СТ-генов и генов ДНК-метилтрансфераз в тканях КРР и нормальной ткани толстой кишки. а ‒ Кратность экспрессии СТ-генов (n = 61); б ‒ кратность копийности и экспрессии генов ДНК-метилтрансфераз. * р < 0.05.
Применение кластерного анализа (Hierarchical Clustering, Euclidean distance) позволило выделить 2 кластера образцов КРР, отличающихся по экспрессии СТ-генов и генов ДНК-метилтрансфераз (рис. 3).
Рис. 3.
Кластерный анализ (Hierarchical Clustering, Euclidean distance) экспрессии СТ-генов и генов ДНК-метилтрансфераз у больных КРР. Обозначения: row max ‒ максимальное значение в ряду, row min ‒ минимальное значение в ряду.
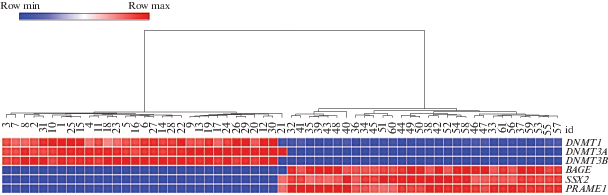
Согласно данным кластерного анализа, в образцах из первого кластера повышена экспрессия следующих генов: DNMT1, DNMT3A и DNMT3B, ‒ кодирующих ДНК-метилтрансферазы, и снижена экспрессия СТ-генов BAGE, SSX2 и PRAME1; в образцах из второго кластера картина была обратная: снижена экспрессия DNMT1, DNMT3A, DNMT3B и повышена экспрессия BAGE, SSX2 и PRAME1.
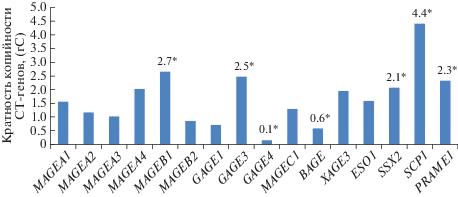
В результате анализа копийности СТ-генов в опухолевой и нормальной ткани толстой кишки (n = 61) обнаружено статистически значимое (р < < 0.05) различие этого показателя в опухолевой ткани относительно нормальной для ряда генов. Так, увеличилась копийность генов MAGEB1 (в 2.7 раза), GAGE3 (в 2.5 раза), SSX2 (в 2.1 раза), SCP1 (в 4.4 раза) и PRAME1 (в 2.3 раза) и снизилась для генов GAGE4 (в 6.8 раза) и BAGE (в 1.7 раза). Обращает на себя внимание разнонаправленность изменений копийности двух генов одного семейства СТА: GAGE3 и GAGE4. Статистически значимое (р < 0.05) изменение экспрессии трех СТ-генов: SSX2, PRAME1 и BAGE ‒ в опухолевой ткани относительно нормальной ткани толстой кишки соотносится с увеличением копийности SSX2 и PRAME1 соответственно в 2.1 и 2.3 раза, а также снижением копийности гена BAGE в 1.7 раза в опухолевой ткани относительно нормальной (рис. 4).
Рис. 4.
Кратность различий в копийности СТ-генов в опухолевой ткани относительно нормальной. *р < 0.05.
При сравнении относительной копийности и экспрессии 16 исследованных СТ-генов наблюдалась слабая положительная корреляция (r = 0.305, р < 0.05); сильная положительная корреляция по копийности и экспрессии выявлена для генов SSX2, PRAME1 и BAGE (r = 0.901, r = 0.895 и r = = 0.942, соответственно, для р < 0.05).
В результате множественного параллельного секвенирования 10 образцов обнаружены следующие категории РНК (приведен средний показатель в процентах): микроРНК (0.5%), мРНК (0.03%), антисмысловая мРНК (0.01%), тРНК (86.7%), транскрипты неясного значения (0.03%), другие РНК (12.7%) (табл. 5).
Таблица 5.
Содержание различных РНК, обнаруженных в опухолевых и нормальных тканях толстой кишки*
| тип РНК | #10 | #14 | #16 | #17 | #20 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | T | N | T | N | T | N | T | N | T | |
| мРНК (антисмысловая) | 0.01 | 0.01 | 0.01 | 0.01 | 0.00 | 0.01 | 0.03 | 0.00 | 0.01 | 0.02 |
| мРНК | 0.04 | 0.04 | 0.04 | 0.03 | 0.02 | 0.02 | 0.01 | 0.06 | 0.00 | 0.01 |
| тРНК | 84.86 | 85.15 | 88.21 | 83.92 | 95.14 | 87.25 | 83.54 | 82.92 | 90.78 | 85.23 |
| рРНК | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 | 0.00 |
| другие РНК | 14.73 | 14.67 | 11.61 | 15.85 | 4.56 | 12.56 | 14.54 | 16.14 | 8.71 | 13.96 |
| н/к РНК | 0.07 | 0.06 | 0.05 | 0.05 | 0.02 | 0.05 | 0.00 | 0.00 | 0.00 | 0.00 |
| микроРНК | 0.30 | 0.08 | 0.07 | 0.13 | 0.26 | 0.10 | 1.88 | 0.85 | 0.50 | 0.79 |
При биогенезе из одного и того же предшественника микроРНК может генерироваться много разных последовательностей, потенциально имеющих разные мишени и противоположные изменения в экспрессии. Эти вариативные формы микроРНК, отличающиеся от эталонной последовательности, принято называть iso-микроРНК (изоформы микроРНК). На рис. 5 представлена информация о классах обнаруженных iso-микроРНК. Наиболее представленными были классы exact (прочтения соответствуют канонической последовательности miRBase), lv3pT (3'-укороченное прочтение: начинается с той же позиции, что и каноническая последовательность, т.е. тот же 5'-конец, но короче канонической последовательности), nta-T (non-templated T addition; неканоническое добавление к последовательности Т), lv3pE (3'-удлиненное прочтение: начинается с той же позиции, что и каноническая последовательность, т.е. тот же 5'-конец, но длиннее канонической последовательности) и mv (multiple variant, множественные изменения в последовательности), менее представленными были классы nta-A, G, C (non-templated addition A, G, C; неканоническое добавление к последовательности A, G или C) и lv5pT (5'-укороченное прочтение: короче канонической последовательности с 5'-конца).
Рис. 5.
Доля разных классов iso-микроРНК, обнаруженных в опухолевых и нормальных тканях толстой кишки методом множественного параллельного секвенирования.
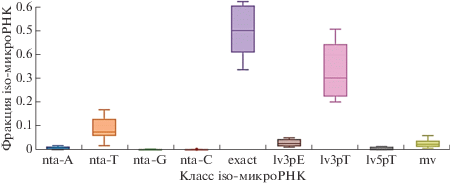
В среднем число обнаруженных микроРНК составляло 30.4 на образец, при минимальном значении 11.0 и максимальном ‒ 54.0 (табл. 6).
С использованием алгоритма DEseq2 проведен анализ дифференциальной экспрессии обнаруженных микроРНК в опухолевой и нормальной ткани толстой кишки пяти пациентов (табл. 7, рис. 6). Обнаружено шесть дифференциально экспрессирующихся микроРНК: экспрессия двух, hsa-miR-143-3p и hsa-miR-26a-5p, была снижена, а четырех, hsa-miR-25-3p, hsa-miR-92a-3p, hsa-miR-21-5p и hsa-let-7i-5p, повышена в опухолевой ткани по сравнению с нормальной.
Таблица 7.
Экспрессия микроРНК в нормальных и опухолевых тканях*
| микроРНК | Нормальная ткань | Опухолевая ткань | FC | log2FC | p-Value | Padj |
|---|---|---|---|---|---|---|
| hsa-let-7i-5p | 1.83 | 8.20 | 4.481 | 2.164 | 0.02 | 0.04 |
| hsa-miR-10a-5p | 36.91 | 37.00 | 1.002 | 0.004 | 0.99 | 0.99 |
| hsa-miR-10b-5p | 301.14 | 283.17 | 0.940 | ‒0.089 | 0.87 | 0.96 |
| hsa-miR-126-5p | 4.02 | 10.09 | 2.510 | 1.328 | 0.03 | 0.05 |
| hsa-miR-143-3p* | 382.87 | 159.01 | 0.415 | ‒1.268 | 0.02 | 0.04 |
| hsa-miR-192-5p | 174.70 | 258.4 | 1.479 | 0.565 | 0.34 | 0.42 |
| hsa-miR-21-5p* | 15.41 | 57.40 | 3.725 | 1.897 | 0.0 | 0.0 |
| hsa-miR-25-3p* | 1.44 | 7.05 | 4.896 | 2.292 | 0.01 | 0.02 |
| hsa-miR-26a-5p* | 220.06 | 61.45 | 0.279 | ‒1.840 | 0.01 | 0.02 |
| hsa-miR-27b-3p | 60.77 | 39.16 | 0.644 | ‒0.634 | 0.3 | 0.41 |
| hsa-miR-92a-3p* | 1.35 | 9.65 | 7.148 | 2.838 | 0.0 | 0.02 |
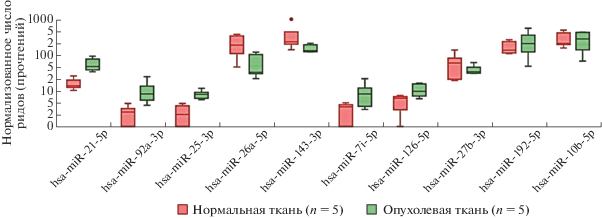
Наибольший уровень экспрессии в нормальной ткани обнаружен для hsa-miR-143-3p, наименьший ‒ для hsa-miR-92a-3p, в опухолевой ткани это были соответственно hsa-miR-10b-5p и hsa-miR-25-3p (табл. 7). Самая большая разница в экспрессии микроРНК в опухолевой ткани относительно нормальной зарегистрирована для hsa-miR-92a-3p (7.2 раза, p = 0.02), наименьшая ‒ для hsa-miR-26a-5p (3.6 раза, p = 0.01) (табл. 7, рис. 6).
Для микроРНК, дифференциально экспрессирующихся в опухолях и в нормальной ткани, проведен поиск целевых генов с использованием алгоритма TarPmiR по базе данных miRWalk 2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2). TarPmiR использует подход на основе метода “случайного леса” (random forest) ‒ алгоритма машинного обучения, заключающегося в использовании ансамбля решающих деревьев для прогнозирования сайта-мишени микроРНК, который сочетает в себе две основные идеи: метод бэггинга Бреймана, и метод случайных подпространств. Результатом модели “случайного леса” является предсказанная вероятность того, что целевой сайт-кандидат – истинный целевой сайт. TarPmiR интегрирует 13 традиционных функций для прогнозирования сайтов-мишеней микроРНК, включая доступность сайтов связывания и термодинамические свойства [13].
Для шести микроРНК, статистически значимо изменяющих экспрессию в опухолях толстой кишки, с помощью алгоритма TarPmiR предсказано 5360 генов-мишеней, из них валидированы в базах данных TargetScan mirDB и miRTarBase взаимодействия микроРНК‒мРНК для 116 генов-мишеней. Выявлено 97 генов-мишеней с минимальной свободной энергией взаимодействия микроРНК‒мРНК, в том числе гены GAGE1, SSX2, PRAME1, SCP и DNMT3A.
Стоит обратить внимание на семь микроРНК, экспрессия которых статистически значимо не отличалась в опухолевой и нормальной тканях (p-value в интервале 0.34‒0.99): hsa-miR-100-5p, hsa-miR-145-5p, hsa-miR-17-3p, hsa-miR-18a-5p, hsa-miR-663a, hsa-miR-143-5p и hsa-miR-15a-5p. Однако у двух из пяти пациентов в опухолевой ткани экспрессия hsa-miR-100-5p была повышена в 2.0 раза, hsa-miR-145-5p понижена в 2.0 раза, hsa-miR-17-3p повышена в 1.5 раза, hsa-miR-18a-5p повышена в 2.2 раза, hsa-miR-663a повышена в 2.0 раза, hsa-miR-143-5p понижена в 2.5 раза, hsa-miR-15a-5p повышена в 1.4 раза. Для этих микроРНК также был проведен поиск генов-мишеней по описанному выше алгоритму (табл. 8).
Таблица 8.
Сопоставление изменений, происходящих в экспрессии микроРНК и их генов-мишеней, в опухолевой ткани по сравнению с нормальной
| микроРНК | Ген-мишень | Изменение экспрессии микроРНКa | Изменение экспрессии гена-мишениb |
|---|---|---|---|
| hsa-miR-143-3p | GAGE1 | Сниженаc | Не отличается от нормы |
| hsa-miR-21-5p | SSX2 | Повышенаc | Повышенаc |
| hsa-miR-92a-3p | PRAME | Повышенаc | Повышенаc |
| hsa-let-7i-5p | SCP1 | Повышенаc | Повышена |
| hsa-miR-25-3p | DNMT3A | Повышенаc | Повышенаc |
| hsa-miR-100-5p | MAGEB1 | Повышена | Не отличается от нормы |
| hsa-miR-145-5p | MAGEC1 | Понижена | Повышена |
| hsa-miR-17-3p | CTAGE1 | Повышена | не отличается от нормы |
| hsa-miR-18a-5p | MAGEA2 MAGEA1 |
Повышена | Понижена не отличается от нормы |
| hsa-miR-663a | SSX2 MAGEA1 |
Повышена | Повышенаc не отличается от нормы |
| hsa-miR-143-5p | DNMT3A | Понижена | Повышенаc |
| hsa-miR-15a-5p | DNMT3B | Повышена | Пониженаc |
Как видно из полученных данных, четкой взаимосвязи между изменением экспрессии микроРНК у пяти пациентов и изменением экспрессии целевых СТ-генов на всей выборке (61 пациент) нет. Однако проведенный ранее кластерный анализ показал, что имеющаяся выборка гетерогенна по показателям экспрессии генов (рис. 3) и, скорее всего, по микроРНК. В связи с этим целесообразно сопоставить данные по экспрессии микроРНК, экспрессии и копийности СТ-генов, а также уровню метилирования LINE-1 на выборке из пяти пациентов (образцы которых использованы во множественном параллельном секвенировании).
Отметим, что уровень метилирования LINE-1 в опухолях толстой кишки составил 65.8 ± 0.89%, что статистически значимо (р < 0.001) ниже уровня, определяемого в нормальной ткани ‒ 74.0 ± ± 0.45% (рис. 7).
Рис. 7.
Пирограмма, полученная после анализа CpG-метилирования LINE-1 в опухоли (а) и в нормальных тканях (б). Значения соотношения С/Т указаны в процентах над каждым анализируемым CpG-сайтом.
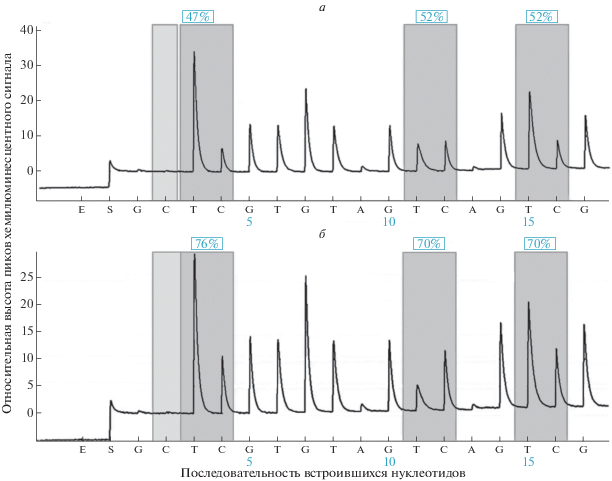
Следует также отметить, что выявлена положительная корреляционная связь между гипометилированием LINE-1 и снижением экспрессии DNMT1, DNMT3A, DNMT3B (r = 0.976, r = 0.989, r = 0.988, р < 0.05) и отрицательная корреляционная связь между гипометилированием LINE-1 и повышенной экспрессией СТ-генов BAGE, SSX2 и PRAME1 (r = ‒0.981, r = ‒0.984, r = ‒0.967, р < < 0.01) у части пациентов (n = 30, кластер 2 ‒ правая часть рис. 3)
На рис. 8 представлены результаты сопоставления данных по экспрессии микроРНК с экспрессией и копийностью СТ-генов, а также со степенью метилирования LINE-1 на выборке из пяти пациентов.
Рис. 8.
Сопоставление данных по уровням экспрессии микроРНК с экспрессией и копийностью (CNV) СТ-генов, а также со степенью метилирования LINE-1 у больных КРР (n = 5). Цифрами 1‒5 обозначены клинические образцы пациентов, соответствующие номерам #10, #14, #16, #17 и #20 в табл. 5 и 6. (row max ‒ максимальное значение в ряду, row min ‒ минимальное значение в ряду; значения r даны для p < 0.05.
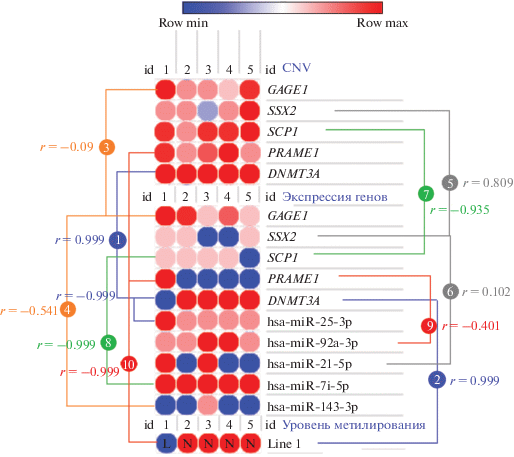
Как видно из данных, представленных на рис. 8, степень метилирования LINE-1 положительно коррелирует с уровнем экспрессии и копийностью гена DNMT3A (r = 0.999 и r = 0.999 соответственно, p < 0.05) и отрицательно коррелирует с уровнем экспрессии hsa-miR-25-3p (r = ‒0.999, p < 0.05). Между экспрессией и копийностью гена GAGE1 значимая корреляционная связь отсутствует, а вот между экспрессией GAGE1 и hsa-miR-143-3p наблюдается отрицательная корреляция (r = ‒0.541, p < 0.05). Экспрессия SSX2 положительно коррелирует с его копийностью (r = 0.809, p < 0.05) и слабо коррелирует с экспрессией hsa-miR-21-5p (r = 0.102, p < 0.05). Экспрессия SCP1 положительно коррелирует с его копийностью (r = 0.935, p < 0.05) и отрицательно ‒ с уровнем экспрессии hsa-let-7i-5p (r = ‒0.999, p < 0.05). Экспрессия PRAME1 не коррелирует с его копийностью (r = ‒0.057, p < 0.05), но отрицательно коррелирует с уровнем экспрессии hsa-miR-92a-3p (r = ‒0.401, p < 0.05) и уровнем метилирования LINE-1 (r = ‒0.999, p < 0.05). Экспрессия генов GAGE1, SSX2 и SCP1 также отрицательно коррелировала с уровнем метилирования LINE-1 (r = = ‒0.496, ‒0.408 и ‒0.250, соответственно, для p < < 0.05).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В проведенном исследовании проанализирована экспрессия 16 CТ-генов в тканях опухоли и в нормальной ткани толстой кишки 61 больного КРР. Однако только для трех генов: BAGE, SSX2 и PRAME1 ‒ получены статистически значимые различия по их экспрессии в опухолевой ткани и нормальной; причем изменения в экспрессии этих CТ-генов в опухолевой ткани соотносились с изменениями их копийности. В отличие от данных литературы [14, 15] для CТ-генов семейства MAGE не обнаружено статистически значимого изменения экспрессии, хотя зарегистрировано увеличение копийности гена MAGEB1. В опухолях изменена и копийность генов GAGE3, GAGE4 и SCP1. При сравнении относительной копийности и экспрессии 16 CТ-генов на всей выборке больных (n = 61) выявлена слабая положительная корреляция (r = 0.305, p < 0.05). Изменение числа копий генов ‒ вид генетического полиморфизма, возникающий в результате несбалансированных хромосомных перестроек, приводящих к делециям и дупликациям фрагментов ДНК размером больше 50 п.н. В результате число копий определенного гена может уменьшаться или увеличиваться. Как следствие, может изменяться и экспрессия этого гена и его продукта – белка или некодирующей РНК (в том числе микроРНК) [10].
Ген BAGE (B melanoma antigen) кодирует опухолевый антиген, распознаваемый аутологичными цитотоксическими T-лимфоцитами. Гипометилирование BAGE характерно для опухолей толстой кишки [16]. Ген BAGE наиболее интенсивно экспрессируется в меланоме, а также в опухолях мочевого пузыря, легкого и молочной железы и не экспрессируется при КРР (по данным https:// www.uniprot.org), хотя, согласно сервису GIANT (Genome-scale Integrated Analysis of gene Networks in Tissues, http://giant.princeton.edu/), BAGE экспрессируется в тканях толстой кишки. Следовательно, полученные нами результаты согласуются с данными литературы и баз данных: низкая относительно нормальной ткани экспрессия гена BAGE в опухолях толстой кишки.
Ген PRAME кодирует антиген, который экспрессируется преимущественно в меланоме человека и распознается цитотоксическими Т-лимфоцитами. Сверхэкспрессия PRAME в опухолевых тканях и относительно низкий уровень в нормальных соматических тканях позволяют рассматривать его как потенциальную мишень для иммунотерапии злокачественных опухолей [17]. Белок PRAME ингибирует дифференцировку клеток и апоптоз [18]. Сверхэкспрессия PRAME способствует подвижности опухолевых клеток посредством индукции эпителиально-мезенхимального перехода [19]. Наблюдаемое 8-кратное увеличение экспрессии гена PRAME хорошо согласуется с клинической картиной течения заболевания ‒ у 80% этих пациентов обнаружены метастазы в лимфатические узлы и печень.
Продукт гена SSX2 способен вызывать спонтанные гуморальные и клеточные иммунные реакции у онкологических больных и считается потенциальной мишенью в иммунотерапии на основе дендритноклеточных вакцин. Гены SSX1, SSX2 и SSX4 участвуют в транслокации t (X; 18), характерной для всех синовиальных сарком [20]. Также ген SSX2 может выступать в роли модулятора транскрипции и протоонкогена (по данным UniProtKB, Q16385 (SSX2_HUMAN)). Нами не выявлено корреляции повышенной транскрипционной активности этого гена у больных КРР с клиническими особенностями течения заболевания (метастазами в лимфатические узлы и печень).
Полученные нами данные по экспрессии BAGE, SSX2 и PRAME1 можно использовать для формирования панелей иммунотерапевтических мишеней (например, для применения дендритноклеточных вакцин) и биомаркеров КРР.
Продукты генов BAGE, SSX2 и PRAME1 различаются как по структуре молекул, так и по локализации в клетке. С использованием автоматизированного сервера моделирования гомологии белковой структуры ‒ SWISS-MODEL ‒ были получены пространственные структуры кодируемых этими CТ-генами белков [21], а с использованием сервиса COMPARTMENTS [22] установлена их локализация (рис. 9)
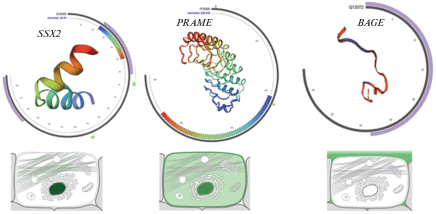
Согласно этим данным, белок SSX2 (188 а.о.) локализуются в ядре, PRAME (509 а.о.) ‒ в ядре и в клеточной мембране, а BAGE (43 а.о.) ‒ на поверхности клеточной мембраны. Таким образом, СТА BAGE и PRAME1 можно считать наиболее перспективными для использования в иммунотерапии КРР, так как они расположены на клеточной мембране, но, если учесть гипоэкспрессию гена BAGE в опухолевых клетках, в качестве потенциального кандидата для иммунотерапии КРР можно рассматривать только ген PRAME1.
В экспериментах по трансфекции репортерных генов, контролирующих промоторы СТ-генов, и в экспериментах по метилированию/деметилированию in vitro получены доказательства того, что регуляция экспрессии генов СТА определяется статусом метилирования их промоторов [23]. Метилирование ДНК ‒ эпигенетическая модификация, которая реализуется ДНК-метилтрансферазами путем ковалентного присоединения метильной группы к цитозину в составе CpG-островков [24], что приводит к подавлению транскрипционной активности генов вследствие нарушения связывания специфических факторов транскрипции с ДНК или из-за присоединения метилсвязывающих белков [25‒28]. В нормальных соматических тканях промоторы СТ-генов, как правило, метилированы, а в семенниках активируются путем деметилирования при сперматогенезе [29]. Для разных членов семейств MAGEA и NY-ESO показана ассоциация экспрессии их генов со статусом гипометилирования их промоторов в опухолях разных нозологий и в клеточных линиях [30]. Обнаруженное нами усиление экспрессии и копийности гена DNMT3A и снижение экспрессии и копийности гена DNMT3B в опухолевой ткани толстой кишки может свидетельствовать о разной роли этих ДНК-метилтрансфераз в регуляции экспрессии СТ-генов. На основании полученных данных можно предположить, что экспрессия генов DNMT1, DNMT3A и DNMT3B зависит от их копийности.
DNMT3A кодирует фермент ДНК-(цитозин-5)-метилтрансферазу 3A, которая осуществляет метилирование de novo. Мутации в этом гене часто наблюдаются при злокачественных опухолях [31]. Метилирование ДНК de novo обеспечивает ключевые эпигенетические модификации, необходимые для таких процессов, как клеточная дифференцировка, транскрипционная регуляция, образование гетерохроматина и стабильность генома [32]. DNMT1 отвечает за поддержание метилирования ДНК, в то время как DNMT3A и DNMT3B осуществляют исправление ошибок DNMT1 и метилирование ДНК de novo. Обнаружено, что после нокаута DNMT1 в раковых клетках человека эти клетки сохраняют унаследованный паттерн метилирования [33], что свидетельствует о поддерживающей активности экспрессируемых DNMT3. DNMT3 имеют одинаковое сродство к неметилированным и гемиметилированным ДНК-субстратам, в то время как DNMT1 предпочтительно взаимодействует с гемиметилированной ДНК (аффинность в 40 раз выше, чем к неметилированной ДНК) [34]. Анализируя полученные нами данные, можно сделать вывод, что в опухолевой ткани толстой кишки наблюдается разнонаправленная дестабилизация транскрипционной активности DNMT3A и DNMT3B, ассоциированная с копийностью соответствующих генов. Проанализированные опухоли можно разделить на две группы: в первой повышена экспрессия генов ДНК-метилтрансфераз DNMT1, DNMT3A и DNMT3B и снижена экспрессия СТ-генов BAGE, SSX2 и PRAME1; во второй снижена экспрессия генов ДНК-метилтрансфераз DNMT1, DNMT3A и DNMT3B и повышена экспрессия СТ-генов BAGE, SSX2 и PRAME1. По-видимому, экспрессия этих СТ-генов в опухолевой ткани толстой кишки зависит от транскрипционной активности генов ДНК-метилтрансфераз.
Нами выявлено, что снижение экспрессии DNMT1, DNMT3A и DNMT3B в опухолях коррелирует со снижением метилирования LINE-1 (r = = 0.987). Статус метилирования ретротранспозонов, в частности LINE-1, играет важную роль в поддержании геномной стабильности и используется в качестве показателя степени метилирования всего генома при различных патологических состояниях, в том числе в злокачественных опухолях [35, 36].
Согласно полученным данным, важным механизмом в регуляции экспрессии СТ-генов может быть изменение экспрессии микроРНК. МикроРНК – некодирующие одноцепочечных РНК длиной порядка 22 нуклеотидов. Они играют важную роль в регуляции трансляции и деградации мРНК, связываясь с комплементарными сайтами в нетранслируемых участках молекул последних, служащих их мишенями.
В результате множественного параллельного секвенирования 10 образцов от пяти больных КРР обнаружено шесть статистически значимо дифференциально экспрессирующихся микроРНК: 2 со сниженной экспрессией (hsa-miR-143-3p и hsa-miR-26a-5p) и четыре с повышенной экспрессией (hsa-miR-25-3p, hsa-miR-92a-3p, hsa-miR-21-5p и hsa-let-7i-5p) в опухоли относительно нормы, ‒ а также семь микроРНК, экспрессия которых статистически значимо не отличалась в опухолевой ткани от нормальной (p в интервале 0.34‒0.99): hsa-miR-100-5p, hsa-miR-145-5p, hsa-miR-17-3p, hsa-miR-18a-5p, hsa-miR-663a, hsa-miR-143-5p и hsa-miR-15a-5p. Среди множества генов-мишеней этих 13 микроРНК есть СТ-гены: GAGE1, SSX2, PRAME, SCP1, ‒ а также ген ДНК-метилтрансферазы 3A (DNMT3A).
Соответственно полученным данным, регуляция экспрессии СТ-генов в опухолях толстой кишки реализуется при помощи разных молекулярных механизмов, включающих изменения в экспрессии микроРНК hsa-miR-143-3p, hsa-miR-26a-5p, hsa-miR-25-3p, hsa-miR-92a-3p,hsa-miR-21-5p,hsa-let-7i-5p (и возможно hsa-miR-100-5p, hsa-miR-145-5p, hsa-miR-17-3p, hsa-miR-18a-5p, hsa-miR-663a, hsa-miR-143-5p и hsa-miR-15a-5p), генов ДНК-метилтрансфераз (DNMT1, DNMT3A, DNMT3B), а также копийности генов ДНК-метил-трансфераз (DNMT1, DNMT3A и DNMT3B) и СТ-генов.
ЗАКЛЮЧЕНИЕ
Проведенное исследование позволило получить важные данные по регулированию транскрипционной активности СТ-генов в злокачественных опухолях толстой кишки. Показано, что в опухолевой ткани толстой кишки наблюдается разнонаправленная дестабилизация транскрипционной активности DNMT3A и DNMT3B, ассоциированная с их копийностью и изменением экспрессии СТ‑генов BAGE, SSX2 и PRAME1. Корреляционный анализ выявил сильную положительную корреляцию между копийностью и экспрессией генов BAGE, SSX2 и PRAME1. В результате множественного параллельного секвенирования обнаружено 6 дифференциально экспрессирующихся микроРНК: hsa-miR-143-3p, hsa-miR-26a-5p, hsa-miR-25-3p, hsa-miR-92a-3p, hsa-miR-21-5p и hsa-let-7i-5p, ‒ таргетирующих СТ-гены GAGE1, SSX2, PRAME и SCP1, а также ген ДНК-метилтрансферазы 3A (DNMT3A). Полученные результаты также имеют важное значение для коррекции (при использовании агентов, изменяющих уровень метилирования) иммунотерапевтических подходов (дендритноклеточных вакцин) в лечении опухолей толстой кишки, направленных на конкретный паттерн СТА.
Работа выполнялась в рамках государственного задания “Исследование аберрантного регулирования транскрипционной активности паттерна генов при развитии малигнизации тканей различных нозологий” (№ АААА-А18-118072790013-1, финансирование министерства здравоохранения Российской Федерации).
Все процедуры, выполненные в данной работе, соответствуют этическим стандартам институционального комитета по исследовательской этике и Хельсинкской декларации 1964 года и ее последующим изменениям или сопоставимым нормам этики.
Автор заявляет об отсутствии конфликта интересов.
Список литературы
Кит О.И., Солдатова К.И., Кутилин Д.С., Водолажский Д.И. (2018) Раково-тестикулярные антигены в диагностике опухолей толстой кишки. Современные проблемы науки и образования. 2, 1‒10. https://doi.org/10.17513/spno.27449
Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68(6), 394‒424.
Водолажский Д.И., Кутилин Д.С., Могушкова Х.А., Кит О.И. (2017) Транскрипционный профиль раково-тестикулярных антигенов у больных раком тела матки. Успехи молекулярной онкологии. 4(4), 135.
Mahmoud A.M. (2018). Cancer testis antigens as immunogenic and oncogenic targets in breast cancer. Immunotherapy. 10(9), 769–778. https://doi.org/10.2217/imt-2017-0179
van der Bruggen P., Traversari C., Chomez P., Lurquin C., De Plaen E., Van den Eynde B., Knuth A., Boon T.A. (2007) Gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. J. Immunol. 178(5), 2617‒2621.
Водолажский Д.И., Кутилин Д.С., Могушкова Х.А., Кит О.И. (2018) Особенности транскрипционной активности раково-тестикулярных антигенов у больных метастатическим и неметастатическим раком молочной железы. Бюллетень экспериментальной биологии и медицины. 165(3), 360‒363.
Кутилин Д.С., Никитин И.С., Кит О.И. (2019) Особенности экспрессии генов некоторых транскрипционных факторов при малигнизации тканей тела матки. Успехи молекулярной онкологии. 6(1), 57‒62.
Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3(7), 1‒12.
Водолажский Д.И., Кутилин Д.С., Могушкова Х.А., Кит О.И. (2018) Транскрипционный профиль раково-тестикулярных антигенов у больных раком молочной железы. Медицинская иммунология. 20(3), 383‒390.
Кутилин Д.С., Айрапетова Т.Г., Анистратов П.А., Пыльцин С.П., Лейман И.А., Карнаухов Н.С., Кит О.И. (2019) Изменение копийности генов в опухолевых клетках и внеклеточной ДНК у больных аденокарциномой легкого. Бюллетень экспериментальной биологии и медицины. 167(6), 731‒738.
Hackenberg M., Sturm M., Langenberger D., Falcón-Pérez J.M., Aransay A.M. (2009) miRanalyzer: a microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res. 37, 68‒76.
Love M.I., Huber W., Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550.
Ding J., Li X., Hu H. (2016) TarPmiR: a new approach for microRNA target site prediction. Bioinformatics. 32(18), 2768‒2775. https://doi.org/10.1093/bioinformatics/btw318
Shantha Kumara H.M., Grieco M.J., Caballero O.L., Su T., Ahmed A., Ritter E., Gnjatic S., Cekic V., Old L.J., Simpson A.J., Cordon-Cardo C., Whelan R.L. (2012) MAGE-A3 is highly expressed in a subset of colorectal cancer patients. Cancer Immun. 12, 16.
Li M., Yuan Y.H., Han Y., Liu Y.X., Yan L., Wang Y., Gu J. (2005) Expression profile of cancer-testis genes in 121 human colorectal cancer tissue and adjacent normal tissue. Clin. Cancer Res. 11(5), 1809‒1814.
Grunau C., Brun M.E., Rivals I., Selves J., Hindermann W., Favre-Mercuret M., Granier G., De Sario A. (2008) BAGE hypomethylation, a new epigenetic biomarker for colon cancer detection. Cancer Epidemiol. Biomarkers Prev. 17(6), 1374‒1379.
Al-Khadairi G., Decock J. (2019) Cancer testis antigens and immunotherapy: where do we stand in the targeting of PRAME? Cancers (Basel). 11(7), E984.
Epping M.T., Wang L., Edel M.J., Carlée L., Hernandez M., Bernards R. (2005) The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell. 122(6), 835–847.
Al-Khadairi G., Naik A., Thomas R., Al-Sulaiti B., Rizly S., Decock J. (2019) PRAME promotes epithelial-to-mesenchymal transition in triple negative breast cancer. J. Transl. Med. 17(1), 9.
de Bruijn D.R., dos Santos N.R., Kater-Baats E., Thijssen J., van den Berk L., Stap J., Balemans M., Schepens M., Merkx G., van Kessel A.G. (2002) The cancer-related protein SSX2 interacts with the human homologue of a Ras-like GTPase interactor, RAB3IP, and a novel nuclear protein, SSX2IP. Genes Chromosomes Cancer. 34(3), 285‒298.
Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., Lepore R., Schwede T. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, 296‒303.
Binder J.X., Pletscher-Frankild S., Tsafou K., Stolte C., O’Donoghue S.I., Schneider R., Jensen L.J. (2014) COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database (Oxford). 2014, bau012. https://doi.org/10.1093/database/bau012
Sigalotti L., Fratta E., Coral S., Tanzarella S., Danielli R., Colizzi F., Fonsatti E., Traversari C., Altomonte M., Maio M. (2004) Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylationregulated and functionally reverted by 5-aza-20-deoxycytidine. Cancer Res. 64, 9167‒9171.
Bestor T.H. (2000) The DNA methyltransferases of mammals. Hum. Mol. Genet. 9(16), 2395‒2402.
Robertson K.D. (2002) DNA methylation and chromatin unraveling the tangled web. Oncogene. 35, 5361‒5379.
Kohli R.M., Zhang Y.(2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 502(7472), 472‒479.
Zemach A., McDaniel I.E., Silva P., Zilberman D. (2010) Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 328(5980), 916‒919.
Wade P.A. (2001) Methyl CpG binding proteins: coupling chromatin architecture to gene regulation. Oncogene. 20, 3166‒3173.
De Smet C., Lurquin C., Lethe B., Martelange V., Boon T. (1999) DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell Biol. 19, 7327‒7335.
Sigalotti L., Coral S., Nardi G. et al. (2002) Promoter methylation controls the expression of MAGE2, 3 and 4 genes inhumancutaneous melanoma. Int. J. Cancer. 25, 16‒26.
Brenner C., Deplus R., Didelot C., Loriot A., Viré E., De Smet C., Gutierrez A., Danovi D., Bernard D., Boon T., Pelicci P. G., Amati B., Kouzarides T., de Launoit Y., Di Croce L., Fuks F. (2005) Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 24(2), 336‒346.
Jia Y., Li P., Fang L., Zhu H., Xu L., Cheng H., Zhang J., Li F., Feng Y., Li Y., Li J., Wang R., Du J.X., Li J., Chen T., Ji H., Han J., Yu W., Wu Q., Wong J. (2016) Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discov. 2, 16007.
Rhee I., Jair K.W., Yen R.W., Lengauer C., Herman J.G., Kinzler K.W., Vogelstein B., Baylin S.B., Schuebel K.E. (2000). CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 404 (6781), 1003‒1007
Pradhan S., Bacolla A., Wells R.D., Roberts R.J. (1999). Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 274(46), 33002‒33010.
Vasilyev S.A., Tolmacheva E.N., Lebedev I.N. (2016) Epigenetic regulation and role of LINE-1 retrotransposon in embryogenesis. Russ. J. Genet. 52(12), 1219‒1226.
Мустафин Р.Н., Хуснутдинова Э.К. (2017) Эпигенетика канцерогенеза. Креативная хирургия и онкология. 7(3), 60‒67.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология