

Молекулярная биология, 2020, T. 54, № 5, стр. 750-775
Связь матричных процессов I и II рода: амилоиды и стабильность генома
Ю. В. Андрейчук a, *, С. П. Задорский a, b, А. С. Жук c, Е. И. Степченкова a, b, С. Г. Инге-Вечтомов a, b
a Институт общей генетики им. Н.И. Вавилова Российской академии наук, Санкт-Петербургский филиал
199034 Санкт-Петербург, Россия
b Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Россия
c Университет ИТМО
197101 Санкт-Петербург, Россия
* E-mail: yullinnabk@yandex.ru
Поступила в редакцию 11.04.2020
После доработки 06.05.2020
Принята к публикации 08.05.2020
Аннотация
Классические представления о механизмах наследственности рассматривают в качестве матриц линейные молекулы нуклеиновых кислот – ДНК и РНК, генетическая информация в которых закодирована в виде последовательности азотистых оснований. Матричный принцип, воплощенный в центральной догме молекулярной биологии, описывает разрешенные пути переноса генетической информации, а именно, от нуклеиновых кислот к белкам. Открытие прионов выявило дополнительный механизм наследственности – передачу пространственной структуры от одной молекулы белка к другой независимо от последовательности азотистых оснований в структурных генах. Одновременное существование линейных (I-го рода) и конформационных (II-го рода) матриц в одной клетке предполагает неизбежность их взаимодействия. В нашем обзоре проведен анализ современных данных, подтверждающих возможность влияния амилоидизации белков на стабильность генома, а также возможных молекулярных механизмов взаимного влияния матричных процессов I и II рода. Особое внимание уделено обсуждению совместного вклада этих процессов в “эволюцию” раковых опухолей, механизмов дестабилизации генома на фоне амилоидизации белков при болезнях Альцгеймера и Паркинсона, а также при синдроме Дауна.
МАТРИЧНЫЙ ПРИНЦИП. МАТРИЦЫ ПЕРВОГО И ВТОРОГО РОДА (ЛИНЕЙНЫЕ И ПРОСТРАНСТВЕННЫЕ)
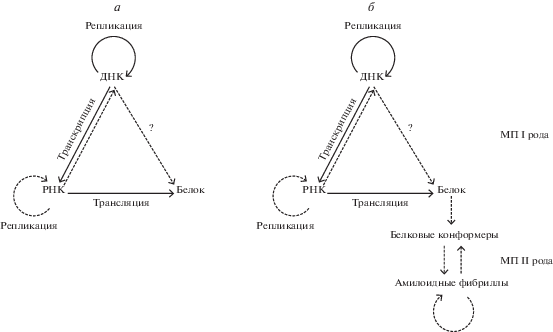
Сегодня матричный принцип – это основная парадигма генетики (и биологии в целом). Матричный принцип находит свое воплощение в центральной догме (ЦД) молекулярной биологии Ф. Крика (рис. 1а) [1, 2]. Ф. Крик писал о переносе генетической информации в клетке (стрелки на рис. 1a, иллюстрирующем ЦД, обозначают разрешенные пути переноса этой информации). Преклоняясь перед гением Крика, стоит, тем не менее, призвать читать смысл, а не только слова. Поток информации осуществляется в действительности и в обратном направлении – от белков к РНК и ДНК. Это – регуляция. А вот копирование носителей информации, осуществляемое посредством матричных процессов (МП), действительно происходит только в соответствии с направлениями, которые указывают стрелки на схеме ЦД. Подробнее о значении и месте ЦД в современной биологии написано в [3–5].
Детальное изучение всех трех МП (репликации, транскрипции, трансляции), осуществляющих копирование носителей генетической информации, вскрыло их общие (универсальные) характеристики. Все МП протекают в три этапа – инициация, элонгация (копирование) и терминация. Неотъемлемыми характеристиками МП служат их неоднозначность и репарация, или коррекция, что впервые отметили в 30–40-е годы ХХ в. Н.В. Тимофеев-Ресовский в его принципе “конвариантной редупликации”, предложенном в начале 30-х гг. ХХ в. для описания появления мутаций в ходе воспроизведения генетического материала, и М.Е. Лобашев, связавший понятия “мутация” и “репарация” в своей физиологической гипотезе мутационного процесса [6]. Представления о взаимодополняющих свойствах процесса удвоения генетического материала (неоднозначности и репарации) оформились в заочной дискуссии этих двух ученых [6, 7]. Подробнее об этом можно прочесть в нашей более ранней публикации [8]. Сегодня очевидно, что неоднозначность и коррекция, уровень которых находится под генетическим контролем (т.е. предопределен биологически, подчеркнем это!), свойственны любым МП, не только репликации, но также транскрипции (см., например, [9]) и трансляции [10]. Для транскрипции это показано сравнительно недавно, поскольку мРНК нестабильна, и работать с признаками, характеризующими мРНК, затруднительно. При изучении трансляции еще в 70-е гг. Л. Горини идентифицировал центр рибосомной неоднозначности Ram (Ribosomal ambiguity). Горини писал, что рибосома склонна “подвирать”, а различные внешние воздействия лишь повышают или понижают эту способность рибосомы [11]. Уровень неоднозначности трансляции зависит от активности фактора элогации трансляции EF-Tu (у бактерий) и его ортолога eEF1A (у эукариот), доставляющего аминоацил-тРНК к А-сайту рибосомы и обеспечивающего размещение в А-сайте преимущественно “подходящих” (cognate) аминоацил-тРНК. Чем скорее идет трансляция, тем выше уровень ее неоднозначности [12]. Кроме того, и некоторые аминоацил-тРНК-синтетазы имеют домен коррекции, отслеживающий соответствие тРНК и присоединяемой к ней аминокислоты [10].
Обсуждая неоднозначность МП или их точность, следует говорить не об ошибках МП (это антропоморфизм), а именно о присущем всем МП свойстве неоднозначности, уравновешенном для каждого из МП противоположным свойством – способностью к репарации, или коррекции. Таким образом, конечный уровень точности воспроизведения носителей генетической информации и их экспрессии оптимизирован в эволюции и является результатом взаимодействия неоднозначности и коррекции всех МП. Неслучайно можно получить мутанты как с повышенным, так и с пониженным уровнем неоднозначности любого из трех МП. Отметим, что коррекция происходит не только на стадии элонгации (воспроизведения), но на стадиях инициации и терминации.
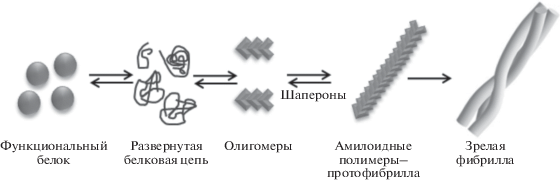
Современное прочтение ЦД, описывающей МП I (репликация, транскрипция, трансляция), требует дополнения в виде МП II (рис. 1б) – оперирующих не с линейными, а с пространственными (белковыми) матрицами. В ходе МП II происходит копирование не первичной структуры белков, а их пространственной укладки. Введение понятия МП II связано с открытием белковой наследственности – описанием инфекционных амилоидов, или прионов [13]. Многие белки могут претерпевать конформационные переходы, обогащаясь β-слоями, и формировать амилоидные олигомеры, образование и рост которых происходит за счет взаимодействия между одними и теми же участками белковых молекул, что ведет в дальнейшем к формированию линейных полимеров – амилоидных протофибрилл [14]. Согласно Добсону, такому переходу потенциально могут подвергаться практически любые белки, поскольку почти все они содержат β-слои, которые фланкированы специфическими аминокислотами – так называемыми привратниками (gatekeepers), препятствующими их экспонированию [15]. В живых организмах агрегации большинства белков препятствуют физико-химические условия внутриклеточной и межклеточной среды, взаимодействие белков с функциональными партнерами и специальные системы, защищающие клетки от нежелательных белковых агрегатов (шапероны, системы протеасомной деградации, аутофагии и др.) [16, 17] (рис. 2). С возрастом эффективность работы этих систем снижается, что приводит к повышению вероятности агрегации различных белков [18]. Более подробно структурные особенности белков, способных к амилоидной агрегации, рассмотрены в работах A. Каявы и соавт. [19–22]. Образование патологических амилоидных белковых структур характерно для более чем 40 тяжелых заболеваний человека, в том числе неизлечимых нейродегенеративных болезней Альцгеймера (БА), Паркинсона (БП), Хантингтона (БХ) и др. [23, 24]. Существуют и адаптивные амилоиды, например, прион, ответственный за вегетативную несовместимость у гриба Podospora anserina [25], но это отдельный предмет обсуждения, и о нем см. далее.
Открытым остается вопрос, насколько характеристики МП I можно распространить и на МП II – по-видимому, можно и нужно, но с учетом специфики этих процессов. Известны варианты (или штаммы) прионов дрожжей, стабильно поддерживающиеся и различающиеся по своим биологическим и физико-химическим свойствам. Неоднозначность МП II проявляется, в частности, в том, что по данным Д.А. Бейтмана и Р.Б. Викнера [26] конкретный штамм приона существует в виде “облака вариантов” (неоднозначность), при этом только один из этих вариантов является преобладающим (коррекция). По-видимому, большая роль в этих процессах принадлежит шаперонам, отвечающим за формирование и воспроизведение конформеров белков, образующих амилоиды [27–29].
Сейчас уже очевидно, что МП I и МП II взаимодействуют между собой. Во-первых, отметим, что из 10 известных прионов грибов почти половина – это факторы транскрипции или трансляции (табл. 1). Кроме того, установлено, что при необратимой остановке рибосомы на транслируемой мРНК (например, при отсутствии кодона-терминатора или обрыве мРНК) одним из механизмов, с помощью которых эукариотическая клетка избавляется от синтезированных аберрантных белков и освобождает рибосому, является образование амилоида, направляющего аберрантный белок в протеасому для деградации [30, 31]. На существование взаимодействия МП I и II может указывать как изменение стабильности генома на фоне амилоидизации некоторых белков, так и обратное – изменение эффективности амилоидизации на фоне геномных мутаций. В данном обзоре, освещая современные представления о взаимодействии МП I и МП II, мы провели анализ экспериментальных данных о взаимном влиянии амилоидизации и процессов, контролирующих стабильность генетического материала.
Таблица 1.
Инфекционные амилоиды (прионы) низших эукариот
| Прион/структур-ный белок | Функция белка в норме | Фенотипическое проявление приона | Организм | Источник |
|---|---|---|---|---|
| [URE3]/Ure2 | Регулятор катаболизма азота | Усвоение бедных источников азота в присутствии богатых | S. cerevisiae | [32, 33] |
| [PSI+]/Sup35 | Фактор терминации трансляции | Супрессия нонсенс-мутаций | S. cerevisiae | [34] |
| [PIN+]/Rnq1 | Неизвестна | Индукция [PSI+] и других дрожжевых прионов de novo | S. cerevisiae | [35, 36] |
| [Het-s]/HET-s | Неизвестна | Вегетативная несовместимость | P. anserina | [25] |
| [SWI+]/Swi1 | Субъединица комплекса ремоделирования хроматина; регулятор транскрипции | Дефект роста на средах с неферментируемыми источниками углерода | S. cerevisiae | [37] |
| [OCT+]/Cyc8 | Субъединица транскрипционного репрессора | Способность к росту на средах с неферментируемыми источниками углерода в штаммах с делецией гена CYC1; дефект споруляции; флоккуляция | S. cerevisiae | [38] |
| [MOT+]/Mot3 | Регулятор транскрипции | Измененная структура клеточной стенки | S. cerevisiae | [39] |
| [MOD+]/Mod5 | тРНК изопентенилтрансфераза | Повышенный уровень эргостерола и резистентности к противогрибковым препаратам | S. cerevisiae | [40] |
АМИЛОИДЫ И ПРИОНЫ
Амилоиды – белковые фибриллярные агрегаты, обладающие кросс-β-структурой. Само понятие “амилоид” претерпело значительную трансформацию за почти два века с момента первого упоминания. Термин “амилоид” (крахмалоподобный) предложен в 1839 году М. Шлейденом для обозначения скоплений крахмала, в норме присутствующих в клетках растений. Позднее Рудольф Вирхов стал использовать этот термин для описания макроскопических аномалий ткани, которые синели при окрашивании йодом, поэтому он ошибочно полагал, что эти структуры включают в себя крахмалоподобное вещество. В 1859 г. Г.М. Фрейндлих и Ф.А. Кекуле показали, что окрашивающиеся йодом патологические отложения в органах не содержат веществ, сходных с крахмалом или целлюлозой, но имеют в своем составе азот и сходны с белками. Вскоре стало ясно, что амилоидные отложения в основном образованы специфическими белками, а окрашивание йодом объясняется тем, что в отложениях присутствуют протеогликаны и глюкозаминогликаны (по [41]). В конце 19–начале 20-го века методология исследования амилоидов активно развивалась: появились методы гистохимической детекции амилоидов. Структурные и биохимические исследования амилоидов берут свое начало во второй половине 20-го века с появлением методов электронной микроскопии, рентгеновской дифракции и обнаружением важнейшего биохимического свойства амилоидов – нерастворимости в ряде денатурирующих агентов. До недавнего времени амилоидные агрегаты выявляли, как правило, при исследовании различных патологий человека и животных или обнаруживали случайно; при этом не существовало методов целенаправленного систематического выявления белков с амилоидными свойствами. В 2000-х годах появились методы изучения амилоидов in situ – компьютерный анализ, позволяющий предсказывать амилоидные свойства белка, основываясь на его аминокислотной последовательности [21, 42, 43]. И только совсем недавно появились методы выявления амилоидов in vivo в масштабе протеома [44–46].
Согласно современным молекулярно-биологическим представлениям, амилоидные агрегаты – это высокоупорядоченные фибриллярные белковые структуры, состоящие из бета-складчатых слоев, формирующихся за счет образования межмолекулярных водородных связей и расположенных перпендикулярно продольной оси фибриллы. При присоединении к амилоидному олигомеру (матрице II-го рода) новых мономеров белка последние изменяют свою конформацию в соответствии с конформацией мономеров в составе олигомера (рис. 2). Этот процесс при его многократном повторении приводит к образованию протофибрилл, которые затем объединяются в фибриллы и крупные амилоидные агрегаты (рис. 2). Упорядоченность амилоидных агрегатов определяется тем, что межмолекулярные связи возникают между одними и теми же последовательностями взаимодействующих мономеров [20]. Как правило, при переходе в амилоидную конформацию доля β-слоев во вторичной структуре белка резко увеличивается. Например, доля β-слоев в прионной (амилоидной) форме белка PrP млекопитающих (PrPSc) составляет около 43%, что примерно в 14 раз больше, чем в мономерной форме PrPC этого белка (3%). Еще большее содержание β-слоев (54%) характерно для прионной формы фрагмента PrP (27–30), образующего протеазоустойчивое ядро приона [47]. Для амилоидных агрегатов характерна повышенная устойчивость к обработке ионными детергентами (такими, как додецилсульфат натрия (SDS)), протеазами, действию повышенных температур и другим воздействиям (см. [24, 48]).
Следует отметить, что в медицинской литературе до сих пор преобладает несколько отличающаяся трактовка понятия “амилоид”: в соответствии с современной медицинской классификацией амилоидами принято считать лишь межклеточные агрегаты, которые окрашиваются амилоид-специфическими красителями [49, 50]. Амилоидные фибриллы обладают специфической способностью к связыванию некоторых красителей, в частности, анилинового красителя Конго красного (CR – Congo Red). Результатом этого связывания является двойное лучепреломление в поляризованном свете, дающее яблочно-зеленое свечение [51]. Другие специфичные к амилоидным фибриллам красители – флуоресцентные красители тиофлавин-Т и тиофлавин-S. При связывании тиофлавина-Т с амилоидными фибриллами происходит смещение спектра излучения в красную область, а в случае тиофлавина-S усиливается флуоресценция в зеленой области без сдвига спектра [52].
Прионы (от proteinaceous infectious (particles)) – особый класс амилоидов. Главное отличие инфекционных амилоидов (прионов) от неинфекционных заключается в том, что в случае инфекционных амилоидов происходит дробление амилоидных фибрилл с образованием амилоидных олигомеров (так называемых “затравок”, или “семян” приона) (рис. 2), что и обусловливает их способность к стабильному поддержанию приона в ходе клеточных делений и к инфицированию клеток (организмов), не несущих прион. Лучше всего процесс дробления прионных агрегатов исследован у дрожжей-сахаромицетов. Он происходит при участии шаперонов (система Hsp104–Hsp70–Hsp40), при этом главную роль играет дезагрегаза – шаперон Hsp104 [53]. При делеции гена HSP104 большая часть дрожжевых прионов теряет инфекционность и вскоре теряется в ходе митотических делений [53–55]. Некоторые варианты приона [PSI+], в отличие от остальных дрожжевых прионов, эффективно элиминируются также при сверхпродукции Hsp104 [54, 56]. В случае неинфекционных амилоидов наблюдаются только образование амилоидных агрегатов de novo и их рост, при этом новые “затравки”, способные осуществлять инфекцию, не образуются.
Впервые прионы были описаны у млекопитающих, в том числе у человека, у которых они вызывают ряд неизлечимых нейродегенеративных заболеваний, способных к передаче от человека к человеку. Инфекционным агентом является особая форма белка PrP – PrPSc, характеризующаяся повышенным содержанием бета-структур, устойчивостью к обработке протеиназой К и ионными детергентами [57]. Физиологическая функция белка PrP не установлена. Единственное известное фенотипическое проявление делеции гена этого белка у животных – невосприимчивость к инфицированию прионом. PrP синтезируется во многих клетках, таких как нейроны, клетки гладких мышц кровеносных сосудов, моноциты и лимфоциты, островковые клетки поджелудочной железы, и практически не синтезируется в печени и почках [58]. Прионная конверсия белка PrP является причиной нейродегенерации при болезни куру, связанной с ритуальным каннибализмом [59], болезни Крейтцфельда–Якоба, синдрома Герстмана–Штраусслера–Шейнкера, фатальной семейной бессонницы у людей и сходных заболеваний у млекопитающих – овец, коз, мышей, хомяков, норок и т.д. (по [60]). К этой же группе заболеваний относится “коровье бешенство”, или губчатая энцефалопатия крупного рогатого скота (BSE от Bovine Spongiform Encephalopathy) (по [61]).
Прионы низших эукариот (дрожжей и других грибов) – это нехромосомные наследственные детерминанты белковой природы. Согласно одному из распространенных определений, прионы – это инфекционные изоформы белков, способные к автокаталитическому поддержанию [55, 62]. Следует отметить, что, хотя под данное определение прионов подпадают некоторые неамилоидные наследственные факторы белковой природы [63–65], в основе формирования большинства известных в данный момент прионов высших и низших эукариот лежат процессы амилоидогенеза. В дальнейшем мы будем рассматривать только прионы, имеющие амилоидную природу. Обычно прионными свойствами обладает определенный конформационный вариант нормального клеточного белка в составе амилоидных агрегатов, которые служат конформационными матрицами для присоединения и укладки подобных белков. Один прионогенный белок может образовывать несколько наследуемых конформационных вариантов агрегатов с различными биологическими свойствами, которые называются “прионными штаммами”, или вариантами приона [66–69]. История изучения дрожжевых прионов началась в 1965 году, когда Брайан Кокс открыл нехромосомный наследственный детерминант, названный [PSI+], который повышал эффективность прочтения кодонов – терминаторов трансляции в мРНК как значащих (вызывал нонсенс-супрессию или повышал ее эффективность) [34, 70]. Немного позже обнаружили другой нехромосомный наследственный детерминант – [URE3], благодаря которому клетки дрожжей могут поглощать уреидосукцинат из среды, что позволяет мутантам ura2, у которых заблокирован синтез этого интермедиата, расти на среде с уреидосукцинатом [33]. Позже выяснилось, что [URE3] и [PSI+] – прионные формы белков Ure2 и Sup35 соответственно [32]. На сегодняшний день известно около 10 прионов дрожжей-сахаромицетов и других низших эукариот (табл. 1).
Прионизация белков может приводить к наследуемому изменению их функциональной активности, которое сопровождается различными фенотипическими проявлениями. Этот феномен получил название “белковая наследственность”, т.е. наследование признаков, определяемых конформацией специфических белков, без изменения нуклеотидной последовательности кодирующих их генов. Здесь важно отметить также, что при переходе белка в амилоидную форму клетка может приобретать фенотип, характерный для мутанта по соответствующему гену [71], однако почти всегда проявление делеции (инактивации) гена и прионизации его продукта не идентичны. В ряде случаев амилоидные агрегаты могут выполнять какие-либо новые функции в клетке, что приводит к появлению нового фенотипа [25, 35, 72–74].
Довольно долгое время работы по изучению амилоидов были связаны с их патологической ролью в организме человека и млекопитающих, однако в последние годы обнаруживается все больше и больше белков, способных к формированию амилоидных фибрилл и не связанных с патологиями, а выполняющих свою функцию в амилоидной форме. Функциональные амилоиды обнаружены почти во всех основных группах организмов. Благодаря своим уникальным характеристикам, таким как высокое сопротивление деформации, эластичность и стабильность при экстремальных условиях, они способны выполнять разнообразные функции. Среди многообразных функций амилоидов и амилоидоподобных белков можно отметить следующие: формирование биопленок у бактерий [75–77], регуляцию биогенеза и структуры клеточной стенки у дрожжей [78–82], участие в контроле оогенеза и сперматогенеза у позвоночных и беспозвоночных [83], регуляцию долговременной памяти [84–86], контроль полимеризации меланина у животных [87] и др. По-видимому, большинство известных функциональных амилоидов представлено структурными белками, амилоидная агрегация которых необходима для биогенеза различных клеточных или внеклеточных структур на определенных стадиях жизненного цикла или в определенных условиях. Вместе с тем, появляющиеся в последние годы данные говорят о том, что функциональные амилоидные агрегаты могут принимать непосредственное участие и в регуляции МП I. Примеры таких функциональных амилоидов представлены в табл. 2.
Таблица 2.
Примеры функциональных амилоидов и амилоидоподобных белков, регулирующих матричные процессы первого рода
| Амилоидный белок | Организм | Функция белка/регулируемый процесс | Источник |
|---|---|---|---|
| Rim4 | S. cerevisiae | Трансляционный репрессор/регуляция расхождения хромосом в процессе мейоза | [88] |
| CPEB/Orb2/CPEB3 | A. californica/Drosophila melanogaster/Mus musculus | Трансляционный регулятор/активация трансляции мРНК в синапсах нейронов | [84–86] |
| Fxr1 | Rattus norvegicus | Трансляционный регулятор | [89] |
| RepA | Pseudomonas aeruginosa | Белок, контролирующий инициацию репликации плазмиды pPS10 | [90, 91] |
| LD, FPA и FCA | Arabidopsis thaliana | Транскрипционные факторы/регуляция индукции цветения | [92] |
Имеется несколько примеров РНК-связывающих амилоидных и амилоидоподобных белков, агрегаты и/или олигомеры которых регулируют трансляцию специфических РНК. Так, белок CPEB моллюска Aplysia californica активирует трансляцию мРНК в синапсах нейронов, что может быть одним из механизмов долговременной памяти [84]. Подобной активностью обладают и гомологи CPEB дрозофилы и мыши [85, 86]. Амилоидные агрегаты белка Rim4 S. cerevisiae, появляющиеся в клетке при ее переходе к мейозу, репрессируют трансляцию мРНК гена CLB3, кодирующего циклин, и некоторых других специфических мРНК, что определяет характер расхождения хромосом в первом делении мейоза. При переходе ко 2-му делению мейоза агрегаты Rim4 разбираются [88]. Открытый недавно функциональный амилоид млекопитающих Fxr1 [89] контролирует трансляцию специфических мРНК в мозге млекопитающих [93, 94]. Амилоидоподобные свойства выявлены у ряда транскрипционных факторов A. thaliana, участвующих в регуляции индукции цветения [92, 95]. Число РНК-связывающих белков, обладающих функциональной активностью в амилоидной конформации, может оказаться достаточно большим, так как многие РНК-связывающие белки различных организмов содержат домены, богатые аспарагином и глутамином, что указывает на их потенциальную склонность к амилоидогенезу [96, 97].
Функциональные амилоиды могут принимать участие и в контроле репликации ДНК, что имеет непосредственное отношение к поддержанию стабильности генома. Так, белок RepA бактерии P. aeruginosa, кодируемый геном в составе плазмидного репликона pPS10, непосредственно участвует в контроле инициации репликации этой плазмиды. Мономеры и амилоидные агрегаты этого белка выполняют противоположные функции в данном процессе. Мономеры, связывающиеся со специфическими повторами ДНК в ориджине репликации (итеронами), инициируют репликацию плазмиды. После инициации репликации взаимодействие RepA с итеронами индуцирует сборку амилоидных олигомеров RepA, которые прочно связывают между собой две синтезированные копии pPS10 в области ориджина репликации и репрессируют инициацию репликации в этой области до начала нового раунда репликации [90, 91].
Напомним, что почти половина известных белков низших эукариот, способных к прионному превращению, участвуют в регуляции МП I – это транскрипционные регуляторы и фактор терминации трансляции eRF3 (табл. 1). Этот факт лишний раз указывает на несомненное взаимодействие МП I и МП II. В связи с бурным развитием исследований по идентификации новых амилоидных белков у самых разных организмов есть основания полагать, что число таких примеров может возрасти в ближайшие годы.
ОБЪЕКТЫ И МОДЕЛЬНЫЕ СИСТЕМЫ ДЛЯ ИЗУЧЕНИЯ ВЗАИМНОГО ВЛИЯНИЯ МАТРИЧНЫХ ПРОЦЕССОВ ПЕРВОГО И ВТОРОГО РОДА
Успех изучения молекулярных механизмов, опосредующих взаимодействие между амилоидизацией белков и системами поддержания стабильности генома, определяется выбором объекта исследования и наличием соответствующих методов и подходов. Изучение клеток и тканей животных и человека с амилоидными заболеваниями позволило получить большой объем экспериментальных данных по проблеме взаимного влияния амилоидизации и факторов поддержания стабильности генома. В пораженных тканях таких пациентов выявлено повышение частоты клеток с анеуплоидным набором хромосом, а также уровня окислительных повреждений ядерной и мтДНК, зафиксирована активация клеточного цикла в нейронах, которые в норме являются неделящимися постмитотическими клетками [98] (табл. 3).
Таблица 3.
Нарушения генетического материала в тканях пациентов с амилоидными заболеваниями
| Тип нарушения генетического материала | Амилоидные заболевания, при которых обнаружены соответствующие нарушения |
|---|---|
| Изменение набора хромосом | БА – увеличение доли клеток с дополнительными хромосомами 21, 17, 18, Х |
| Повышенная частота образования микроядер в периферических тканях | БА – микроядра содержат участки центромерной ДНК
и образуются в результате неправильного расхождения целых хромосом БП – микроядра не содержат центромерную ДНК, образуются преимущественно в результате поломок хромосом |
| Окислительные повреждения ядерной и мтДНК |
БА, БП, БАС |
| Окислительные повреждения РНК | БА, БП – в клетках мозга и крови |
| Одно- и двухцепочечные разрывы ДНК | БА, БАС |
| Делеции и точковые мутации мтДНК нейронов | БА, БП, БАС |
В мозге людей с БА клетки с одной (моносомия) или тремя (трисомия) хромосомами 21 встречаются в 6 раз чаще, чем у здоровых людей. Доля нервных клеток, анеуплоидных по 21-й хромосоме, в норме составляет примерно 1.7%, тогда как при БА доля таких клеток возрастает до 10.7% [99]. Кроме того, отклонения от нормального набора хромосом при БА включают как гипер-, так и гипоплоидию по 13, 18 и 17 хромосомам [100]. У женщин с БА в 2 раза увеличена частота клеток с лишней Х-хромосомой. Кроме того, обнаружено увеличение частоты клеток (фибробластов и лимфоцитов) с микроядрами при БА [101]. Микроядра формируются из фрагментов хромосом, которые не попали в основное ядро при расхождении хромосом в митозе. Микроядра могут образовываться при разрыве хромосомы с сопутствующим образованием фрагментов без центромеры, или же в результате неправильного прикрепления микротрубочек веретена деления к хромосомам. С использованием флуоресцентных проб к центромерным участкам ДНК показано, что микроядра, образующиеся в фибробластах как при спорадических, так и при наследственных формах БА, содержат центромерные участки и, скорее всего, их возникновение обусловлено потерей целых хромосом, а не их поломкой [100]. Зрелые дифференцированные нейроны в норме не делятся, а клетки с анеуплоидным набором хромосом могут появиться только в результате полной репликации ДНК и расхождения хромосом в митозе (т.е. только в делящихся клетках). Отклонение от нормального набора хромосом в нейронах предположительно может иметь два источника происхождения [98]. Во-первых, некоторые индивиды могут обладать генетической предрасположенностью к неправильному расхождению хромосом, и образование анеуплоидных клеток у них могло произойти еще на ранних этапах созревания и дифференцировки нейронов. В этом случае одна из стволовых клеток, которая образовалась в результате неправильного расхождения хромосом, делясь, становится источником зрелых анеуплоидных нейронов. Во-вторых, во взрослом мозге млекопитающих есть стволовые клетки, которые могут замещать погибшие нейроны новыми клетками. Поэтому у взрослых лиц анеуплоидные нейроны могут возникать в результате деления и дифференцировки стволовых клеток. В связи с этим важно отметить, что процессы нейрогенеза усиленно происходят в мозге людей с БА, вероятно, в результате значительной апоптотической гибели нейронов при нейродегенеративных процессах [102].
При БП в лейкоцитах также наблюдается повышение частоты образования микроядер. Кроме того, применение маркеров центромерной ДНК выявило значительную долю микроядер, не содержащих центромеры. Это свидетельствует о том, что при БП микроядра в лимфоцитах формируются преимущественно в результате хромосомных поломок [103]. С помощью метода комет обнаружено значительное накопление двухцепочечных разрывов в нескольких участках мозга (фронтальная, темпоральная и окципетальная кора, путамен, гиппокамп, таламус, средний мозг и мозжечок) при БП [104]. Таким образом, дестабилизация генома в клетках пациентов с амилоидными нейродегенеративными заболеваниями указывает на то, что на фоне амилоидизации по крайней мере некоторых белков происходит нарушение МП I, а на модели нейродегенеративных и, возможно, других амилоидных заболеваний можно исследовать тонкие молекулярные механизмы взаимодействия МП I и МП II в норме и при патологии.
Одна из наиболее удобных моделей для исследования МП и первого, и второго рода и их взаимодействия – дрожжи S. cerevisiae. Различные штаммы S. cerevisiae используют для изучения фундаментальных аспектов биологии эукариот, в том числе механизмов репликации, транскрипции, трансляции, репарации и мутагенеза, возникновения анеуплоидии и ее проявлений, начиная с 50-х годов 20 века. С использованием S. cerevisiae изучают сигнальные и метаболические пути, регуляцию клеточного цикла, моделируют процессы старения, онкологические и другие заболевания человека. Дрожжи позволяют совместить преимущества работы с микроорганизмами с возможностями исследования эукариотических клеток. Хорошо разработаны разнообразные методы частной и молекулярной генетики дрожжей, основанные на классических микробиологических и современных молекулярных методах и подходах [105]. С помощью инструментов функциональной геномики созданы коллекции делеционных мутантов дрожжей [106, 107] и различные библиотеки векторов для сверхэкспрессии генов дрожжей, покрывающие 97% генома [108]. К настоящему времени с использованием геномных коллекций накоплен огромный массив данных об изменениях транскриптома [109] и протеома [110] на фоне различных мутаций в различных условиях культивирования.
Дрожжи S. cerevisiae широко используют для изучения процесса амилоидогенеза, белков, ассоциированных с нейродегенеративными заболеваниями, а также для разработки и тестирования потенциальных терапевтических средств, эффективных при этих болезнях [111]. С использованием в качестве модели дрожжевых прионов [PSI+], [PIN+], [URE3] и других изучены основные механизмы белок–белковых взаимодействий при амилоидогенезе. Обнаружено взаимодействие прионов и амилоидов между собой, и сформулировано понятие “прионных сетей”. Показано, какие белковые домены участвуют в образовании амилоидов [24, 112]. Разработаны дрожжевые модели, позволяющие изучать основные молекулярные механизмы развития БА, БХ, БП, сахарного диабета типа 2, БАС, а также наследственных болезней, связанных с нарушением метаболизма аминокислот и приводящих к возникновению амилоидоподобных структур [113].
Дрожжевая модель хорошо зарекомендовала себя при изучении нестабильности генома. На этой модели изучены многие фундаментальные аспекты механизмов возникновения генных, хромосомных и геномных мутаций, а также вклад систем репарации в процесс мутагенеза. Это стало возможным благодаря хорошо разработанным методам выявления точковых мутаций, мутаций сдвига рамки считывания, двухцепочечных разрывов, делеций, хромосомных перестроек и других нарушений генетического материала. Наиболее эффективные и популярные методы изучения различных аспектов мутагенеза у дрожжей детально описаны в обзоре [114]. С использованием дрожжей на кафедре генетики и биотехнологии Санкт-Петербургского государственного университета разработан метод, получивший название альфа-тест [115, 116], который, в отличие от многих других тест-систем, позволяет учитывать генетические нарушения всех типов: первичные повреждения, генные мутации, гомологичную рекомбинацию и конверсию, а также потерю целой хромосомы или ее плеча [116–120]. С использованием альфа-теста нам удалось показать, что присутствие приона [PSI+] может снижать частоту генных мутаций и потери целой хромосомы III, но повышать частоту конверсии кассеты HMRa в локус MATα [121].
Таким образом, благодаря глубокой проработке методических подходов, применяемых для изучения как мутагенеза, так и амилоидов, дрожжи используют в качестве удобной модели для исследования влияния амилоидизации белков на стабильность генетического материала.
АМИЛОИДЫ И СТАБИЛЬНОСТЬ ГЕНОМА: ВОЗМОЖНЫЕ МЕХАНИЗМЫ ВЗАИМНОГО ВЛИЯНИЯ
Существование связи между эффективностью амилоидогенеза в клетке и уровнем стабильности генома не вызывает сомнений, однако открытым остается вопрос о том, какой из этих факторов первичный. Не ясно, является ли амилоидизация некоторых белков первопричиной дестабилизации генома или, наоборот, нарушения генетического материала приводят к повышению эффективности амилоидогенеза. Современные данные указывают на то, что оба механизма могут быть реализованы в живых организмах, однако соотношение процессов может различаться в разных клетках и в разных условиях. Далее мы обсудим экспериментальные данные, подтверждающие каждую из возможных моделей взаимодействия амилоидов и системы контроля стабильности генома.
Прежде всего, необходимо пояснить, что к признакам дестабилизации генома относится появление мутаций различных типов. Традиционно мутации делят на три основные группы: 1) генные, или точковые (транзиции, трансверсии, вставки и выпадения нескольких нуклеотидов, изменение копийности коротких повторов); 2) хромосомные (крупные делеции, дупликации, транслокации, инверсии); 3) геномные (анеуплоидия и полиплоидия) [122–124]. Мутации каждого типа возникают посредством разных механизмов, которые, однако, могут взаимодействовать между собой. Так, точковые мутации возникают в результате ошибок синтеза ДНК в ходе репликации, репарации или рекомбинации, точность которого зависит от повреждений в матричной цепи, точности ДНК-полимераз, осуществляющих синтез, активности всех белков, регулирующих активность ДНК-полимераз, эффективности систем репарации, а также наличия сбалансированного пула нуклеотидов в клетке [123, 125, 126].
Хромосомные мутации возникают в результате нарушения процесса репарации ДНК с двухцепочечными разрывами и другими серьезными повреждениями, индуцированными мутагенами или возникшими в ходе естественных процессов, например, при кроссинговере, переключении типа спаривания у дрожжей или созревании генов иммуноглобулинов у млекопитающих [123, 127]. Геномные мутации возникают при нарушении расхождения хромосом в мейозе или митозе, чаще всего связанном с аномалиями белков веретена деления клетки [128, 129].
Для выявления причин дестабилизации генома на фоне амилоидизации каких-либо белков необходимо в первую очередь учитывать тип нарушений генетического материала, а в качестве наиболее вероятной мишени рассматривать белки, вносящие наибольший вклад в предотвращение возникновения мутаций данного типа. Опубликованные данные указывают на то, что амилоиды могут взаимодействовать с факторами, контролирующими возникновение мутаций всех трех типов: генных, хромосомных и геномных. Этот вопрос будет освещен в следующем разделе. Поэтому можно предположить, что амилоиды могут влиять на процессы возникновения повреждений генетического материала и репарации поврежденной ДНК, модифицировать точность матричного синтеза ДНК или РНК, а также влиять на процесс рекомбинации и сегрегации хромосом. Некоторые из этих предположений уже получили экспериментальное подтверждение.
Амилоиды и повреждения ДНК
Экзогенные и эндогенные факторы постоянно индуцируют возникновение первичных повреждений в ДНК, которые, в свою очередь, в ходе нетождественной репарации могут превращаться в мутации. Первичные повреждения ДНК бывают различного типа. Повреждаться могут как азотистые основания, так и сахарофосфатный остов. К числу наиболее частых первичных повреждений относятся продукты дезаминирования и метилирования азотистых оснований, апуриновые сайты (АП-сайты), крупные аддукты, одно- и двухцепочечные разрывы и поперечные сшивки цепей ДНК [130]. Ежедневно в каждой клетке млекопитающих могут возникать десятки и сотни тысяч различных первичных повреждений, например, 50 000–200 000 АП-сайтов и от 10 000 до 86 000 окислительных повреждений [131–133]. Системы репарации бесследно устраняют абсолютное большинство этих повреждений, а небольшая их доля превращается в мутации. О точности процесса репарации можно судить, сравнивая данные о десятках тысяч первичных повреждений, возникающих в клетке ежедневно, и скорости мутагенеза, составляющей у разных видов примерно 0.001–1 мутации на геном за одно деление [134]. Повреждения ДНК за счет эндогенных факторов происходят при вступлении ДНК в химические реакции, например, в реакции гидролиза, окисления, метилирования, дезаминирования, при воздействии метаболитов или ДНК-модифицирующих ферментов, таких как эндонуклеазы, гликозилазы, цитозиндезаминазы, топоизомеразы, принимающих участие в репликации, репарации, транскрипции и структурной реорганизации хроматина [122, 130]. К экзогенным факторам, повреждающим ДНК, относят ультрафиолетовое и ионизирующее излучение, а также различные химические вещества: аналоги азотистых оснований, алкилирующие агенты, тяжелые металлы, ароматические амины, полициклические ароматические углеводороды, вторичные метаболиты растений и грибов [122, 130]. Таким образом, первичные повреждения ДНК нарушают МП I, затрудняя репликацию генетического материала и экспрессию генетической информации. Они могут снижать точность репликации, служить сигналами запуска систем репарации, существенным этапом которых является гомологичная и негомологичная рекомбинация. Первичные повреждения ДНК вносят вклад в процессы старения и развитие многих болезней, таких как рак, наследственные и нейродегенеративные заболевания [135, 136].
При нейродегенеративных заболеваниях в нейронах обнаруживают накопление огромного количества окислительных повреждений, индуцированных активными формами кислорода (АФК). АФК способны повреждать различные клеточные компоненты, мембраны, органеллы, липиды, белки и нуклеиновые кислоты. При повреждении ДНК и растворимых нуклеотидов АФК образуется около 20 различных окисленных продуктов, наиболее частый из которых 8-гидроксигуанин (8-OHG) и его производное 8-гидрокси-2′-дезоксигуанин (8-OHdG), по увеличению уровня которых определяют повышение уровня окислительного стресса [98]. В аутоптатах мозга больных БА увеличен уровень окисленных продуктов: 8-OHG, 8-гидроксиаденина, 5-гидроксицитозина и 5-гидроксиурацила [136]. При БА окислительному повреждению подвергаются также РНК. Показано, что 30–70% мРНК, выделенной из постмортального мозга лиц с БА, содержат окисленные основания, тогда как в контроле доля окисленных мРНК составляет 2% [137]. Трехкратное увеличение содержания 8-OHdG обнаружено в мтДНК из нейронов индивидов с БА [138]. Окислительные повреждения ДНК найдены даже в периферических тканях больных БА. С помощью метода комет показано значительное (по сравнению с контролем) увеличение доли окисленных пуринов и пиримидинов в ДНК лимфоцитов крови при БА [139, 140]. Повышение уровня повреждений ДНК обнаруживается и в постмортальных препаратах мозга на ранних стадиях БА, для которых характерны небольшие нарушения процессов запоминания, памяти, проблемы с вычислениями, когда симптомы деменции еще не проявляются [141]. При БА содержание двухцепочечных разрывов ДНК в мозге также возрастает в 2 раза [142]. Скорее всего, появление двухцепочечных разрывов в ДНК при БА обусловлено незавершенными попытками устранения поврежденных азотистых оснований, так как в нейронах ферменты системы репарации работают с недостаточной эффективностью.
Повышение уровня окислительного стресса и увеличение числа повреждений в ядерной и мтДНК обнаружено и в постмортальных образцах мозга при БП. С помощью газовой хроматографии показано, что в образцах тканей мозга больных БП количество 8-OHdG выше, чем в контрольных образцах, причем наибольшая концентрация 8-OHdG зафиксирована в черной субстанции – участке мозга, поврежденном при БП [143]. Увеличение количества одноцепочечных разрывов и окисленных пуриновых оснований обнаружено также в образцах периферической крови пациентов с БП и в культуре лимфоцитов из крови пациентов с БП [103]. Как и при БА, окислительные повреждения ДНК находят даже на ранних стадиях БП [144].
В ДНК нейронов, выделенных из моторной коры мозга пациентов с БАС, содержание 8-OHdG также выше, чем в контрольной группе [145]. Концентрация 8-OHdG при БАС повышена также в плазме крови, моче и спинномозговой жидкости, причем по мере прогрессирования заболевания концентрация 8-OHdG увеличивается [146]. Увеличение частоты окислительных повреждений ядерной и мтДНК, в частности 8-OHdG, зафиксировано также и в моторных нейронах мышей с мутацией G93A в гене Sod1 [147, 148]. Мутации в этом гене находят в 20% случаев семейных форм БАС [149].
Некоторые исследования доказывают непосредственную роль амилоидных агрегатов в образовании свободных радикалов. Взаимодействие агрегированной формы β-амилоида с ионами таких металлов, как медь, железо или цинк, может катализировать образование АФК. Образовавшиеся таким образом АФК, в частности наиболее реактивный гидроксильный радикал, могут способствовать окислительному повреждению как самого β-амилоида, так и любых других белков или ДНК [150, 151]. Показано, что Аβ1-42 влияет непосредственно на процессы образования свободных радикалов, и ключевую роль в этом процессе играет взаимодействие атома серы Met35 с кислородом карбоксильной группы Ile31 [151]. Участие Met35 Аβ1-42 в образовании свободных радикалов подтверждают и результаты исследования трансгенных червей Caenorhabditis elegans. Экспрессия минигена человека, кодирующего Aβ1-42, в C. elegans приводит к повышению уровня окислительного стресса, определяемого по концентрации карбонилированных белков [152, 153]. Согласно гипотезе окислительного стресса, образование токсичных форм Aβ1-42 при БА приводит к повышению уровня окислительного стресса, что, в свою очередь, вызывает гибель клеток. В пользу этого предположения свидетельствует тот факт, что применение витамина Е, обладающего антиоксидантными свойствами, снижает уровень окислительных повреждений в культуре нейронов и предотвращает гибель клеток. Образование АФК наблюдали также в присутствии фрагментов PrP, несущих аминокислотные замены, которые способствуют прионизации [154]. Один из возможных механизмов влияния окислительных повреждений на патогенез нейродегенеративных заболеваний может заключаться в том, что на матрице ДНК с окисленными формами гуанина транскрипция происходит с ошибками, а последующая трансляция приводит к накоплению аномальных белков и нарушению их функций. Существует предположение, что окислительные повреждения ДНК можно рассматривать как одну из причин нарушений в процессе фибриллогенеза белка альфа-синуклеина (α-SYN), которые могут способствовать накоплению его агрегатов, приводящих к развитию БП [155].
Обсуждается возможность прямого повреждающего действия некоторых амилоидов на ДНК. Так, некоторые амилоиды способны непосредственно связываться с молекулами ДНК и РНК. Амилоид Aβ1-42 обладает нуклеазной активностью. Например, известно, что растворимые формы Aβ1-42in vitro связываются с ДНК с образованием ее разрывов [13]. α-Синуклеин также способен связываться с ДНК, при этом такое взаимодействие индуцирует его полимеризацию [156]. Показано, что внутриклеточный Aβ может связываться с промотором гена, кодирующего опухолевый супрессор p53, и активировать его экспрессию. Кроме того, окислительные повреждения ДНК индуцируют перемещение Aβ в ядро клеток морских свинок [157]. Механизмы взаимодействия амилоидогенных белков с ДНК и РНК и значение этих взаимодействий описаны в обзоре [158]. Таким образом, первичные повреждения ДНК, нарушая стабильность генома, не только влияют на МП I, но и затрагивают MП II. В свою очередь, амилоидные агрегаты некоторых белков, связываясь с ДНК, влияют на ее стабильность и модифицируют МП I.
Амилоиды и эффективность репарации
Увеличение частоты повреждений ДНК при нейродегенеративных амилоидозах может быть связано не только с повышенным уровнем окислительного стресса, но и с недостаточной эффективностью ферментов систем репарации ДНК. Участие той или иной системы репарации в процессе восстановления структуры ДНК зависит от типа повреждения и стадии клеточного цикла. Например, эксцизионная репарация оснований, наиболее активная на стадии G1 клеточного цикла, устраняет окислительные повреждения, дезаминированные и алкилированные основания, АП-сайты. Более крупные повреждения, такие как пиримидиновые димеры, 6-4-фотопродукты, ДНК-аддукты, удаляются путем эксцизионной репарации нуклеотидов, активной на стадии G1 [123, 159, 160]. Неспаренности оснований, возникающие главным образом при ошибках ДНК-полимераз в ходе репликации, устраняет система MMR репарации (Mismatch repair) на стадии S клеточного цикла [161]. Двухцепочечные разрывы удаляются путем негомологичного воссоединения концов (NHEJ) в основном на стадии G1 или гомологичной рекомбинации на стадии S или G2 [162]. Процесс устранения повреждений ДНК происходит поэтапно. В общем виде на первом этапе репарации вырезается участок ДНК с повреждением, а затем застраивается образовавшаяся брешь. Возможны также гомологичная рекомбинация и негомологичное воссоединение концов двух молекул ДНК. При репарации могут происходить взаимопревращения повреждений, образуются промежуточные продукты незавершенной репарации, которые представляют собой повреждения ДНК другого типа, служащие субстратом для ферментов других систем репарации. Например, в ходе эксцизионной репарации модифицированных оснований ДНК-гликозилаза вырезает поврежденные основания с образованием АП-сайтов, при устранении которых возникают одноцепочечные разрывы, что стимулирует появление двухцепочечных разрывов и рекомбинацию. Двухцепочечные разрывы могут приводить к потере хромосом или их участков, а в случае ошибочной репарации ДНК с двухцепочечными разрывами посредством рекомбинационной репарации или прямого воссоединения концов могут возникать генные мутации и перестройки хромосом [163, 164]. Избыточные повреждения ДНК в дифференцированных нейронах могут приводить к активации клеточного цикла, а недостаточная эффективность систем репарации, в свою очередь, к невозможности закончить репарацию ДНК с высокой точностью и к активации апоптоза. Так, в мозге людей с БА увеличена доля нейронов, синтезирующих белки-маркеры клеточного цикла, такие как циклины B и D [165]. Поскольку нейроны в норме не делятся, синтез таких белков свидетельствует об активации клеточного цикла.
Уже довольно давно известно о снижении эффективности репарации ДНК в фибробластах, лимфоцитах и лимфобластах при БА [166–168]. Снижение активности ферментов репарации ДНК на разных стадиях БА подтверждают результаты более поздних исследований [169]. Отмечено также снижение активности факторов эксцизионной репарации оснований (BER), в частности 8-оксигуанин-ДНК-гликозилазы (OGG1) и урацил-ДНК-гликозилазы (UDG), специфически удаляющих, соответственно, 8-OHG и урацил из ДНК, а также ДНК-полимеразы β [170, 171]. Именно ферменты BER принимают участие в устранении подавляющего большинства повреждений ДНК, вызванных окислительным стрессом. Существуют генетические заболевания, обусловленные мутациями в генах, кодирующих ферменты репарации ДНК. У человека такие заболевания проявляются повышенным риском развития рака (пигментная ксеродерма) или преждевременного старения (синдром Кокейна); развиваются также нейродегенеративные процессы. Некоторые исследователи предполагают, что мутации в генах, кодирующих ферменты репарации ДНК, могут влиять на процессы нейродегенерации и увеличивают риск развития БА, но данных, подтверждающих это предположение, недостаточно [172].
Можно представить и другие механизмы влияния амилоидов на эффективность репарации, однако они требуют проверки и глубокого экспериментального изучения. Как правило, агрегация амилоидных белков снижает их функциональную активность. Амилоидные агрегаты могут титровать молекулы неамилоидных белков и/или провоцировать их агрегацию. Таким образом может снижаться активность и/или изменяться локализация белков, не обладающих амилоидными свойствами. Такими белками, вовлекаемыми в амилоидные агрегаты, теоретически могут быть и белки, участвующие в контроле стабильности генома. Так, почти половина дрожжевых прионов представляют собой факторы транскрипции, поэтому весьма вероятно, что прионизация таких белков будет приводить к изменению экспрессии тех генов, которые контролируют эти факторы транскрипции, в их число могут входить и гены репарации. Показано, что у мышей с мутантным вариантом гена Htt, содержащим дополнительные повторы триплетов CAG, которые увеличивают вероятность амилоидизации белка хантингтина, появление признаков БХ коррелирует с изменением транскриптома [173].
Амилоиды и геномные мутации
Амилоидизация (прионизация) некоторых белков может индуцировать изменения кариотипа (анеуплоидию), по крайней мере, на определенном генотипическом фоне. Отдельные данные, иллюстрирующие это положение, получены на модели дрожжей-сахаромицетов. Так, штаммы, содержащие неменделевский фактор [ISP+], идентифицированный как прионная форма транскрипционного регулятора Sfp1 [174], в процессе хранения могут терять прион, одновременно сохраняя его фенотипическое проявление за счет изменения копийности хромосомы II. Кроме того, показана индукция потери хромосомы II при сверхпродукции белка Sfp1, приводящей к амилоидизации этого белка [175]. Подобный феномен отмечен и при исследовании неменделевского детерминанта [ASP+], первоначально показавшего характерные для прионов особенности цитоплазматического наследования и, по-видимому, индуцировавшего дисомию по хромосоме VIII, имеющую сходное с [ASP+] фенотипическое проявление [176].
Амилоидизация белков может непосредственно влиять на процессы расхождения хромосом, поскольку переход белков в амилоидное состояние, по-видимому, затрагивает функции микротрубочек. Микротрубочки играют ключевую роль в транспорте внутриклеточных компонентов, а также в движении хромосом во время митоза. Работа микротрубочек регулируется с помощью контроля процессов полимеризации и деполимеризации. Мутации в гене SUP35, кодирующем фактор терминации трансляции, способный к образованию приона [PSI+], приводят к повышению частоты потери хромосомы III [177] и к повышению чувствительности к беномилу, нарушающему полимеризацию микротрубочек [178]. Показано прямое взаимодействие Sup35 с тубулином, опосредованное прионизующим NM-доменом Sup35. Штаммы SUP35-ΔNM также чувствительны к беномилу [179]. Мутации или недостаточность Sup35 вызывают морфологические изменения клеток дрожжей, такие как увеличение размеров, изменения морфологии веретена деления, деполимеризацию актина, которые, вероятно, обусловлены дефектами цитоскелета [180]. У самцов D. melanogaster мутации в гене SUP35 нарушают сборку веретена деления и сегрегацию хромосом, а также цитокинез в мейозе [181].
Каким образом амилоиды могут влиять на стабильность генома в клетках человека? Можно рассмотреть следующий пример. Показано, что накопление агрегированного β-амилоида при БА сопровождается гиперфосфорилированием белка Tau [182–184]. Агрегация Tau, специфичного для нейронов белка микротрубочек, приводит к нарушению цитоскелета и гибели нейронов [183, 185]. Можно предположить, что амилоидные формы белков, влияя на функцию белков цитоскелета, способны индуцировать нерасхождение хромосом при делении и вызывать таким образом нарушение плоидности генома. Это согласуется с данными о повышении частоты анеуплоидии в нейронах при БА. Показана также способность белка PrP в растворимой форме связываться с тубулином, при этом PrP препятствует правильной сборке микротрубочек, индуцируя образование кольцевых, дископодобных или слоистых тубулиновых олигомеров [186]. Мутантный PrP, синтезируемый при наследственных формах болезни Герстмана–Штраусслера–Шейнкера, обладает более сильными ингибирующими свойствами по сравнению с белком дикого типа. Таким образом, амилоидизация белков может индуцировать как первичные повреждения, репарация которых может приводить к нарушению MП I и II, так и структурные нарушения генома.
ВЛИЯНИЕ ИЗМЕНЕНИЙ В ГЕНОМЕ НА АМИЛОИДОГЕНЕЗ
В последние годы накапливается все больше фактов, говорящих о том, что не только амилоиды могут служить первопричиной дестабилизации генома, но и генетические нарушения могут запускать агрегацию белков, хотя известно, что генетическая составляющая амилоидозов – всего около 10% [99]. Поддержание гомеостаза протеома требует координированной работы систем синтеза, контроля качества и деградации белков. Синтезированные пептиды должны быть правильно свернуты, неправильно сложенные белки должны быть повторно свернуты, а необратимо поврежденные белки должны подвергнуться деградации. Кроме того, должны быть правильно собраны мультибелковые комплексы. Молекулярные шапероны играют главную роль в сворачивании и рефолдинге белков и сборке мультибелковых комплексов, а протеасомные и вакуолярные протеазы – в деградации неправильно сложенных белков [16, 17]. Шапероны и убиквитин-протеасомные системы функционируют согласованно, обеспечивая гомеостаз протеома. Когда клетки испытывают протеотоксический стресс, т.е. когда системы контроля качества белка, включающие шапероны и системы протеасомной деградации, работают неэффективно или перегружены, неправильно свернутые белки не устраняются, что провоцирует образование агрегатов [17]. Крупные изменения генома, такие как потери или повышение числа отдельных хромосом, крупные делеции или дупликации могут приводить к дестабилизации протеома из-за изменения дозы генов и повышать вероятность образования белковых агрегатов, в том числе амилоидных.
Действительно, в ряде исследований показано, что анеуплоидные клетки испытывают протеотоксический стресс, вызванный нарушением протеомного баланса, в частности, стехиометрических соотношений компонентов белковых комплексов. Синтез большого количества невостребованных белковых молекул приводит к повышенной нагрузке на системы, осуществляющие деградацию и контроль качества белков [187, 188]. Так, анеуплоидные штаммы дрожжей – дисомные по различным хромосомам, обладают повышенной чувствительностью к высоким температурам, ингибиторам белкового синтеза и сворачивания белка, а также к химическим агентам и мутациям, инактивирующим протеасомную деградацию. При этом повышение эффективности протеасомной деградации при делеции гена, кодирующего деубиквитинирующий фермент Ubp6, повышает жизнеспособность ряда дисомных штаммов дрожжей [189, 190]. Вследствие недостаточности систем контроля качества белка у анеуплоидных штаммов дрожжей повышена эффективность агрегации белков, в том числе образования амилоидных агрегатов. Так, в ряде дисомных штаммов дрожжей, продуцирующих белок хантингтин (Htt) с различной длиной полиглутаминовых повторов, агрегация данного белка происходит с большей эффективностью. При этом токсическим эффектом обладали варианты белка, у которых длина поли-Gln-участка была меньше, чем в соответствующих эугаплоидных штаммах [191]. В этой же работе показано повышение эффективности образования агрегатов модифицированного варианта белка Sup35p дрожжей, склонного к амилоидной агрегации, в ряде анеуплоидных штаммов по сравнению с контрольными гаплоидными штаммами.
Принципиально сходные данные получены и с использованием культур клеток человека и мыши. В культивируемых клетках млекопитающих анеуплоидия также нарушает гомеостаз протеома, приводя к аномальному сворачиванию белковых молекул, накоплению белковых агрегатов, изменению характера аутофагии и накоплению компонентов аутофагосом в лизосомах, что приводит к перегрузке лизосом и, как следствие, к неэффективной деградации аномальных белков в лизосомах [192–195].
Эти и другие данные позволяют предполагать, что анеуплоидия может быть одним из факторов, провоцирующих возникновение нейродегенеративных заболеваний амилоидной природы, таких как БА, БП, БХ и другие. Значительная часть клеток мозга человека представлена анеуплоидными клетками (анеуплоидными являются около 10% нейронов взрослого человека [99, 196–198]). Тот факт, что анеуплоидия вызывает протеотоксический стресс, включая формирование агрегатов поли-Gln-содержащих белков в дрожжевой модели [191], позволяет предполагать, что анеуплоидные нейроны могут иметь повышенную способность к формированию амилоидных агрегатов и / или сниженную способность к их элиминации. Таким образом, анеуплоидные нейроны могут быть предрасположены к развитию амилоидных заболеваний. Так, ранее отмечали, что у мышей, химерных по трисомии по хромосоме 16 (одна из моделей СД), повышена восприимчивость к инфекции. У таких мышей при инъекции прионной формы белка PrP (PrPSc) болезнь развивается быстрее, а симптомы выражены сильнее, чем у нормальных мышей [199].
Наряду с общей дестабилизацией протеома в анеуплоидных клетках, одной из причин повышения эффективности амилоидизации отдельных белков может быть увеличение дозы генов, кодирующих белки, склонные к амилоидной агрегации. На такую возможность указывает обнаружение связи между БА и cиндромом Дауна (СД) [200, 201]. СД – врожденное заболевание, обусловленное наличием лишней копии хромосомы 21. Лица с СД предрасположены к развитию ожирения, лейкозов в детском возрасте, сахарного диабета, БА и ряда других. Симптомы БА появляются при СД уже к 20 годам, а в 35–40 лет практически у всех больных диагностируют БА [202]. Поскольку ген APP, кодирующий белок-предшественник Aβ, расположен в хромосоме 21 человека [203], предполагают, что именно дополнительная копия гена APP при СД вызывает увеличение продукции белка АPP и, соответственно, Aβ1-42 [204, 205]. В пользу этого предположения свидетельствуют данные о развитии БА у лиц с разными вариантами СД, а также у носителей редкой дупликации локуса, содержащего ген APP. Причиной СД может быть не только целая лишняя хромосома 21, но и ее фрагмент. При СД с дополнительным фрагментом хромосомы 21, который не содержит локус APP, отсутствуют симптомы БА [206], а редкая дупликация локуса с геном APP вызывает преждевременное развитие аутосомно-доминантной формы БА [207, 208]. Существование положительной корреляции между СД и БА позволило предположить, что люди, у которых развивается БА, являются мозаиками по хромосоме 21 (частичный, или мозаичный СД), а лишняя копия гена APP даже в небольшом количестве нервных клеток приводит к увеличению продукции Aβ1-42 [209]. Действительно, при БА в мозге повышена частота клеток с анеуплоидным набором хромосом, чаще всего клеток, содержащих дополнительную хромосому 21 [99].
Об общих генетических механизмах БА и СД свидетельствуют также следующие данные. В семьях с наследственной предрасположенностью к развитию БА повышена частота рождения детей с СД, а в семьях, в которых у молодых женщин (до 35 лет) часто рождаются дети с СД, повышена и частота БА [210, 211]. Так как СД связывают, в первую очередь, с неправильным расхождением хромосом при созревании яйцеклеток предположили, что у таких женщин повышен общий уровень нерасхождения хромосом, и они особенно предрасположены к БА [212]. Как оказалось, у молодых матерей детей с СД частота БА в 5 раз выше, чем в среднем по популяции [213]. В число генетических факторов, способствующих развитию БА при СД, входят мутации в генах APP, PS1, PS2, ACT и APOE [214]. Ген ACT кодирует α1-антихимотрипсин, компонент амилоидных бляшек, взаимодействующий с Aβ. Предполагается, что аллель ACT-17A обуславливает предрасположенность к БА [215, 216]. Ген APP кодирует белок-предшественник Аβ, который подвергается посттрансляционной модификации с образованием зрелого β-амилоида при участии α- и γ-секретазных комплексов. В состав этих комплексов входят пресенилины, кодируемые генами PSEN1 (PS1) и PSEN2 (PS2) [217]. Ген APOE кодирует три изоформы аполипопротеина Е (ε2, ε3, ε4). Носительство аллеля APOE4, кодирующего изоформу ε4, определяет предрасположенность к БА [218]. У молодых матерей детей с СД чаще встречается генотип APOE4/APOE4 [219]. Изоформа ε4 сильнее других способствует образованию олигомеров Aβ [220]. Кроме того, ε4 способен связываться с такими белками микротрубочек, как MAP (microtubule-assosiated proteins), что может влиять на работу микротрубочек цитоскелета и приводить к неправильному расхождению хромосом [221].
ЗНАЧЕНИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ ВЗАИМОДЕЙСТВИЯ МП I И МП II ДЛЯ МЕДИЦИНЫ: P53, АМИЛОИДЫ И КАНЦЕРОГЕНЕЗ
Онкологические и амилоидные нейродегенеративные заболевания – две самые распространенные группы заболеваний с высокой смертностью. Возникновение и прогрессия онкологических заболеваний тесно связаны с накоплением мутаций, а нейродегенеративные заболевания рассматривают, прежде всего, в связи с амилоидизацией белков. Необычную, преимущественно обратную, корреляцию между этими двумя группами патологий обнаружили еще в 1954 г., когда заметили, что у пациентов с БП реже диагностируют рак [222]. В дальнейшем это наблюдение подтвердили более чем в 30 исследованиях [223–229]. Неоднократно показано снижение риска развития таких видов рака, как рак легкого, кожи, молочной и предстательной желез у пациентов с БП [230, 231]. Единственное исключение представляет злокачественная меланома, частота которой увеличивается при БП. Интересно, что меланоциты кожи и нейроны черной субстанции это пигментированные клетки, имеющие общее происхождение. У пациентов с диагнозом БА риск развития опухолей снижен на 50%, а у индивидов, у которых был диагностирован рак, БА встречается на 35% реже, чем в контрольной группе лиц соответствующего возраста (средняя популяционная частота) [232]. Эти данные подтверждаются и результатами других исследований [233, 234]. Сходные корреляции обнаружены также между онкологическими и такими нейродегенеративными заболеваниями, как БХ и БАС [227, 235].
Механизм такой необычной зависимости не установлен, однако выдвинуто несколько предположений. И БА, и разные виды рака относятся к довольно агрессивным заболеваниям с высокой смертностью, поэтому можно предположить, что обратная зависимость между частотами этих патологий вызвана тем, что пациенты одной группы не доживают до манифестации другого заболевания. Чтобы уменьшить влияние смертности на частоту заболеваний, нужно использовать данные, полученные в результате длительного наблюдения. Довольно хорошо исследованную группу лиц, находящихся под длительным медицинским наблюдением, представляют жители Японии, пережившие атомную бомбардировку, у которых повышен риск развития рака. В одном из исследований сообщается, что у входящих в эту группу лиц с диагнозом БА рак развивается на 70% реже, чем у лиц того же возраста без признаков деменции [236]. Согласно результатам эпидемиологического исследования [234], обнаруженная обратная зависимость между частотой БА и рака сохраняется, даже если исключить из анализа людей, не доживших до 80 лет. Стоит отметить, что подобной корреляции между частотой онкологических заболеваний и сосудистых деменций (группа нейродегенеративных заболеваний неамилоидной природы) не обнаружено.
Еще одно объяснение обратной корреляции между риском онкологических и амилоидных заболеваний может заключаться в том, что терапия, применяемая при одном из указанных заболеваний, может снизить риск развития другого. Так, например, при раке применяют лучевую или химиотерапию, а для снижения симптомов БП используют леводопу. Известно, что предварительное облучение малыми дозами ионизирующего облучения способствует лучшей переносимости высоких доз радиации [237–239]. Механизм этого действия связан с тем, что небольшие дозы мутагена вызывают активацию клеточных систем репарации, например, синтез ферментов систем репарации ДНК, и клеточные повреждения, вызванные последующим облучением большими дозами, быстрее устраняются. Возможно, снижение частоты БА у онкологических больных связано с применением препаратов, блокирующих деление клеток. Предполагается, что воздействие противоопухолевой терапии способствует активации как системы репарации ДНК, так и системы восстановления белков (шапероны и убиквитин-протеазы), что, с одной стороны, ускоряет устранение окислительных повреждений, вызванных токсичными формами амилоидных белков, а с другой, способствует удалению белковых молекул с измененной пространственной структурой. Снижение частоты опухолевых заболеваний у пациентов с амилоидозами может быть связано с активацией систем репарации ДНК в результате окислительного стресса, вызванного присутствием токсичных молекул белков, и ускоренным устранением повреждений ДНК, что препятствует образованию раковых клеток. Это теоретическое предположение требует экспериментальной проверки с использованием модельных организмов и мутагенов. Некоторые работы в этом направлении уже проводятся. Так, недавно показали, что у трансгенных мух D. melanogaster, синтезирующих Aβ1-42 человека (модель БА), небольшие дозы ионизирующего облучения подавляют развитие дефектов глаз и крыльев, вызванных накоплением амилоидных агрегатов [240].
Получены свидетельства того, что переход белков в амилоидное состояние может способствовать возникновению и росту раковых опухолей. Известны некоторые мутации в гене-супрессоре опухолей p53, приводящие к синтезу мутантного белка с амилоидными свойствами [241]. Мутантные молекулы р53 склонны к агрегации и формируют амилоидоподобные полимеры, в которые включаются также нормальные молекулы p53, что приводит к снижению концентрации функционального белка в цитоплазме. Такие мутации доминантны, они приводят к развитию рака у их носителя, несмотря на присутствие копии гена дикого типа [242].
Таким образом, имеющиеся данные позволяют сделать вывод о сложном взаимном влиянии механизмов, лежащих в основе развития онкологических и амилоидных заболеваний, и о возможности как прямой, так и обратной корреляции между развитием амилоидозов и канцерогенезом. Аналогичные выводы были сделаны и по результатам изучения данных почти 4 млн. пациентов. Оказалось, что риск смерти от БА у больных с различными формами рака в разной степени зависит от таких факторов, как раса, тип опухоли, возраст, в котором был диагностирован рак, время, прошедшее с постановки диагноза, а также от применения химио- и радиотерапии [243]. В данный момент мы можем только предполагать, что взаимодействие МП I и МП II может иметь как различные формы, так и различные последствия для пациентов. В настоящее время эта область науки находится на этапе накопления сведений и формирования гипотез.
В связи с обсуждаемой темой представляет интерес блок экспериментальных данных о том, что при некоторых опухолевых заболеваниях изменяется экспрессия генов, кодирующих белки-предшественники прионов и амилоидов. Увеличение продукции белка PrP отмечено в клетках остеосаркомы, глиобластомы, меланомы, рака желудка, поджелудочной железы, легких и других опухолей [58]. Экспрессия гена APP, кодирующего Аβ, а также его гомолога APLP2 (APP-like protein 2), повышена в клетках рака молочной железы, легкого, щитовидной и предстательной железы, а также меланомы и саркомы Юинга [244–249]. Повышение продукции белков APP и PrP во многих клеточных линиях опухолевых клеток коррелирует с усиленной пролиферацией [244, 250] и повышенной миграцией клеток [251, 252], что придает опухолевым клеткам способность к метастазированию. При этом экспериментальное снижение концентрации этих белков приводит к удлинению клеточного цикла, клетки реже делятся и даже синтезируют маркеры апоптоза [245, 253]. Кроме того, снижение продукции белка APP в клеточных линиях меланомы человека повышает их чувствительность к химиотерапевтическим препаратам [248, 249], которые блокируют вилку репликации и действуют в основном на активно делящиеся клетки. Этот факт указывает на то, что в клетках с низкой продукцией APP ДНК не синтезируется.
Необходимо отметить, что в работах [58, 242–251] оценивали только уровень экспрессии генов, кодирующих амилоидогенные белки, но не определяли присутствие самих амилоидов. Поэтому невозможно сделать однозначный вывод о прямом или обратном влиянии именно амилоидов на канцерогенез. Возможно, одно из объяснений существования обратной корреляции между онкологическими и нейродегенеративными заболеваниями заключается в том, что повышение концентрации амилоидогенного белка (APP или PrP) в цитоплазме является фактором, способствующим повышению вероятности амилоидизации. А появление в клетке амилоидных агрегатов снижает концентрацию растворимого активного белка, что равнозначно снижению продукции этого белка, и может приводить к замедлению клеточного цикла и даже апоптозу [245, 253]. Конечно, это предположение нуждается в подтверждении, но уже сейчас изучают возможность терапии рака молочной железы с помощью ингибирования APP [254 ] . В любом случае, эти данные имеют важное значение для поиска молекулярных механизмов, обеспечивающих взаимодействие факторов контроля стабильности протеома и генома.
ЗАКЛЮЧЕНИЕ
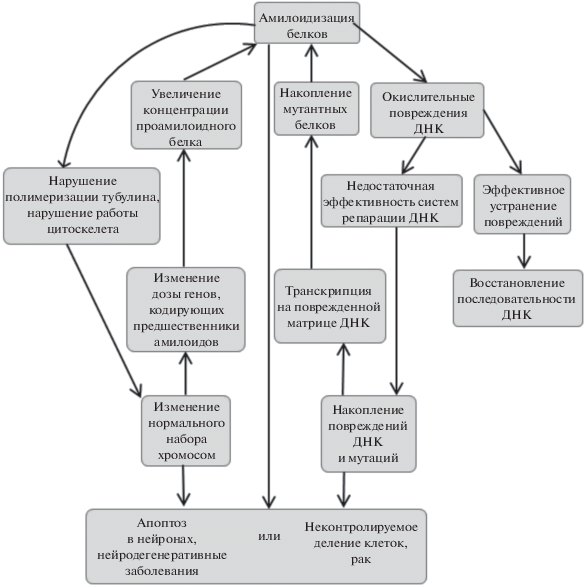
Настоящий обзор представляет собой, насколько нам известно, первую попытку рассмотрения геномно-протеомных взаимодействий с единой позиции матричного принципа, согласно которому МП подразделяются на две категории – МП I, оперирующие с линейными матрицами, характерными для процессов воспроизведения носителей генетической информации (репликации) и реализации генетической информации (транскрипции и трансляции), с одной стороны, и МП II, оперирующие с пространственными, или конформационными (белковыми) матрицами, с другой (рис. 1). С этой точки зрения наследственность представляет собой комбинацию признаков, зашифрованных как в виде линейной последовательности нуклеиновых кислот (геном), так и в виде пространственной организации белковых молекул (протеом). Стоит отметить, что первые данные, свидетельствующие о возможном взаимном влиянии МП I и МП II, появились давно, около 20–30 лет назад, и стали стремительно накапливаться, особенно в последние 5 лет. Нами рассмотрены имеющиеся на сегодняшний момент данные о взаимодействии МП I и МП II, а также вероятные механизмы этих взаимодействий (рис. 3). Постоянно получаемые новые факты взаимного влияния МП I и МП II имеют значение как для понимания фундаментальных механизмов наследственности и изменчивости, так и для медицинских приложений в связи с обнаружением прямой и обратной корреляции между онкологическими и амилоидными заболеваниями. На основе приведенных в обзоре сведений мы можем предположить, что при изучении генетических механизмов и последствий взаимодействия МП I и МП II необходимо помнить об общности свойств этих процессов, таких как неоднозначность и способность к коррекции ошибок. Несомненно, эта область сулит нам много открытий, и мы находимся в начале многообещающего пути выяснения механизмов рассмотренных взаимодействий.
Исследование выполнено при финансовой поддержке РФФИ в рамках научного проекта № 19-14-50230 и при государственной финансовой поддержке ведущих университетов Российской Федерации в рамках программы ITMO Fellowship and Professorship Program.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы сообщают об отсутствии конфликта интересов.
Список литературы
Crick F.H. (1958) On protein synthesis. Symp. Soc. Exp. Biol. 12, 138–163.
Crick F. (1970) Central dogma of molecular biology. Nature. 227, 561–563.
Кольцов Н.К. (1936) Наследственные молекулы. В кн.: Организация клетки. М.-Л.: Гос. изд. биол. и мед. лит., pp. 585–622.
Инге-Вечтомов С.Г. (2013) Матричный принцип как парадигма современной генетики. Генетика. 49, 9–15.
Инге-Вечтомов С.Г. (2015) От хромосомной теории к матричному принципу. Генетика. 51, 397–408.
Лобашев М.Е. (1947) Физиологическая (паранекротическая) гипотеза мутационного процесса. Вестн. Лен. гос. унив. 8, 10–29.
Timofeeff-Ressovsky N.W., Zimmer K.G., Delbrück M. (1935) Über die Natur der Genmutation und der Genstruktur. Nachr. Ges. Wiss. Gottingen. Fachr. 13, 189–245.
Инге-Вечтомов С.Г. (2015) Ретроспектива генетики. Санкт-Петербург: Изд-во Н-Л, 336 с.
Sydow J.F., Cramer P. (2009) RNA polymerase fidelity and transcriptional proofreading. Curr. Opin. Struct. Biol. 19, 732–739.
Roy H., Ibba M. (2006) Phenylalanyl-tRNA synthetase contains a dispensable RNA-binding domain that contributes to the editing of noncognate aminoacyl-tRNA. Biochemistry. 45, 9156–9162.
Gorini L. (1974) Streptomycin and misreading of the genetic code. In: Ribosomes. Eds Nomura M., Missieres A., Lengyel P. Cold Spring Harbor, N.Y.: Cold Spring Harbor Lab., pp. 791–803.
Hopfield J.J. (1974) Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl. Acad. Sci. USA. 71, 4135–4139.
Prusiner S.B. (1998) Prions. Proc. Natl. Acad. Sci. USA. 95, 13363–13383.
Wickner R.B., Shewmaker F., Edskes H., Kryndushkin D., Nemecek J., McGlinchey R., Bateman D., Winchester C.L. (2010) Prion amyloid structure explains templating: how proteins can be genes. FEMS Yeast Res. 10, 980–991.
Dobson C.M. (2004) Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol. 15, 3–16.
Tyedmers J., Mogk A., Bukau B. (2010) Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell. Biol. 11, 777–788.
Houck S.A., Singh S., Cyr D.M. (2012) Cellular responses to misfolded proteins and protein aggregates. Methods Mol. Biol. 832, 455–461.
Schnabel J. (2010) Protein folding: the dark side of proteins. Nature. 464, 828–829.
Kajava A.V., Aebi U., Steven A.C. (2005) The parallel superpleated beta-structure as a model for amyloid fibrils of human amylin. J. Mol. Biol. 348, 247–252.
Kajava A.V., Baxa U., Steven A.C. (2010) Beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 24, 1311–1319.
Ahmed A.B., Znassi N., Chateau M.T., Kajava A.V. (2015) A structure-based approach to predict predisposition to amyloidosis. Alzheimers Dement. 11, 681–690.
Bondarev S.A., Bondareva O.V., Zhouravleva G.A., Kajava A.V. (2018) BetaSerpentine: a bioinformatics tool for reconstruction of amyloid structures. Bioinformatics. 34, 599–608.
Eisenberg D., Jucker M. (2012) The amyloid state of proteins in human diseases. Cell. 148, 1188–1203.
Нижников А.А., Антонец К.С., Инге-Вечтомов С.Г. (2015) Амилоиды: от патогенеза к функции (обзор). Биохимия. 80, 1356–1375.
Coustou V., Deleu C., Saupe S., Begueret J. (1997) The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA. 94, 9773–9778.
Bateman D.A., Wickner R.B. (2013) The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genet. 9, e1003257.
Borchsenius A.S., Muller S., Newnam G.P., Inge-Vechtomov S.G., Chernoff Y.O. (2006) Prion variant maintained only at high levels of the Hsp104 disaggregase. Curr. Genet. 49, 21–29.
Barbitoff Y.A., Matveenko A.G., Moskalenko S.E., Zemlyanko O.M., Newnam G.P., Patel A., Chernova T.A., Chernoff Y.O., Zhouravleva G.A. (2017) To CURe or not to CURe? Differential effects of the chaperone sorting factor Cur1 on yeast prions are mediated by the chaperone Sis1. Mol. Microbiol. 105, 242–257.
Matveenko A.G., Barbitoff Y.A., Jay-Garcia L.M., Chernoff Y.O., Zhouravleva G.A. (2018) Differential effects of chaperones on yeast prions: CURrent view. Curr. Genet. 64, 317–325.
Choe Y.J., Park S.H., Hassemer T., Korner R., Vincenz-Donnelly L., Hayer-Hartl M., Hartl F.U. (2016) Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature. 531, 191–195.
Sitron C.S., Brandman O. (2019) CAT tails drive degradation of stalled polypeptides on and off the ribosome. Nat. Struct. Mol. Biol. 26, 450–459.
Wickner R.B. (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 264, 566–569.
Lacroute F. (1971) Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J. Bacteriol. 106, 519–522.
Cox B.S. (1965) [PSI], a cytoplasmic suppressor of super-suppressor in yeast. Heredity. 20, 505–521.
Derkatch I.L., Bradley M.E., Zhou P., Chernoff Y.O., Liebman S.W. (1997) Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 147, 507–519.
Sondheimer N., Lindquist S. (2000) Rnq1: an epigenetic modifier of protein function in yeast. Mol. Cell. 5, 163–172.
Du Z., Park K.W., Yu H., Fan Q., Li L. (2008) Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 40, 460–465.
Patel B.K., Gavin-Smyth J., Liebman S.W. (2009) The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat. Cell Biol. 11, 344–349.
Alberti S., Halfmann R., King O., Kapila A., Lindquist S. (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 137, 146–158.
Suzuki G., Shimazu N., Tanaka M. (2012) A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science. 336, 355–359.
Sipe J.D., Cohen A.S. (2000) Review: history of the amyloid fibril. J. Struct. Biol. 130, 88–98.
Hamodrakas S.J. (2011) Protein aggregation and amyloid fibril formation prediction software from primary sequence: towards controlling the formation of bacterial inclusion bodies. FEBS J. 278, 2428–2435.
Tsolis A.C., Papandreou N.C., Iconomidou V.A., Hamodrakas S.J. (2013) A consensus method for the prediction of 'aggregation-prone' peptides in globular proteins. PLoS One. 8, e54175.
Kryndushkin D., Pripuzova N., Burnett B.G., Shewmaker F. (2013) Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells. J. Biol. Chem. 288, 27100–27111.
Nizhnikov A.A., Alexandrov A.I., Ryzhova T.A., Mitkevich O.V., Dergalev A.A., Ter-Avanesyan M.D., Galkin A.P. (2014) Proteomic screening for amyloid proteins. PLoS One. 9, e116003.
Nizhnikov A.A., Ryzhova T.A., Volkov K.V., Zadorsky S.P., Sopova J.V., Inge-Vechtomov S.G., Galkin A.P. (2016) Interaction of prions causes heritable traits in Saccharomyces cerevisiae. PLoS Genet. 12, e1006504.
Pan K.M., Baldwin M., Nguyen J., Gasset M., Serban A., Groth D., Mehlhorn I., Huang Z., Fletterick R.J., Cohen F.E., Prusiner S.B. (1993) Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA. 90, 10962–10966.
Kryndushkin D.S., Alexandrov I.M., Ter-Avanesyan M.D., Kushnirov V.V. (2003) Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278, 49636–49643.
Sipe J.D., Benson M.D., Buxbaum J.N., Ikeda S., Merlini G., Saraiva M.J., Westermark P. (2014) Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 21, 221–224.
Галкин А.П., Велижанина М.Е., Сопова Ю.В., Шенфельд А.А., Задорский С.П. (2018) Прионы и неинфекционные амилоиды млекопитающих — сходства и отличия. Биохимия. 83, 1476–1489.
Steensma D.P. (2001) “Congo” red: out of Africa? Arch. Pathol. Lab. Med. 125, 250–252.
LeVine H., 3rd. (1999) Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284.
Kushnirov V.V., Ter-Avanesyan M.D. (1998) Structure and replication of yeast prions. Cell. 94, 13–16.
Chernoff Y.O., Lindquist S.L., Ono B., Inge-Vechtomov S.G., Liebman S.W. (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [PSI+]. Science. 268, 880–884.
Liebman S.W., Chernoff Y.O. (2012) Prions in yeast. Genetics. 191, 1041–1072.
Dergalev A.A., Alexandrov A.I., Ivannikov R.I., Ter-Avanesyan M.D., Kushnirov V.V. (2019) Yeast Sup35 prion structure: two types, four parts, many variants. Int. J. Mol. Sci. 20.
Prusiner S.B. (1989) Scrapie prions. Annu. Rev. Microbiol. 43, 345–374.
Yang X., Cheng Z., Zhang L., Wu G., Shi R., Gao Z., Li C. (2017) Prion protein family contributes to tumorigenesis via multiple pathways. Adv. Exp. Med. Biol. 1018, 207–224.
Gajdusek D.C. (1991) The transmissible amyloidoses: genetical control of spontaneous generation of infectious amyloid proteins by nucleation of configurational change in host precursors: kuru-CJD-GSS-scrapie-BSE. Eur. J. Epidemiol. 7, 567–577.
Prusiner S.B., Scott M.R. (1997) Genetics of prions. Annu. Rev. Genet. 31, 139–175.
Prusiner S.B. (2001) Shattuck lecture–neurodegenerative diseases and prions. N. Engl. J. Med. 344, 1516–1526.
Wickner R.B., Edskes H.K., Son M., Bezsonov E.E., DeWilde M., Ducatez M. (2018) Yeast prions compared to functional prions and amyloids. J. Mol. Biol. 430, 3707–3719.
Roberts B.T., Wickner R.B. (2003) Heritable activity: a prion that propagates by covalent autoactivation. Genes Dev. 17, 2083–2087.
Brown J.C., Lindquist S. (2009) A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev. 23, 2320–2332.
Chakravarty A.K., Smejkal T., Itakura A.K., Garcia D.M., Jarosz D.F. (2020) A non-amyloid prion particle that activates a heritable gene expression program. Mol. Cell. 77, 251–265 e259.
Derkatch I.L., Chernoff Y.O., Kushnirov V.V., Inge-Vechtomov S.G., Liebman S.W. (1996) Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 144, 1375–1386.
Schlumpberger M., Prusiner S.B., Herskowitz I. (2001) Induction of distinct [URE3] yeast prion strains. Mol. Cell. Biol. 21, 7035–7046.
King C.Y. (2001) Supporting the structural basis of prion strains: induction and identification of [PSI] variants. J. Mol. Biol. 307, 1247–1260.
Prusiner S.B. (2013) Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 47, 601–623.
Liebman S.W., Sherman F. (1979) Extrachromosomal psi+ determinant suppresses nonsense mutations in yeast. J. Bacteriol. 139, 1068–1071.
Wickner R.B., Masison D.C., Edskes H.K. (1995) [PSI] and [URE3] as yeast prions. Yeast. 11, 1671–1685.
Derkatch I.L., Bradley M.E., Hong J.Y., Liebman S.W. (2001) Prions affect the appearance of other prions: the story of [PIN(+)]. Cell. 106, 171–182.
Антонец К.С., Кливер С.Ф., Полев Д.Е., Шувалова А.Р., Андреева Е.А., Инге-Вечтомов С.Г., Нижников А.А. (2017) Различные механизмы фенотипических эффектов инактивации и прионизации белка Swi1 дрожжей Saccharomyces cerevisiae. Биохимия. 82, 1497–1509.
Malovichko Y.V., Antonets K.S., Maslova A.R., Andreeva E.A., Inge-Vechtomov S.G., Nizhnikov A.A. (2019) RNA sequencing reveals specific transcriptomicsignatures distinguishing effects of the [SWI(+)] prion and SWI1 deletion in yeast Saccharomyces cerevisiae. Genes (Basel). 10(3), 212. https://doi.org/10.3390/genes10030212
Chapman M.R., Robinson L.S., Pinkner J.S., Roth R., Heuser J., Hammar M., Normark S., Hultgren S.J. (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 295, 851–855.
Alteri C.J., Xicohtencatl-Cortes J., Hess S., Caballero-Olin G., Giron J.A., Friedman R.L. (2007) Mycobacterium tuberculosis produces pili during human infection. Proc. Natl. Acad. Sci. USA. 104, 5145–5150.
Dueholm M.S., Petersen S.V., Sonderkaer M., Larsen P., Christiansen G., Hein K.L., Enghild J.J., Nielsen J.L., Nielsen K.L., Nielsen P.H., Otzen D.E. (2010) Functional amyloid in Pseudomonas. Mol. Microbiol. 77, 1009–1020.
Kalebina T.S., Plotnikova T.A., Gorkovskii A.A., Selyakh I.O., Galzitskaya O.V., Bezsonov E.E., Gellissen G., Kulaev I.S. (2008) Amyloid-like properties of Saccharomyces cerevisiae cell wall glucantransferase Bgl2p: prediction and experimental evidences. Prion. 2, 91–96.
Ramsook C.B., Tan C., Garcia M.C., Fung R., Soybelman G., Henry R., Litewka A., O’Meally S., Otoo H.N., Khalaf R.A., Dranginis A.M., Gaur N.K., Klotz S.A., Rauceo J.M., Jue C.K., Lipke P.N. (2010) Yeast cell adhesion molecules have functional amyloid-forming sequences. Eukaryot. Cell. 9, 393–404.
Ryzhova T.A., Sopova J.V., Zadorsky S.P., Siniukova V.A., Sergeeva A.V., Galkina S.A., Nizhnikov A.A., Shenfeld A.A., Volkov K.V., Galkin A.P. (2018) Screening for amyloid proteins in the yeast proteome. Curr. Genet. 64, 469–478.
Sergeeva A.V., Sopova J.V., Belashova T.A., Siniukova V.A., Chirinskaite A.V., Galkin A.P., Zadorsky S.P. (2019) Amyloid properties of the yeast cell wall protein Toh1 and its interaction with prion proteins Rnq1 and Sup35. Prion. 13, 21–32.
Калебина Т.С., Рекстина В.В. (2019) Молекулярная организация клеточной поверхности дрожжей. Молекул. биология. 53, 968–981.
Hewetson A., Do H.Q., Myers C., Muthusubramanian A., Sutton R.B., Wylie B.J., Cornwall G.A. (2017) Functional amyloids in reproduction. Biomolecules. 7.
Si K., Choi Y.B., White-Grindley E., Majumdar A., Kandel E.R. (2010) Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell. 140, 421–435.
Majumdar A., Cesario W.C., White-Grindley E., Jiang H., Ren F., Khan M.R., Li L., Choi E.M., Kannan K., Guo F., Unruh J., Slaughter B., Si K. (2012) Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell. 148, 515–529.
Stephan J.S., Fioriti L., Lamba N., Colnaghi L., Karl K., Derkatch I.L., Kandel E.R. (2015) The CPEB3 protein is a functional prion that interacts with the actin cytoskeleton. Cell. Rep. 11, 1772–1785.
Fowler D.M., Koulov A.V., Alory-Jost C., Marks M.S., Balch W.E., Kelly J.W. (2006) Functional amyloid formation within mammalian tissue. PLoS Biol. 4, e6.
Berchowitz L.E., Kabachinski G., Walker M.R., Carlile T.M., Gilbert W.V., Schwartz T.U., Amon A. (2015) Regulated formation of an amyloid-like translational repressor governs gametogenesis. Cell. 163, 406–418.
Sopova J.V., Koshel E.I., Belashova T.A., Zadorsky S.P., Sergeeva A.V., Siniukova V.A., Shenfeld A.A., Velizhanina M.E., Volkov K.V., Nizhnikov A.A., Kac-hkin D.V., Gaginskaya E.R., Galkin A.P. (2019) RNA-binding protein FXR1 is presented in rat brain in amyloid form. Sci. Rep. 9, 18983.
Giraldo R. (2007) Defined DNA sequences promote the assembly of a bacterial protein into distinct amyloid nanostructures. Proc. Natl. Acad. Sci. USA. 104, 17388–17393.
Molina-Garcia L., Gasset-Rosa F., Moreno-Del Alamo M., Fernandez-Tresguerres M.E., Moreno-Diaz de la Espina S., Lurz R., Giraldo R. (2016) Functional amyloids as inhibitors of plasmid DNA replication. Sci. Rep. 6, 25425.
Chakrabortee S., Kayatekin C., Newby G.A., Mendillo M.L., Lancaster A., Lindquist S. (2016) Luminidependens (LD) is an Arabidopsis protein with prion behavior. Proc. Natl. Acad. Sci. USA. 113, 6065–6070.
Vasudevan S., Steitz J.A. (2007) AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell. 128, 1105–1118.
Majumder M., House R., Palanisamy N., Qie S., Day T.A., Neskey D., Diehl J.A., Palanisamy V. (2016) RNA-binding protein FXR1 regulates p21 and TERC RNA to bypass p53-mediated cellular senescence in OSCC. PLoS Genet. 12, e1006306.
Antonets K.S., Nizhnikov A.A. (2017) Predicting amyloidogenic proteins in the proteomes of plants. Int. J. Mol. Sci. 18(10), 2155. https://doi.org/10.3390/ijms18102155
Harrison A.F., Shorter J. (2017) RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 474, 1417–1438.
Li L., McGinnis J.P., Si K. (2018) Translational control by prion-like proteins. Trends Cell. Biol. 28, 494–505.
Coppede F., Migliore L. (2015) DNA damage in neurodegenerative diseases. Mutat. Res. 776, 84–97.
Iourov I.Y., Vorsanova S.G., Liehr T., Yurov Y.B. (2009) Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobio.l Dis. 34, 212–220.
Migliore L., Botto N., Scarpato R., Petrozzi L., Cipriani G., Bonuccelli U. (1999) Preferential occurrence of chromosome 21 malsegregation in peripheral blood lymphocytes of Alzheimer disease patients. Cytogenet. Cell. Genet. 87, 41–46.
Trippi F., Botto N., Scarpato R., Petrozzi L., Bonuccelli U., Latorraca S., Sorbi S., Migliore L. (2001) Spontaneous and induced chromosome damage in somatic cells of sporadic and familial Alzheimer’s disease patients. Mutagenesis. 16, 323–327.
Yurov Y.B., Vorsanova S.G., Liehr T., Kolotii A.D., Iourov I.Y. (2014) X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 7, 20. https://doi.org/10.1186/1755-8166-7-20
Migliore L., Scarpato R., Coppede F., Petrozzi L., Bonuccelli U., Rodilla V. (2001) Chromosome and oxidative damage biomarkers in lymphocytes of Parkinson’s disease patients. Int. J. Hyg. Environ. Hlth. 204, 61–66.
Hegde M.L., Gupta V.B., Anitha M., Harikrishna T., Shankar S.K., Muthane U., Subba Rao K., Jagannatha Rao K.S. (2006) Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Arch. Biochem. Biophys. 449, 143–156.
Hanson P.K. (2018) Saccharomyces cerevisiae: A unicellular model genetic organism of enduring importance. Curr. Prot. Essential Lab. Techn.
Giaever G., Chu A.M., Ni L., Connelly C., Riles L., Veronneau S., Dow S., Lucau-Danila A., Anderson K., Andre B., Arkin A.P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K.D., Flaherty P., Foury F., Garfinkel D.J., Gerstein M., Gotte D., Guldener U., Hegemann J.H., Hempel S., Herman Z., Jaramillo D.F., Kelly D.E., Kelly S.L., Kotter P., LaBonte D., Lamb D.C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S.L., Revuelta J.L., Roberts C.J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D.D., Sookhai-Mahadeo S., Storms R.K., Strathern J.N., Valle G., Voet M., Volckaert G., Wang C.Y., Ward T.R., Wilhelmy J., Winzeler E.A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J.D., Snyder M., Philippsen P., Davis R.W., Johnston M. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature. 418, 387–391.
Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., Chu A.M., Connelly C., Davis K., Dietrich F., Dow S.W., El Bakkoury M., Foury F., Friend S.H., Gentalen E., Giaever G., Hegemann J.H., Jones T., Laub M., Liao H., Liebundguth N., Lockhart D.J., Lucau-Danila A., Lussier M., M’Rabet N., Menard P., Mittmann M., Pai C., Rebischung C., Revuelta J.L., Riles L., Roberts C.J., Ross-MacDonald P., Scherens B., Snyder M., Sookhai-Mahadeo S., Storms R.K., Veronneau S., Voet M., Volckaert G., Ward T.R., Wysocki R., Yen G.S., Yu K., Zimmermann K., Philippsen P., Johnston M., Davis R.W. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 285, 901–906.
Jones G.M., Stalker J., Humphray S., West A., Cox T., Rogers J., Dunham I., Prelich G. (2008) A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat. Methods. 5, 239–241.
Ng P.C., Wong E.D., MacPherson K.A., Aleksander S., Argasinska J., Dunn B., Nash R.S., Skrzypek M.S., Gondwe F., Jha S., Karra K., Weng S., Miyasato S., Simison M., Engel S.R., Cherry J.M. (2020) Transcriptome visualization and data availability at the Saccharomyces Genome Database. Nucl. Acids Res. 48, D743–D748.
Schmidt A. (2018) Merged map of the yeast proteome. Cell Syst. 6, 150–152.
Chernova T.A., Chernoff Y.O., Wilkinson K.D. (2019) Yeast models for amyloids and prions: environmental modulation and drug discovery. Molecules. 24, 3388. https://doi.org/10.3390/molecules24183388
Инге-Вечтомов С.Г. (2011) Прионы дрожжей как модель нейродегенеративных инфекционных амилоидозов человека. Онтогенез. 42, 337–345.
Rencus-Lazar S., DeRowe Y., Adsi H., Gazit E., Laor D. (2019) Yeast models for the study of amyloid-associated disorders and development of future therapy. Front. Mol. Biosci. 6, 15.
Klein H.L., Bacinskaja G., Che J., Cheblal A., Elango R., Epshtein A., Fitzgerald D.M., Gomez-Gonzalez B., Khan S.R., Kumar S., Leland B.A., Marie L., Mei Q., Mine-Hattab J., Piotrowska A., Polleys E.J., Putnam C.D., Radchenko E.A., Saada A.A., Sakofsky C.J., Shim E.Y., Stracy M., Xia J., Yan Z., Yin Y., Aguilera A., Argueso J.L., Freudenreich C.H., Gasser S.M., Gordenin D.A., Haber J.E., Ira G., Jinks-Robertson S., King M.C., Kolodner R.D., Kuzminov A., Lambert S.A., Lee S.E., Miller K.M., Mirkin S.M., Petes T.D., Rosenberg S.M., Rothstein R., Symington L.S., Zawadzki P., Kim N., Lisby M., Malkova A. (2019) Guidelines for DNA recombination and repair studies: cellular assays of DNA repair pathways. Microb. Cell. 6, 1–64.
Inge-Vechtomov S.G., Repnevskaya M.V. (1989) Phenotypic expression of primary lesions of genetic material in Saccharomyces yeasts. Genome. 31, 497–502.
Инге-Вечтомов С.Г., Репневская М.В., Карпова Т.С. (1986) Изучение скрещивание клеток одинакового типа спаривания у дрожжей сахаромицетов. Генетика. 22, 2625–2636.
Степченкова Е.И., Коченова О.В., Жук А.С., Андрейчук Ю.В., Инге-Вечтомов С.Г. (2011) Фенотипическое проявление и взаимопревращение первичных повреждений генетического материала, учитываемых в альфа-тесте, у дрожжей Saccharomyces cerevisiae. Гигиена и санитария. 6, 64–69.
Жук А.С., Ширяева А.А., Коченова О.В., Андрейчук Ю.В., Степченкова Е.И., Инге-Вечтомов С.Г. (2013) Альфа-тест – система для оценки генетически активных факторов. Актуальные проблемы гуманитарных и естественных наук. 11, 54–60.
Степченкова Е.И., Коченова О.В., Инге-Вечтомов С.Г. (2009) “Незаконная” гибридизация и “незаконная” цитодукция у гетероталличных дрожжей Saccharomyces cerevisiae как система для анализа генетической активности экзогенных и эндогенных факторов в “альфа-тесте”. Вестник Санкт-Петербургского государственного университета. 3, 129–140.
Репневская М.В., Кашкин П.К., Инге-Вечтомов С.Г. (1989) Модификационные изменения генетического материала у дрожжей сахаромицетов. Генетика. 25, 425–436.
Андрейчук Ю.В., Ширяева А.А., Жук А.А., Степченкова Е.И., Инге-Вечтомов С.Г. (2015) Влияние прионизации белка Sup35 [PSI+] на частоту генетических нарушений, учитываемых в альфа-тесте у дрожжей Saccharomyces cerevisiae. Экологическая генетика. 13, 22–24.)
Абилев С.К., Глазер В.М. (2015) Мутагенез с основами генетоксикологии: учебное пособие. СПб.: Нестер-История.
Friedberg E.C., Walker G.C., Siede W., Wood R.D., Schultz R.A., Ellenberger T. (2006) DNA repair and mutagenesis. Sec. Ed. Washington, D.C.: ASM Press.
Инге-Вечтомов С.Г. (2015) Генетика с основами селекции. СПб.: Изд-во Н-Л.
Waisertreiger I.S., Liston V.G., Menezes M.R., Kim H.M., Lobachev K.S., Stepchenkova E.I., Tahirov T.H., Rogozin I.B., Pavlov Y.I. (2012) Modulation of mutagenesis in eukaryotes by DNA replication fork dynamics and quality of nucleotide pools. Environ. Mol. Mutagen. 53, 699–724.
Kunkel T.A. (2004) DNA replication fidelity. J. Biol. Chem. 279, 16895–16898.
Ferguson D.O., Alt F.W. (2001) DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene. 20, 5572–5579.
Dey P. (2004) Aneuploidy and malignancy: an unsolved equation. J. Clin. Pathol. 57, 1245–1249.
Matsuura S., Ito E., Tauchi H., Komatsu K., Ikeuchi T., Kajii T. (2000) Chromosomal instability syndrome of total premature chromatid separation with mosaic variegated aneuploidy is defective in mitotic-spindle checkpoint. Am. J. Hum. Genet. 67, 483–486.
Chatterjee N., Walker G.C. (2017) Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 58, 235–263.
Martin L.J. (2008) DNA damage and repair: relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 67, 377–387.
Bernstein C., Prasad A.R., Nfonsam V., Bernstein H. (2013) DNA damage, DNA repair and cancer. In: New Res. Directions DNA Repair. Chen, C.: InTech, 413–465.
Bernstein C., Bernstein H. (2015) Epigenetic reduction of DNA repair in progression to gastrointestinal cancer. W. J. Gastrointest. Oncol. 7, 30–46.
Drake J.W. (1999) The distribution of rates of spontaneous mutation over viruses, prokaryotes, and eukaryotes. Ann. N.Y. Acad. Sci. 870, 100–107.
Tiwari V., Wilson D.M., 3rd. (2019) DNA damage and associated DNA repair defects in disease and premature aging. Am. J. Hum. Genet. 105, 237–257.
Lyras L., Cairns N.J., Jenner A., Jenner P., Halliwell B. (1997) An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J. Neurochem. 68, 2061–2069.
Nunomura A., Honda K., Takeda A., Hirai K., Zhu X., Smith M.A., Perry G. (2006) Oxidative damage to RNA in neurodegenerative diseases. J. Biomed. Biotechnol. 2006, 82323.
Mecocci P., MacGarvey U., Beal M.F. (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 36, 747–751.
Lovell M.A., Gabbita S.P., Markesbery W.R. (1999) Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. J. Neurochem. 72, 771–776.
Mecocci P., Polidori M.C., Ingegni T., Cherubini A., Chionne F., Cecchetti R., Senin U. (1998) Oxidative damage to DNA in lymphocytes from AD patients. Neurology. 51, 1014–1017.
Wang J., Markesbery W.R., Lovell M.A. (2006) Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 96, 825–832.
Mullaart E., Boerrigter M.E., Ravid R., Swaab D.F., Vijg J. (1990) Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol. Aging. 11, 169–173.
Alam Z.I., Jenner A., Daniel S.E., Lees A.J., Cairns N., Marsden C.D., Jenner P., Halliwell B. (1997) Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 69, 1196–1203.
Seet R.C., Lee C.Y., Lim E.C., Tan J.J., Quek A.M., Chong W.L., Looi W.F., Huang S.H., Wang H., Chan Y.H., Halliwell B. (2010) Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radic. Biol. Med. 48, 560–566.
Ferrante R.J., Browne S.E., Shinobu L.A., Bowling A.C., Baik M.J., MacGarvey U., Kowall N.W., Brown R.H., Jr., Beal M.F. (1997) Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 69, 2064–2074.
Bogdanov M., Brown R.H., Matson W., Smart R., Hayden D., O’Donnell H., Flint Beal M., Cudkowicz M. (2000) Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 29, 652–658.
Aguirre N., Beal M.F., Matson W.R., Bogdanov M.B. (2005) Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radic. Res. 39, 383–388.
Warita H., Hayashi T., Murakami T., Manabe Y., Abe K. (2001) Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Brain Res. Mol. Brain Res. 89, 147–152.
Ferri A., Cozzolino M., Crosio C., Nencini M., Casciati A., Gralla E.B., Rotilio G., Valentine J.S., Carri M.T. (2006) Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA. 103, 13860–13865.
Cheignon C., Tomas M., Bonnefont-Rousselot D., Faller P., Hureau C., Collin F. (2018) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464.
Butterfield D.A., Swomley A.M., Sultana R. (2013) Amyloid beta-peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid. Redox Signal. 19, 823–835.
Yatin S.M., Varadarajan S., Link C.D., Butterfield D.A. (1999) In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1-42). Neurobiol. Aging. 20, 325–330; discussion 339–342.
Butterfield D.A., Yatin S.M., Link C.D. (1999) In vitro and in vivo protein oxidation induced by Alzheimer’s disease amyloid beta-peptide (1-42). Ann. N.Y. Acad. Sci. 893, 265–268.
Turnbull S., Tabner B.J., Brown D.R., Allsop D. (2003) Generation of hydrogen peroxide from mutant forms of the prion protein fragment PrP121-231. Biochemistry. 42, 7675–7681.
Scudamore O., Ciossek T. (2018) Increased oxidative stress exacerbates alpha-synuclein aggregation in vivo. J. Neuropathol. Exp. Neurol. 77, 443–453.
Cherny D., Hoyer W., Subramaniam V., Jovin T.M. (2004) Double-stranded DNA stimulates the fibrillation of alpha-synuclein in vitro and is associated with the mature fibrils: an electron microscopy study. J. Mol. Biol. 344, 929–938.
Ohyagi Y., Asahara H., Chui D.H., Tsuruta Y., Sakae N., Miyoshi K., Yamada T., Kikuchi H., Taniwaki T., Murai H., Ikezoe K., Furuya H., Kawarabayashi T., Shoji M., Checler F., Iwaki T., Makifuchi T., Takeda K., Kira J., Tabira T. (2005) Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 19, 255–257.
Nizhnikov A.A., Antonets K.S., Bondarev S.A., Inge-Vechtomov S.G., Derkatch I.L. (2016) Prions, amyloids, and RNA: pieces of a puzzle. Prion. 10, 182–206.
Chaudhry M.A. (2007) Base excision repair of ionizing radiation-induced DNA damage in G1 and G2 cell cycle phases. Cancer Cell Int. 7, 15.
Branzei D., Foiani M. (2008) Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell. Biol. 9, 297–308.
Schroering A.G., Edelbrock M.A., Richards T.J., Williams K.J. (2007) The cell cycle and DNA mismatch repair. Exp. Cell Res. 313, 292–304.
Zhao X., Wei C., Li J., Xing P., Li J., Zheng S., Chen X. (2017) Cell cycle-dependent control of homologous recombination. Acta Biochim. Biophys. Sin. (Shanghai). 49, 655–668.
Strathern J.N., Shafer B.K., McGill C.B. (1995) DNA synthesis errors associated with double-strand-break repair. Genetics. 140, 965–972.
Varga T., Aplan P.D. (2005) Chromosomal aberrations induced by double strand DNA breaks. DNA Repair (Amst). 4, 1038–1046.
McPhie D.L., Coopersmith R., Hines-Peralta A., Chen Y., Ivins K.J., Manly S.P., Kozlowski M.R., Neve K.A., Neve R.L. (2003) DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J. Neurosci. 23, 6914–6927.
Li J.C., Kaminskas E. (1985) Deficient repair of DNA lesions in Alzheimer’s disease fibroblasts. Biochem. Biophys. Res. Commun. 129, 733–738.
Robison S.H., Munzer J.S., Tandan R., Bradley W.G. (1987) Alzheimer’s disease cells exhibit defective repair of alkylating agent-induced DNA damage. Ann. Neurol. 21, 250–258.
Jones S.K., Nee L.E., Sweet L., Polinsky R.J., Bartlett J.D., Bradley W.G., Robison S.H. (1989) Decreased DNA repair in familial Alzheimer’s disease. Mutat. Res. 219, 247–255.
Coppede F., Migliore L. (2009) DNA damage and repair in Alzheimer’s disease. Curr. Alzheimer Res. 6, 36–47.
Weissman L., Jo D.G., Sorensen M.M., de Souza-Pinto N.C., Markesbery W.R., Mattson M.P., Bohr V.A. (2007) Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucl. Acids Res. 35, 5545–5555.
Canugovi C., Misiak M., Ferrarelli L.K., Croteau D.L., Bohr V.A. (2013) The role of DNA repair in brain related disease pathology. DNA Repair (Amst). 12, 578–587.
Coppede F. (2011) Variants and polymorphisms of DNA base excision repair genes and Alzheimer’s disease. J. Neurol. Sci. 300, 200–201; author reply 201.
Gallardo-Orihuela A., Hervas-Corpion I., Hierro-Bujalance C., Sanchez-Sotano D., Jimenez-Gomez G., Mora-Lopez F., Campos-Caro A., Garcia-Alloza M., Valor L.M. (2019) Transcriptional correlates of the pathological phenotype in a Huntington’s disease mouse model. Sci. Rep. 9, 18696.
Rogoza T., Goginashvili A., Rodionova S., Ivanov M., Viktorovskaya O., Rubel A., Volkov K., Mironova L. (2010) Non-Mendelian determinant [ISP+] in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1. Proc. Natl. Acad. Sci. USA. 107, 10573–10577.
Drozdova P., Mironova L., Zhouravleva G. (2016) Haploid yeast cells undergo a reversible phenotypic switch associated with chromosome II copy number. BMC Genet. 17, 152.
Zadorsky S.P., Sopova Y.V., Andreichuk D.Y., Startsev V.A., Medvedeva V.P., Inge-Vechtomov S.G. (2015) Chromosome VIII disomy influences the nonsense suppression efficiency and transition metal tolerance of the yeast Saccharomyces cerevisiae. Yeast. 32, 479–497.
Borchsenius A.S., Tchourikova A.A., Inge-Vechtomov S.G. (2000) Recessive mutations in SUP35 and SUP45 genes coding for translation release factors affect chromosome stability in Saccharomyces cerevisiae. Curr. Genet. 37, 285–291.
Tikhomirova V.L., Inge-Vechtomov S.G. (1996) Sensitivity of sup35 and sup45 suppressor mutants in Saccharomyces cerevisiae to the anti-microtubule drug benomyl. Curr. Genet. 30, 44–49.
Li X., Rayman J.B., Kandel E.R., Derkatch I.L. (2014) Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol. Cell. 55, 305–318.
Valouev I.A., Kushnirov V.V., Ter-Avanesyan M.D. (2002) Yeast polypeptide chain release factors eRF1 and eRF3 are involved in cytoskeleton organization and cell cycle regulation. Cell. Motil. Cytoskeleton. 52, 161–173.
Basu J., Williams B.C., Li Z., Williams E.V., Goldberg M.L. (1998) Depletion of a Drosophila homolog of yeast Sup35p disrupts spindle assembly, chromosome segregation, and cytokinesis during male meiosis. Cell. Motil. Cytoskeleton. 39, 286–302.
Wu H.Y., Kuo P.C., Wang Y.T., Lin H.T., Roe A.D., Wang B.Y., Han C.L., Hyman B.T., Chen Y.J., Tai H.C. (2018) Beta-amyloid induces pathology-related patterns of tau hyperphosphorylation at synaptic terminals. J. Neuropathol. Exp. Neurol. 77, 814–826.
Mao P., Reddy P.H. (2011) Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochim. Biophys. Acta. 1812, 1359–1370.
Julien C., Tomberlin C., Roberts C.M., Akram A., Stein G.H., Silverman M.A., Link C.D. (2018) In vivo induction of membrane damage by beta-amyloid peptide oligomers. Acta Neuropathol. Commun. 6, 131.
Alonso A.D., Cohen L.S., Corbo C., Morozova V., ElIdrissi A., Phillips G., Kleiman F.E. (2018) Hyperphosphorylation of Tau associates with changes in its function beyond microtubule stability. Front. Cell. Neurosci. 12, 338.
Nieznanski K., Podlubnaya Z.A., Nieznanska H. (2006) Prion protein inhibits microtubule assembly by inducing tubulin oligomerization. Biochem. Biophys. Res. Commun. 349, 391–399.
Tang Y.C., Amon A. (2013) Gene copy-number alterations: a cost-benefit analysis. Cell. 152, 394–405.
Mulla W., Zhu J., Li R. (2014) Yeast: a simple model system to study complex phenomena of aneuploidy. FEMS Microbiol. Rev. 38, 201–212.
Torres E.M., Sokolsky T., Tucker C.M., Chan L.Y., Boselli M., Dunham M.J., Amon A. (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 317, 916–924.
Torres E.M., Williams B.R., Tang Y.C., Amon A. (2010) Thoughts on aneuploidy. Cold Spring Harb. Symp. Quant. Biol. 75, 445–451.
Oromendia A.B., Dodgson S.E., Amon A. (2012) Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 26, 2696–2708.
Tang Y.C., Williams B.R., Siegel J.J., Amon A. (2011) Identification of aneuploidy-selective antiproliferation compounds. Cell. 144, 499–512.
Stingele S., Stoehr G., Peplowska K., Cox J., Mann M., Storchova Z. (2012) Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 8, 608.
Donnelly N., Storchova Z. (2014) Dynamic karyotype, dynamic proteome: buffering the effects of aneuploidy. Biochim. Biophys. Acta. 1843, 473–481.
Santaguida S., Vasile E., White E., Amon A. (2015) Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 29, 2010–2021.
Юров Ю.Б., Ворсанова С.Г., Соловьев И.В., Юров И.Ю. (2010) Нестабильность хромосом в нервных клетках человека в норме и при нервно-психических заболеваниях. Генетика. 46, 1352–1355.
Rehen S.K., Yung Y.C., McCreight M.P., Kaushal D., Yang A.H., Almeida B.S., Kingsbury M.A., Cabral K.M., McConnell M.J., Anliker B., Fontanoz M., Chun J. (2005) Constitutional aneuploidy in the normal human brain. J. Neurosci. 25, 2176–2180.
Rehen S.K., McConnell M.J., Kaushal D., Kingsbury M.A., Yang A.H., Chun J. (2001) Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Natl. Acad. Sci. USA. 98, 13361–13366.
Epstein C.J., Foster D.B., DeArmond S.J., Prusiner S.B. (1991) Acceleration of scrapie in trisomy 16 diploid aggregation chimeras. Ann. Neurol. 29, 95–97.
Popovitch E.R., Wisniewski H.M., Barcikowska M., Silverman W., Bancher C., Sersen E., Wen G.Y. (1990) Alzheimer neuropathology in non-Down’s syndrome mentally retarded adults. Acta Neuropathol. 80, 362–367.
Wisniewski K.E., Dalton A.J., McLachlan C., Wen G.Y., Wisniewski H.M. (1985) Alzheimer’s disease in Down’s syndrome: clinicopathologic studies. Neurology. 35, 957–961.
Oliver C., Holland A.J. (1986) Down’s syndrome and Alzheimer’s disease: a review. Psychol. Med. 16, 307–322.
Patterson D., Gardiner K., Kao F.T., Tanzi R., Watkins P., Gusella J.F. (1988) Mapping of the gene encoding the beta-amyloid precursor protein and its relationship to the Down syndrome region of chromosome 21. Proc. Natl. Acad. Sci. USA. 85, 8266–8270.
Oyama F., Cairns N.J., Shimada H., Oyama R., Titani K., Ihara Y. (1994) Down’s syndrome: up-regulation of beta-amyloid protein precursor and tau mRNAs and their defective coordination. J. Neurochem. 62, 1062–1066.
Patterson D., Costa A.C. (2005) Down syndrome and genetics - a case of linked histories. Nat. Rev. Genet. 6, 137–147.
Prasher V.P., Farrer M.J., Kessling A.M., Fisher E.M., West R.J., Barber P.C., Butler A.C. (1998) Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Ann. Neurol. 43, 380–383.
Blom E.S., Viswanathan J., Kilander L., Helisalmi S., Soininen H., Lannfelt L., Ingelsson M., Glaser A., Hiltunen M. (2008) Low prevalence of APP duplications in Swedish and Finnish patients with early-onset Alzheimer’s disease. Eur. J. Hum. Genet. 16, 171–175.
Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerriere A., Vital A., Dumanchin C., Feuillette S., Brice A., Vercelletto M., Dubas F., Frebourg T., Campion D. (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26.
Potter H. (1991) Review and hypothesis: Alzheimer disease and Down syndrome–chromosome 21 nondisjunction may underlie both disorders. Am. J. Hum. Genet. 48, 1192–1200.
Heston L.L., Mastri A.R. (1977) The genetics of Alzheimer’s disease: associations with hematologic malignancy and Down’s syndrome. Arch. Gen. Psychiatry. 34, 976–981.
Heyman A., Wilkinson W.E., Hurwitz B.J., Schme-chel D., Sigmon A.H., Weinberg T., Helms M.J., Swift M. (1983) Alzheimer’s disease: genetic aspects and associated clinical disorders. Ann. Neurol. 14, 507–515.
Geller L.N., Potter H. (1999) Chromosome missegregation and trisomy 21 mosaicism in Alzheimer’s disease. Neurobiol. Dis. 6, 167–179.
Schupf N., Kapell D., Lee J.H., Ottman R., Mayeux R. (1994) Increased risk of Alzheimer’s disease in mothers of adults with Down’s syndrome. Lancet. 344, 353–356.
Potter H., Granic A., Caneus J. (2016) Role of trisomy 21 mosaicism in sporadic and familial Alzheimer’s disease. Curr. Alzheimer Res. 13, 7–17.
Wang X., DeKosky S.T., Luedecking-Zimmer E., Ganguli M., Kamboh M.I. (2002) Genetic variation in alpha(1)-antichymotrypsin and its association with Alzheimer’s disease. Hum. Genet. 110, 356–365.
Ye Z., Ye Q., Shao B., He J., Zhu Z., Cheng J., Chen Y., Chen S., Huang X. (2015) Association between alpha-1 antichymotrypsin gene A/T polymorphism and primary intracerebral hemorrhage: a meta-analysis. Int. J. Clin. Exp. Med. 8, 20796–20804.
Lanoiselee H.M., Nicolas G., Wallon D., Rovelet-Lecrux A., Lacour M., Rousseau S., Richard A.C., Pasquier F., Rollin-Sillaire A., Martinaud O., Quillard-Muraine M., de la Sayette V., Boutoleau-Bretonniere C., Etcharry-Bouyx F., Chauvire V., Sarazin M., le Ber I., Epelbaum S., Jonveaux T., Rouaud O., Ceccaldi M., Felician O., Godefroy O., Formaglio M., Croisile B., Auriacombe S., Chamard L., Vincent J.L., Sauvee M., Marelli-Tosi C., Gabelle A., Ozsancak C., Pariente J., Paquet C., Hannequin D., Campion D., collaborators of the CNR-MAJ project (2017) APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med. 14, e1002270.
Roses A.D. (1996) Apolipoprotein E and Alzheimer’s disease. A rapidly expanding field with medical and epidemiological consequences. Ann. N. Y. Acad. Sci. 802, 50–57.
Strittmatter W.J., Roses A.D. (1995) Apolipoprotein E and Alzheimer disease. Proc. Natl. Acad. Sci. USA. 92, 4725–4727.
Ma J., Yee A., Brewer H.B., Jr., Das S., Potter H. (1994) Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 372, 92–94.
Zekanowski C., Wojda U. (2009) Aneuploidy, chromosomal missegregation, and cell cycle reentry in Alzheimer’s disease. Acta Neurobiol. Exp. (Wars). 69, 232–253.
Doshay L.J. (1954) Problem situations in the treatment of paralysis agitans. J. Am. Med. Assoc. 156, 680–684.
Driver J.A., Kurth T., Buring J.E., Gaziano J.M., Logroscino G. (2007) Prospective case-control study of nonfatal cancer preceding the diagnosis of Parkinson’s disease. Cancer Causes Control. 18, 705–711.
Driver J.A., Logroscino G., Buring J.E., Gaziano J.M., Kurth T. (2007) A prospective cohort study of cancer incidence following the diagnosis of Parkinson’s disease. Cancer Epidemiol. Biomarkers Prev. 16, 1260–1265.
Elbaz A., Peterson B.J., Yang P., Van Gerpen J.A., Bower J.H., Maraganore D.M., McDonnell S.K., Ahlskog J.E., Rocca W.A. (2002) Nonfatal cancer preceding Parkinson’s disease: a case-control study. Epidemiology. 13, 157–164.
Elbaz A., Peterson B.J., Bower J.H., Yang P., Maraganore D.M., McDonnell S.K., Ahlskog J.E., Rocca W.A. (2005) Risk of cancer after the diagnosis of Parkinson’s disease: a historical cohort study. Mov. Disord. 20, 719–725.
Fois A.F., Wotton C.J., Yeates D., Turner M.R., Goldacre M.J. (2010) Cancer in patients with motor neuron disease, multiple sclerosis and Parkinson’s disease: record linkage studies. J. Neurol. Neurosurg. Psychiatry. 81, 215–221.
Olsen J.H., Friis S., Frederiksen K. (2006) Malignant melanoma and other types of cancer preceding Parkinson disease. Epidemiology. 17, 582–587.
Olsen J.H., Friis S., Frederiksen K., McLaughlin J.K., Mellemkjaer L., Moller H. (2005) Atypical cancer pattern in patients with Parkinson’s disease. Br. J. Cancer. 92, 201–205.
Driver J.A. (2014) Inverse association between cancer and neurodegenerative disease: review of the epidemiologic and biological evidence. Biogerontology. 15, 547–557.
Bajaj A., Driver J.A., Schernhammer E.S. (2010) Parkinson’s disease and cancer risk: a systematic review and meta-analysis. Cancer Causes Control. 21, 697–707.
Musicco M., Adorni F., Di Santo S., Prinelli F., Pettenati C., Caltagirone C., Palmer K., Russo A. (2013) Inverse occurrence of cancer and Alzheimer disease: a population-based incidence study. Neurology. 81, 322–328.
Roe C.M., Fitzpatrick A.L., Xiong C., Sieh W., Kuller L., Miller J.P., Williams M.M., Kopan R., Behrens M.I., Morris J.C. (2010) Cancer linked to Alzheimer disease but not vascular dementia. Neurology. 74, 106–112.
Frain L., Swanson D., Cho K., Gagnon D., Lu K.P., Betensky R.A., Driver J. (2017) Association of cancer and Alzheimer’s disease risk in a national cohort of veterans. Alzheimers Dement. 13, 1364–1370.
Sorensen S.A., Fenger K., Olsen J.H. (1999) Significantly lower incidence of cancer among patients with Huntington disease: An apoptotic effect of an expanded polyglutamine tract? Cancer. 86, 1342–1346.
Yamada M., Sasaki H., Mimori Y., Kasagi F., Sudoh S., Ikeda J., Hosoda Y., Nakamura S., Kodama K. (1999) Prevalence and risks of dementia in the Japanese population: RERF’s adult health study Hiroshima subjects. Radiation effects research foundation. J. Am. Geriatr. Soc. 47, 189–195.
Koval L., Proshkina E., Shaposhnikov M., Moskalev A. (2020) The role of DNA repair genes in radiation-induced adaptive response in Drosophila melanogaster is differential and conditional. Biogerontology. 21, 45–56.
Gueguen Y., Bontemps A., Ebrahimian T.G. (2019) Adaptive responses to low doses of radiation or chemicals: their cellular and molecular mechanisms. Cell. Mol. Life Sci. 76, 1255–1273.
Mattson M.P., Calabrese E.J. (2010) Hormesis: what it is and why it matters. In: Hormesis: A Revolution in Biology, Toxicology and Medicine. Totowa US: Humana Press Inc, pp. 1–13.
Hwang S., Jeong H., Hong E.H., Joo H.M., Cho K.S., Nam S.Y. (2019) Low-dose ionizing radiation alleviates Abeta42-induced cell death via regulating AKT and p38 pathways in Drosophila Alzheimer’s disease models. Biol. Open. 8(2), bio036657. https://doi.org/10.1242/bio.036657
Ishimaru D., Andrade L.R., Teixeira L.S., Quesado P.A., Maiolino L.M., Lopez P.M., Cordeiro Y., Costa L.T., Heckl W.M., Weissmuller G., Foguel D., Silva J.L. (2003) Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry. 42, 9022–9027.
Ano Bom A.P., Rangel L.P., Costa D.C., de Oliveira G.A., Sanches D., Braga C.A., Gava L.M., Ramos C.H., Cepeda A.O., Stumbo A.C., De Moura Gallo C.V., Cordeiro Y., Silva J.L. (2012) Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J. Biol. Chem. 287, 28152–28162.
Lim S., Yoo B.K., Kim H.S., Gilmore H.L., Lee Y., Lee H.P., Kim S.J., Letterio J., Lee H.G. (2014) Amyloid-beta precursor protein promotes cell proliferation and motility of advanced breast cancer. BMC Cancer. 14, 928.
Itoh H., Kataoka H., Koita H., Nabeshima K., Inoue T., Kangawa K., Koono M. (1991) Establishment of a new human cancer cell line secreting protease nexin-II/amyloid beta protein precursor derived from squamous-cell carcinoma of lung. Int. J. Cancer. 49, 436–443.
Yang Z., Fan Y., Deng Z., Wu B., Zheng Q. (2012) Amyloid precursor protein as a potential marker of malignancy and prognosis in papillary thyroid carcinoma. Oncol. Lett. 3, 1227–1230.
Miyazaki T., Ikeda K., Horie-Inoue K., Inoue S. (2014) Amyloid precursor protein regulates migration and metalloproteinase gene expression in prostate cancer cells. Biochem. Biophys. Res. Commun. 452, 828–833.
Botelho M.G., Wang X., Arndt-Jovin D.J., Becker D., Jovin T.M. (2010) Induction of terminal differentiation in melanoma cells on downregulation of beta-amyloid precursor protein. J/Invest/Dermatol. 130, 1400–1410.
Peters H.L., Yan Y., Nordgren T.M., Cutucache C.E., Joshi S.S., Solheim J.C. (2013) Amyloid precursor-like protein 2 suppresses irradiation-induced apoptosis in Ewing sarcoma cells and is elevated in immune-evasive Ewing sarcoma cells. Cancer Biol. Ther. 14, 752–760.
Liang J., Pan Y., Zhang D., Guo C., Shi Y., Wang J., Chen Y., Wang X., Liu J., Guo X., Chen Z., Qiao T., Fan D. (2007) Cellular prion protein promotes proliferation and G1/S transition of human gastric cancer cells SGC7901 and AGS. FASEB J. 21, 2247–2256.
Malaga-Trillo E., Solis G.P., Schrock Y., Geiss C., Luncz L., Thomanetz V., Stuermer C.A. (2009) Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 7, e55.
Du L., Rao G., Wang H., Li B., Tian W., Cui J., He L., Laffin B., Tian X., Hao C., Liu H., Sun X., Zhu Y., Tang D.G., Mehrpour M., Lu Y., Chen Q. (2013) CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res. 73, 2682–2694.
Cheng Y., Tao L., Xu J., Li Q., Yu J., Jin Y., Chen Q., Xu Z., Zou Q., Liu X. (2014) CD44/cellular prion protein interact in multidrug resistant breast cancer cells and correlate with responses to neoadjuvant chemotherapy in breast cancer patients. Mol. Carcinog. 53, 686–697.
Danish Rizvi S.M., Hussain T., Subaiea G.M., Shakil S., Ahmad A. (2018) Therapeutic targeting of amyloid precursor protein and its processing enzymes for breast cancer treatment. Curr. Protein Pept. Sci. 19, 841–849.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология