

Молекулярная биология, 2020, T. 54, № 5, стр. 725-749
Молекулярно-биологические аспекты депрессивных состояний: современный взгляд на проблему
В. М. Ушакова a, b, *, А. Ю. Морозова a, d, А. М. Резник c, Г. П. Костюк d, В. П. Чехонин a, e
a Национальный медицинский исследовательский центр психиатрии и наркологии им. В.П. Сербского
Министерства здравоохранения России
119034 Москва, Россия
b Московский государственный университет им. М.В. Ломоносова
119234 Москва, Россия
c Московский государственный университет пищевых производств
125080 Москва, Россия
d Психиатрическая больница №1 им. Н.А. Алексеева
117152 Москва, Россия
e Российский национальный исследовательский медицинский университет им. Н.И. Пирогова
117997 Москва, Россия
* E-mail: ushakovavm@yandex.ru
Поступила в редакцию 21.04.2020
После доработки 15.05.2020
Принята к публикации 16.05.2020
Аннотация
Депрессия – серьезное психическое расстройство, поразившее более 300 миллионов человек. Неоднозначная этиология депрессии и недостаток эффективных методов ее терапии делают необходимым изучение патогенеза этой болезни для понимания механизмов ее развития и поиска новых методов лечения. В данном обзоре рассмотрены основные представления о развитии депрессивных расстройств, такие как моноаминергическая гипотеза, роль нарушений гипоталамо-гипофизарно-надпочечниковой системы, снижение выработки нейротрофинов, нейровоспаление. Проанализированы генетические корреляции депрессивных расстройств, известные полиморфизмы генов, а также эпигенетические механизмы. Сопоставлены и проанализированы сходства и различия в этиологии депрессивного расстройства и депрессивных состояний, ассоциированных с другими болезнями (шизофренией, болезнью Паркинсона, болезнью Альцгеймера). Также рассмотрены современные экспериментальные подходы, позволяющие исследовать молекулярно-биологические механизмы развития депрессивных состояний. В частности, обсуждены методы нокаута генов у лабораторных животных, подходы, основанные на использовании системы CRISPR/Сas, обосновано применение оптогенетических и хемогенетических методов. Рассмотрены методики, основанные на анализе генетических полиморфизмов и их ассоциаций. Приведенные данные помогут сформировать более полную картину современных представлений о патогенезе депрессии в качестве и самостоятельного заболевания, и сопутствующей патологии, а также наметить перспективы исследования механизмов депрессивных состояний.
ВВЕДЕНИЕ
Депрессивные расстройства, которые диагностируют почти у 350 миллионов человек [1], приводят к значительному снижению трудоспособности и качества жизни пациентов, а депрессивная симптоматика часто сопровождает другие психические и неврологические заболевания. Например, симптомы депрессии наблюдаются у 40–60% больных шизофренией [2, 3], а такие нейродегенеративные заболевания, как болезнь Паркинсона (БП) и болезнь Альцгеймера (БА), в 30–50% случаев бывают ассоциированы с аффективными нарушениями [4]. Таким образом, понятие “депрессия” затрагивает широкий круг патологических проявлений [5].
Депрессия – сложное гетерогенное нарушение функционирования головного мозга, в основе которого лежит ряд биохимических, молекулярно-генетических и анатомических изменений. Имеющиеся данные свидетельствуют о связи аффективной патологии с дисфункцией моноаминергических систем мозга, нарушением работы гипоталамо-гипофизарно-адреналовой (ГГА) системы, снижением выработки трофических факторов мозга и нарушением нейропластичности [6]. Установлены ассоциации с полиморфизмом ряда генов, вовлеченных в работу центральной нервной системы (ЦНС) [7], а также с влиянием эпигенетических преобразований [8]. Находит подтверждение связь аффективных нарушений с нейровоспалительными процессами и повреждающим действием окислительного стресса [6].
Тем не менее, несмотря на большой объем накопленных знаний о развитии и течении заболевания, современные методы терапии депрессивных расстройств остаются недостаточно эффективными [9].
Благодаря последним достижениям в разработке новых методологических подходов, ассоциированных с генетическими манипуляциями, появляется возможность воссоздания молекулярно-биологических каскадов, вовлеченных в патогенез депрессивных расстройств. Детальное исследование механизмов данных нарушений позволит разработать персонализированные подходы к терапии заболеваний, ассоциированных с депрессивной симптоматикой.
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ ПАТОГЕНЕЗА ДЕПРЕССИВНЫХ СОСТОЯНИЙ
В процессе развития депрессивного расстройства в организме изменяются различные физиологические (сердечный ритм, кожно-гальваническая реакция, биоэлектрическая активность мышц и др.) и нейрохимические (концентрация гормонов, содержание нейромедиаторов) показатели [10]. Нарушения затрагивают и лимбическую систему мозга, тесно связанную с эмоциональной сферой. Зафиксированы изменения структуры гиппокампа, миндалины, области передней фронтальной коры, поясной извилины и стриатума [11]. Данные магнитно-резонансной томографии (МРТ) выявляют общее уменьшение объема мозга при депрессии [11] и атрофию серого вещества [12].
Накопленные экспериментальные и клинические данные позволяют предположить значительное число причин развития депрессивных состояний, которые можно подразделить на несколько основных.
Нарушение ГГА-оси как механизм патогенеза депрессивных состояний
Стресс является ключевой причиной развития депрессии. Ответ организма на стресс регулируется посредством оси ГГА. Под влиянием факторов стресса клетки паравентрикулярного ядра гипоталамуса высвобождают аргинин-вазопрессин и кортикотропин-рилизинг-фактор, которые поступают в гипофиз [13]. Стимуляция рецепторов гипофиза индуцирует выработку адренокортикотропного гормона, доставляемого с током крови к надпочечникам. Под влиянием гормона в надпочечниках синтезируются глюкокортикоиды и минералокортикоиды: у человека, главным образом, кортизол, а у грызунов – кортикостерон. Рецепторы к этим гормонам экспрессируются в головном мозге и реагируют на изменение концентрации веществ по механизму отрицательной обратной связи [13].
Кортикостероиды играют важную роль в ответе на стресс, поддержании гомеостаза, регуляции эндокринного и иммунного ответа, энергетического обмена [14]. В норме глюкокортикоиды в небольшом количестве активируют преимущественно рецепторы минералокортикоидов, в условиях же стресса их уровень значительно повышается и обеспечивает активацию рецепторов глюкокортикоидов (Glucocorticoid receptor, GR) [15]. Продолжительный стресс приводит к их десенситизации и снижению активности, нарушению системы обратной связи и появлению поведенческих изменений [15, 16]. При повышении уровня глюкокортикоидов GR начинают функционировать в качестве транскрипционного фактора, поступая из цитозоля в ядро [17]. Этот процесс регулируется, главным образом, молекулярным комплексом белка 51, связывающего FK506 (FK506 binding protein 51, FKBP51). Данный комплекс кодируется геном FKBP5 и функционирует как кошаперон через связывание с головкой шаперона Hsp 90 во время формирования GR [17]. Как следствие, GR имеют сниженное сродство к глюкокортикоидам. Этот механизм подтверждается результатами исследования, показавшими снижение уровня GR при депрессии, а также увеличение экспрессии FKBP5 [18].
Ассоциацию нарушения работы оси с депрессивной симптоматикой подтверждают клинические и доклинические данные. В слюне и плазме крови пациентов с депрессией наблюдается повышение уровня кортизола, а в лимбических структурах мозга – увеличение кортикотропин-рилизинг-фактора [19]. Кроме того, в посмертных образцах мозга пациентов с историей депрессии регистрируется увеличение содержания в паравентрикулярном ядре нейронов, экспрессирующих аргинин-вазопрессин, более чем на 50% [20]. В экспериментах на лабораторных грызунах показано, что длительное введение глюкокортикоидов индуцирует депрессивно-подобное поведение, а также вызывает снижение экспрессии нейротрофического фактора мозга (Brain derived neurotrophic factor, BDNF) в гиппокампе [21]. Блокада рецепторов аргинина-вазопрессина типа 1a (V1a) оказывает противодепрессивное и противотревожное действие на животных, что делает антагонисты рецепторов потенциальными препаратами для терапии тревожных и депрессивных расстройств [22].
Моноаминергическая гипотеза патогенеза депрессивных состояний
Дефицит моноаминов (серотонина, норадреналина и дофамина) в структурах головного мозга лежит в основе гипотезы патогенеза депрессивных расстройств [23]. Моноаминергические системы вовлечены в регуляцию множества функций организма. Так, серотонин связан с контролем болевой чувствительности, нейрогормональной регуляцией, вовлечен в развитие тревожных и фобических расстройств [24]. Норадреналин ассоциирован с вегетативными реакциями и эмоциями, участвует в контроле уровня бодрствования, процессов селективного внимания [25]. Дофамин тесно связан с моторной системой и системой подкрепления и вовлечен в патогенез таких расстройств, как шизофрения и БП [26].
Десенситизация и снижение числа рецепторов серотонинергической системы лежат в основе моноаминергической теории депрессии [15]. В первую очередь, это относится к рецепторам 5-HT1A, часть которых функционирует в качестве соматодендрических ауторецепторов [27]. Методами позитронно-эмиссионной томографии (ПЭТ) показано, что активация ауторецепторов снижает частоту пульсации серотонинергических нейронов в области ядер шва [28], что может вызывать протревожные и продепрессивные эффекты. Показано, что полиморфизм рецепторов данного типа ассоциирован с развитием расстройств настроения, что подтверждает его участие в патогенезе заболевания. Однонуклеотидный полиморфизм (SNP) С(–1019)G в промоторе гена рецептора 5-HT1A приводит к угнетению транскрипционного репрессора NUDR/DEAF-1 (Deformed epidermal autoregulatory factor 1 homolog) и фактора транскрипции 5 из семейства Hes (HES5), что обуславливает усиление экспрессии рецептора и опосредованное снижение импульсации серотонинергических нейронов [29]. Гомозиготный полиморфизм связан с повышенной тревожностью, развитием клинической депрессии и сниженным ответом на терапию антидепрессантами [29]. Одновременное носительство гомозиготного полиморфизма С(–1019)G гена 5-HT1A и гена BDNF с заменой валина на метионин в кодоне 66 (Val66Met) хотя бы в одном из аллелей повышает риск развития депрессии в 3 раза [30].
С развитием депрессивных состояний ассоциированы и другие рецепторы, например, 5-HT1B и 5-HT2C [23]. Рецепторы 5-HT4 также связаны с развитием депрессивных состояний, что подтверждается данными об ассоциации локальных полиморфизмов гена 5-HTR4, в области, кодирующей С-концевую часть рецептора, с риском развития униполярной депрессии [31]. Показано, что агонист рецептора (RS67333) вызывает быстрый ответ на терапию антидепрессантами и увеличение экспрессии мРНК BDNF в области гиппокампа [32]. В целом, выявлена тесная ассоциация системы серотонина с нейротрофинами [33]. Вклад медиатора в развитие депрессивных состояний подтверждается ассоциацией транспортера серотонина (Serotonin transporter, SERT) с течением патологии. Показано, что короткий аллель (14 повторов) функционального промоторного полиморфизма гена SERT в гомозиготном состоянии снижает выработку транспортера, повышает чувствительность к стрессу и вероятность развития депрессии [34].
Система норадреналина тесно связана с ответом организма на стрессовое воздействие [35]. Так, у экспериментальных грызунов в депрессивно-подобном состоянии обнаружено повышение активности норадренергических нейронов в области голубого пятна (locus coeruleus). Эти эффекты могут быть связаны с подавлением ингибиторных α2-адренорецепторов [36], так как использование их агонистов снижает импульсацию нейронов и уменьшает проявление депрессивно-подобного поведения.
Также система норадреналина тесно связана с ответом на терапию антидепрессантами. В частности, положительный эффект препаратов может быть следствием деактивации α2-адренорецепторов в области locus coeruleus и β1-адренорецепторов в гиппокампе [37]. Кроме того, лечебное действие антидепрессантов опосредуют и β3-адренорецепторы [37]. Блокаторы обратного захвата норадреналина и его транспортера повышают количество норадреналина в синаптической щели, что приводит к активации β3-адренорецепторов. Действие этих рецепторов опосредуется Gs-белками, активирующими аденилатциклазу. Аденилатциклаза катализирует реакцию образования cAMP, который активирует протеинкиназу А (PKA). PKA, в свою очередь, фосфорилирует белок, связывающийся с cAMP-зависимым элементом (cAMP response element-binding protein, CREB), индуцируя транскрипцию BDNF [37]. Кроме того, белок Gs активирует сигнальный путь, опосредованный фосфоинозитид-3-киназой (PI3K), который приводит к фосфорилированию серин/треонин-специфичной протеинкиназы RAC-α (Akt). Активированная Akt фосфорилирует белок mTOR (мишень рапамицина млекопитающих), что стимулирует пролиферацию клеток [37, 38]. За счет взаимодействия с тропомиозиновым тирозинкиназным рецептором В (TrkB) BDNF способствует повышению экспрессии мРНК субъединиц GluR1 и GluR2 рецептора AMPA и их синаптической локализации, что играет ключевую роль в формировании долговременной потенциации (LTP) и в процессах синаптической пластичности. Таким образом, система норадреналина играет важную роль в противодепрессивном действии медикаментозных препаратов, что подтверждает его участие в патогенезе депрессивных состояний.
Дофамин – еще один моноамин ЦНС, который также вовлечен в процессы развития депрессии [39]. Изменения его выработки, ассоциированные с патогенезом депрессии, связаны с функционированием дофаминергических нейронов в вентральной области покрышки, области вентромедиальной префронтальной коры, нарушением работы механизмов “системы вознаграждения” и формированием ангедонии [39].
С помощью ПЭТ показано снижение связывания транспортера дофамина у больных с депрессией по сравнению со здоровыми испытуемыми [40]. Кроме того, изучение посмертных образцов мозга пациентов с клинической депрессией, выявило повышение уровня рецепторов D2 в области стриатума и D2/D3 в области центрального и базального ядер миндалины [39]. Эти рецепторы, выполняющие тормозную функцию, опосредуют снижение уровня дофамина [39]. Роль рецепторов этого типа в депрессивных расстройствах подтверждают и данные о регуляции сигнального пути Akt и киназы 3 гликоген-синтазы (GSK3, Akt-GSK3) за счет D2 [41]. В частности, D2 оказывают тормозный эффект на Akt и вызывают опосредованное этим увеличение уровня GSK3 [41]. Изменение GSK3β показано при психических заболеваниях, таких как шизофрения и биполярное аффективное расстройство (БАР) [42], а ингибирование GSK3β оказывает противодепрессивное действие [43].
На моделях депрессивных состояний у грызунов подтверждено нарушение функционирования мезолимбической системы дофамина при эмоциональных расстройствах. Например, снижение экспрессии дофамина в лимбических структурах мозга выявлено в модели выученной беспомощности [44]. Тесно ассоциирован с депрессивными состояниями и мезокортикальный дофаминергический путь, связанный с когнитивными нарушениями, наблюдаемыми при депрессии [45]. Предполагается, что снижение активности дофаминергической системы в вентромедиальной коре опосредуется нейровоспалительными процессами, сопровождающими длительное стрессорное воздействие. Повторяющийся стресс вызывает выброс простагландина E2 (PGE2), который действует на свои рецепторы EP1 [45]. Активация рецепторов подавляет работу мезокортикальной системы дофамина и индуцирует поведенческие продепрессивные эффекты.
В пользу связи дофамина с депрессивными расстройствами свидетельствуют и данные о взаимодействии глутаматергической и дофаминергической систем, и участие нейромедиатора в формировании LTP [46], которая играет важную роль в процессах синаптической пластичности.
Таким образом, роль моноаминов в патогенезе депрессии хорошо изучена. Однако нарушение функционирования этих систем является лишь частью комплекса изменений, и их дисфункция сама по себе не способна полноценно индуцировать возникновение клинической депрессии.
Помимо моноаминергических систем мозга существенный вклад в патогенез депрессивных состояний вносят и другие нейромедиаторы. Баланс основных возбуждающих и тормозных систем в головном мозге значительно нарушается при депрессии, что указывает на вовлечение в патологические процессы систем глутамата и ГАМК [47]. Так, при помощи протонной МРТ выявлено снижение метаболитов глутамата в вентромедиальной префронтальной коре [47], а в постмортальных исследованиях установлено снижение числа синапсов и синаптических маркеров в дорсолатеральной префронтальной коре при депрессии [48]. Исследования ГАМКергической системы показали снижение ГАМК в спинномозговой жидкости больных депрессией [47], а также подавление активности системы в кортикальных областях мозга, нормализующееся при ремиссии. Доклинические эксперименты выявили снижение синаптических маркеров глутамата, таких как GluA1, постсинаптический плотный белок 95 (Postsynaptic density protein 95, PSD95) и синапсин 1 в вентромедиальной префронтальной коре [49]. Показано, что трехнедельный хронический непредсказуемый мягкий стресс (ХНМС) вызывает снижение пре- и постсинаптических маркеров ГАМК, таких как глутаматдекарбоксилаза 65/67 (GAD65/67) и гефрин в префронтальной коре [50]. Мыши, мутантные по гену рецептора ГАМКА, имеют депрессивно-подобную симптоматику, в том числе признаки ангедонии и неофагии [51].
Вовлеченность медиаторных систем в патогенез депрессии однозначно подтверждается противодепрессивными эффектами, вызванными антагонистом NMDA-рецепторов – кетамином [52]. Клиническое действие кетамина обусловлено блокадой NMDA-рецепторов на тормозных ГАМКергических интернейронах, что обеспечивает эффект растормаживания и увеличение выброса глутамата [52]. Активация глутаматергической системы вызывает нейропластические изменения посредством вовлечения рецепторов AMPA, активации BDNF и опосредованным им запуском сигнального пути mTORC1, ассоциированного с пластическими процессами [52]. Наряду с активацией глутаматергической системы кетамин восстанавливает и нарушения системы ГАМК, связанные с депрессией. В частности, показано, что однократное применение кетамина увеличивает содержание пре- и постсинаптических маркеров GAD65/67, везикулярного транспортера ГАМК, гефрина, дефицит которых сопровождает депрессивные состояния [51].
Таким образом, рассмотренные медиаторные системы тесно вовлечены в патогенез депрессии, а также ассоциированы с действием антидепрессантов. Кетамин в виде С-стереомера уже протестирован в фазе III клинических исследований и одобрен Управлением по надзору за качеством пищевых продуктов и медикаментов (Food and drug administration, FDA) в качестве препарата для лечения клинической депрессии [47].
Генетические механизмы патогенеза депрессивных состояний
Большую роль в развитии депрессии играют генетические факторы. Значительное развитие инструментов современной биологии позволяет детально изучать генетические аспекты этого расстройства. Так, показано, что 31–42% случаев депрессии ассоциированы с наследственными факторами [38].
Современные исследования в области медицинской генетики сосредоточены на поиске полиморфизмов, связанных с различными патологиями, в том числе c психическими расстройствами. Так, в промоторе гена транспортера серотонина (SLC6A4) найден полиморфизм 5HTTLRP (44bp Ins/Del) [53], ассоциированный с увеличенным риском развития депрессии и более тяжелой симптоматикой [54, 55]. Кроме SLC6A4, обнаружены полиморфизмы в генах рецептора серотонина (5HT1A), моноаминоксидазы А (МАОА), триптофангидроксилазы 2 (TPH2), нейронального регулятора роста 1 (Neuronal growth regulator 1, NEGR1) и др. [56]. Выявлена ассоциация с депрессией и гена аполипопротеина Е (APOE) [55]. У носителей аллеля ԑ4 выявлена атрофия гиппокампа, церебральный гипометаболизм, накопление бета-амилоида [57]. Этот полиморфизм вовлечен в патогенез БА, а так как заболевание часто сопровождается депрессивной симптоматикой, то эти изменения могут быть вовлечены в формирование аффективных расстройств [55].
Один из наиболее изученных и тесно ассоциированных с депрессивными состояниями полиморфизмов – Val66Met гена BDNF [58]. Показано, что у носителей аллеля Met снижен объем гиппокампа и усилен ответ миндалины на негативные стимулы, изменения секреции BDNF [56]. В патогенез депрессии вовлечен и SNP в гене CREB1, связанном с выработкой нейротрофина [56], а также гены многих других систем: FKBP5, μ-рецептора 1 опиоидов (μ-type opioid receptor 1, OPRM1), рецепторов дофамина (DRD2, DRD4) и др. [56, 59].
Таким образом, в основе предрасположенности к депрессивным состояниям лежит большое число генов, вовлеченных в работу ЦНС. Тем не менее, SNP определяют лишь более высокую вероятность развития депрессии. Под влиянием факторов окружающей среды могут возникать эпигенетические перестройки генома, которые также играют большую роль в развитии депрессивных расстройств [60, 61].
Эпигенетические механизмы
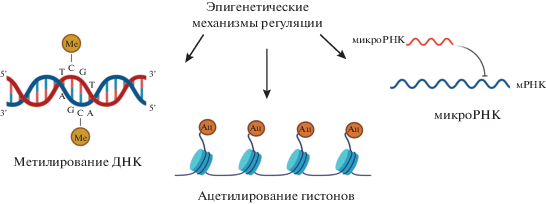
В формирование патологических изменений вносят вклад такие известные эпигенетические механизмы, как метилирование ДНК, ацетилирование гистонов и регуляция некодирующих участков РНК [62] (рис. 1). Эпигенетические модификации изучают на клетках крови, слюны, эпителия, а также на постмортальных образцах мозга [60].
Рис. 1.
Эпигенетические механизмы регуляции транскрипции и трансляции. Эпигенетические механизмы регуляции подразделяют на три класса. 1) Метилирование ДНК – процесс переноса метильной группы от S-аденозин-L-метионина на C5 цитозина в CpG-динуклеотидах [60, 63]. Метилирование промоторного участка гена обычно приводит к подавлению транскрипции. 2) Ацетилирование гистонов – добавление ацетильных групп к остаткам лизина в N-концевых участках, выступающих из гистоновой оболочки нуклеосомы [60, 64]. Процесс контролируется гистон-ацетилтрансферазами (HAT), которые переносят ацетильные группы от ацетил-кофермента А на лизин, и гистондеацетилазами (HDAC), отсоединяющими их. При этом общий заряд гистона изменяется с положительного на нейтральный, что способствует активации транскрипции. 3) Регуляция некодирующих участков РНК (микроРНК). В данном случае микроРНК комплементарно спариваются с участками мРНК, ингибируя их трансляцию, и таким образом снижают активность генов [64, 65]. Эпигенетические преобразования приводят к подавлению или усилению синтеза белков, что вносит вклад в поведенческие реакции и регуляцию патологических процессов. Ме – метильные группы, Ац – ацетильные группы.
К настоящему времени достаточно хорошо изучено метилирование гена BDNF, который локализуется в хромосоме 11p14.1 [66]. Так, обнаружена ассоциация метилирования гена BDNF, выделенного из образцов крови, слюны и тканей ротовой полости пациентов [63], в первую очередь его промоторного участка, с депрессивной симптоматикой. Выявлено также метилирование гена SLC6A4, расположенного в хромосоме 17q11.1–q12, при клинической депрессии [67]. В основном, депрессивная симптоматика связана с гиперметилированием участков промоторной области [67]. Предполагается, что метилирование SLC6A4 приводит к угнетению работы транспортера серотонина, что, в конечном итоге, вызывает десенситизацию рецепторов и дисрегуляцию серотонинергической системы [62].
Эпигенетические изменения выявлены и в генах, принимающих участие в контроле работы ГГА-оси. Например, обнаружено метилирование участков гена NR3C1, кодирующего GR [68]. В целом, показана ассоциация эпигенетических модификаций многих генов с депрессивной симптоматикой, в том числе генов FKBP5, OXTR (рецептор окситоцина, связанный с социальным поведением), LINGO3 (Leucine-rich repeat and immunoglobulin-like domain containing 3, белок 3, взаимодействующий с рецептором Nogo и вовлеченный в функционирование олигодендроцитов) [62].
Изучение модификации гистонов в посмертных образцах мозга пациентов с депрессией и психически здоровых людей выявило увеличение ацетилирования гистонов по остаткам H3K14 в области прилежащих ядер и снижение уровня HDAC2 у больных депрессией [69]. Обнаружено также повышение триметилирования H3K4 гистонов в префронтальной коре посмертных образцов мозга. Кроме того, показано повышение уровня HDAC2 и HDAC5 в периферической крови пациентов, переживших острый эпизод депрессии [70].
Ассоциация микроРНК с депрессивными состояниями, выявленная во многих исследованиях, делает микроРНК потенциальными биомаркерами клинической депрессии и ответа на антидепрессанты [71]. В частности, Gururajan и соавт. показали существенное снижение экспрессии микроРНК let-7c и let-7b у пациентов с лекарственно-устойчивой депрессией [72]. Согласно данным биоинформатических исследований, let-7c и let-7b участвуют в регуляции экспрессии 27 генов, вовлеченных в контроль сигнального пути PI3k–Akt–mTOR, повреждаемого при депрессии [71]. Показано также выраженное снижение профиля экспрессии таких микроРНК, как miR-142-5p, miR-142-3p, miR-494, miR-376a, miR-496, miR-369-3p, miR-23b, в префронтальной коре пациентов с депрессией [71]. Ассоциацию этих типов РНК с патогенезом депрессивных состояний подтверждают и полученные в доклинических исследованиях данные об их связи с ответом на антидепрессанты [73].
Наряду с микроРНК, в последнее время в качестве возможного регулятора депрессивных состояний рассматривают экзосомы – биологические “наноконтейнеры”, содержащие и доставляющие к клетке-реципиенту молекулы ДНК, мРНК, микроРНК и белков. Например, экзосомы, выделяемые астроцитами, могут обеспечивать нейропротекторные эффекты и нейрональную пластичность через выделение прионного белка PrP, который защищает нейроны от гипоксии, окислительного стресса и гипогликемии [71]. Подобные свойства экзосом делают их перспективным инструментом терапии депрессивных состояний.
Нарушение выработки нейротрофинов как механизм патогенеза депрессивных состояний
Реализация функций ЦНС базируется на генерации, трансформации и хранении информации в нейронных сетях, что тесно связано с процессами нейрогенеза, ангиогенеза и синаптической пластичности. Стресс и депрессивные расстройства нарушают динамический баланс работы ЦНС, что лежит в основе нейропластической или нейротрофической теории развития депрессии [74]. Показано, что трофические факторы, такие как BDNF, фактор роста нервов (NGF), фактор роста эндотелия сосудов (VEGF), фактор роста глии (GDNF) и др. обеспечивают процессы нейрогенеза, ангиогенеза, выполняют нейропротекторную функцию и принимают участие в процессах синаптической пластичности [74–76]. Наиболее изучена связь с синаптическими процессами и ассоциация с депрессией фактора BDNF, относящегося к семейству нейротрофинов [77]. BDNF связывается с рецептором TrkB и опосредует через него свои эффекты [77]. Связывание BDNF с рецептором приводит к лиганд-рецепторной димеризации и аутофосфорилированию остатков тирозина во внутриклеточном киназном домене рецептора и может индуцировать три основных внутриклеточных каскада (рис. 2): 1) путь Ras-MAPK; 2) сигнальный путь PLCγ; 3) сигнальный путь PI3K [78, 79].
Рис. 2.
Сигнальные пути BDNF–TrkB. Активация рецепторов TrkB приводит к индукции трех сигнальных путей. 1 – Путь PI3К [80]. PI3K может активироваться за счет Ras-зависимого механизма или независимо за счет трансмембранных рецепторов. В процессе внутриклеточной передачи сигналов PI3K индуцирует активацию PDK1, которая, в свою очередь, фосфорилирует протеинкиназу Akt [79]. Помимо участия в регуляции транскрипции Akt фосфорилирует несколько белков, регулирующих выживаемость клеток, в частности, может ингибировать апоптоз. Фосфорилированная Akt также влияет на сигнальный путь mTOR [78], приводящий к пролиферации клеток. 2 – Путь PLCγ [81]. Фосфорилирование остатков тирозина вызывает активацию PLCγ. Активированная форма расщепляет PIP2 до IP3 и DAG [78]. IP3 индуцирует выброс внутриклеточного Ca2+ и активацию CAMK. DAG стимулирует образование DAG-регулируемой PKC. Внутриклеточный каскад влияет на транскрипционную активность генов и вносит вклад в нейропластические процессы [79]. 3 – Путь Ras-MAPK [80]. Фосфорилирование остатков тирозина в околомембранном пространстве рецептора индуцирует активацию сигнального комплекса околомембранных белков, который, в свою очередь, вызывает активацию GTPазы Ras [79]. Ras связывает и активирует Raf. Raf фосфорилирует MEK, которая фосфорилирует и активирует ERK. Субстраты ERK влияют на транскрипцию ряда генов, ассоциированных с дифференцировкой и выживаемостью нейронов, нейропластическими процессами [82]. В частности, внутриклеточный каскад приводит к фосфорилированию и активации CREB. PI3K – фосфоинозитид-3-киназа, PDK1 – фосфоинозитид-зависимая киназа 1, Akt – серин/треонин-специфичная протеинкиназа RAC, PLCγ – фосфолипаза Сγ, PIP2 – фосфатидилинозитолдифосфат, IP3 – инозитолтрифосфат, DAG – диацилглицерин, CAMK – Сa2+/кальмодулинзависимая протеинкиназа, PKC – протеинкиназа С. MAPK – митоген-активируемая протеинкиназа, ERK – внеклеточная сигнал-регулируемая киназа, Raf – протоонкоген, серин/треонин-специфичная протеинкиназа, MEK – киназа MAPK/ERK.

Показано, что BDNF играет важную роль в процессах синаптической пластичности, в основе которой лежит эффект LTP [78]. Ранняя фаза пластических изменений индуцируется повышением уровня Ca2+ в результате возбуждения NMDA-рецепторов и активации CaMKII и PKC [78]. Генерация активных форм CaMKII и PKC приводит к фосфорилированию рецепторов AMPA, их миграции и встраиванию в постсинаптическую мембрану. Долговременная LTP опосредуется влиянием сигнальных молекул (PKA, ERK и др.) на активацию факторов транскрипции, включая CREB и трехкомпонентный комплексный фактор (Ternary complex factor, TCF) Elk-1 [83]. Эти факторы обеспечивают синтез белков, вызывающих функциональные изменения в синапсах и структурные изменения в шипиках дендритов. Показано, что на ранней фазе LTP BDNF пресинаптически увеличивает экзоцитоз глутамат-содержащих везикул и постсинаптически индуцирует фосфорилирование и встраивание рецепторов AMPA [78]. На длительном этапе LTP BDNF влияет на синтез белков, регулирует миграцию мРНК в дендритах и распаковку компонентов РНК-содержащих гранул [78].
При депрессивных состояниях нейропластические преобразования в мозгу угнетаются, что приводит к морфологическим и физиологическим нарушениям. Показано, что хронический стресс вызывает атрофию апикальных дендритов в пирамидных нейронах гиппокампа, угнетение нейрогенеза в зубчатой извилине [84], значительное снижение длины и числа апикальных веточек дендритов пирамидных нейронов в префронтальной коре [75]. Показано, что тяжелый стресс приводит к нарушению LTP и усилению долговременной депрессии (Long-term depression, LTD) в нейронах [75].
Изменения BDNF при депрессивных состояниях достаточно хорошо изучены [85, 86]. Так, при клинической депрессии снижается содержание BDNF в сыворотке крови [76]. Снижение уровня BDNF выявлено также в гиппокампе и префронтальной коре мозга жертв суицида [75]. Кроме того, снижение экспрессии гена BDNF, вызванное ХНМС, может опосредовать развитие депрессивно-подобного поведения у животных за счет подавления сигнального пути, опосредуемого MAPK и ERK [82]. Влияние антидепрессантов на экспрессию гена BDNF также подтверждают значительную роль этого фактора в патогенезе депрессии. Недавно показали, что длительный прием антидепрессантов приводит к увеличению уровня мРНК BDNF и TrkB в области гиппокампа [87]. Кроме того, установлено, что использование медикаментов приводит к повышению уровня BDNF в плазме крови [77].
В формирование психических расстройств вовлечен не только BDNF, но и ряд других факторов роста. В частности, в развитии депрессивных состояний принимает участие VEGF, экспрессия гена которого изменяется под воздействием стресса. Показано снижение уровня мРНК VEGF в гиппокампе грызунов, подвергавшихся ХНМС [75]. В развитие депрессии вовлечены и другие факторы, такие как фактор роста фибробластов 2 (Fibroblast growth factor-2 – FGF-2) и NGF. Ассоциацию FGF-2 с патогенезом психопатологии подтверждают данные о снижении экспрессии генов FGF-2 и его рецепторов в лимбических структурах мозга при депрессии [88]. Более того, выявлено противодепрессивное действие FGF-2 при его внутрижелудочковом введении в мозг грызунов [89]. Влияние NGF на патогенез депрессии связывают с его ролью в пролиферации и дифференцировке нейронов [75], процессах синаптической пластичности. У крыс линии Флиндерса, используемых в качестве экспериментальной модели депрессивного состояния, наблюдали снижение уровня NGF, корректируемого с помощью электросудорожной терапии [90].
Таким образом, нейропластические процессы и факторы, их опосредующие, активно вовлечены в патогенез депрессии. Существование связи между коррекцией нейропластических нарушений и ремиссией депрессии указывает на необходимость более широкого изучения роли этих процессов в патогенезе депрессии.
Нейровоспаление и окислительный стресс как механизмы патогенеза депрессивных состояний
К настоящему времени убедительно показано, что депрессивные состояния тесно связаны с иммунологическими процессами и могут провоцироваться иммунными реакциями организма [91]. Нервная и иммунная системы контролируют работу друг друга с помощью двунаправленных механизмов. Например, клетки микроглии в головном мозге участвуют в осуществлении процесса нейрональной пластичности и поддержании гомеостаза [92]. В то же время, нейроны влияют на функционирование микроглии. В частности, в нормальных условиях нейроны выделяют хемокин фракталкин (C-X3-C motif chemokine ligand 1, CX3CL1), колониестимулирующий фактор 1 (CSF1) и трансформирующий фактор роста β (TGF-β), действующие на иммунные клетки [92]. Например, CX3CL1 связывается с рецепторами к фракталкину (CX3CR1) на поверхности клеток микроглии и таким образом регулирует ее нормальное морфологическое и гомеостатическое развитие [92]. Интересно, что у трансгенных мышей с нокаутом гена CX3CR1 нарушено развитие нервной системы, угнетены связи гиппокампа и нейрогенез, появляются признаки депрессивно-подобного поведения [93].
В пользу вовлеченности иммунной системы в процесс развития психопатологии свидетельствуют и данные о том, что более чем у 50% больных, принимающих интерферон-α (IFN-α), в течение нескольких месяцев проявляются клинические признаки депрессии [94]. В плазме крови и спинномозговой жидкости пациентов увеличено содержание провоспалительных цитокинов – фактора некроза опухоли α (TNFα), интерлейкинов (IL) 6 и IL-1 [92]. В доклинических исследованиях наряду с этим обнаружено, что введение низких доз липополисахарида и IL-1β индуцирует нарушение поведения у грызунов (снижение социальной, ориентировочно-исследовательской и половой активности), сопровождающееся выбросом провоспалительных цитокинов, таких как IFN-α, TNFα и IL-6 [95].
Внешнее стрессорное воздействие существенно влияет на гомеостаз иммунной системы и провоцирует нейровоспалительные процессы. Симпатическая система, ассоциированная с работой оси ГГА [92], индуцирует выброс норадреналина, действие которого стимулирует прогениторные иммунные клетки в гемопоэтических органах и опосредует рост числа моноцитов и гранулоцитов, поступающих в кровяное русло [96]. В результате воздействия стрессорных агентов в организме активируются индуцированные патогеном молекулярные паттерны (Pathogen-associated molecular pattern, PAMP) и молекулярные фрагменты, ассоциированные с опасностью (Damage-associated molecular pattern, DAMP). Показано, что эти сигнальные пути осуществляют свою работу через активацию Toll-подобных рецепторов (TLR) и рецепторов усиленного гликилирования конечных продуктов (Receptor for advanced glycation end products, RAGE), что провоцирует выброс провоспалительных цитокинов [97].
Влияние стресса нарушает нормальное взаимодействие микроглии и нейронов. На молекулярном уровне подобное воздействие приводит к индукции сигнального пути ядерного фактора NF‑κB [95]; цитокины TNFα и IL-1 связываются со своими рецепторами на клетке и активируют латентный каскад реакций, что приводит к фосфорилированию ингибитора NF-kB (IκBα) за счет IκB-киназы (IKK) и к деградации IκBα, который в норме подавляет NF‑κB. Таким образом, NF‑κB активируется и проникает в ядро, где функционирует в качестве фактора транскрипции и индуцирует транскрипцию генов сотен провоспалительных белков, включая цитокины, хемокины и циклооксигеназу 2 (СOX2) [98].
В иммунном ответе на стресс участвует также молекулярный каскад, затрагивающий инфламмасому NLRP3 [99]. Активация инфламмасомы приводит к высвобождению провоспалительного IL-1β, в чем участвуют несколько процессов. Так, лиганды рецептора стимулируют транскрипцию и трансляцию про-IL-1β. Кроме того, АТР индуцирует сборку комплекса инфламмасомы и активирует каспазу-1, которая опосредует превращение предшественника IL-1β в зрелую форму [99]. Ассоциация системы инфламмасомы с депрессивной симптоматикой подтверждается данными о повышении экспрессии NLRP3 у пациентов с депрессией, не получающих лечение, что коррелирует с уровнем IL-1β в сыворотке крови и тяжестью заболевания [38].
На участие нейровоспаления в патогенезе депрессии указывают данные о влиянии антидепрессантов на иммунную систему. Например, показано, что их применение снижает содержание IL-6, TNFα, IL-10 и хемокина CCL-2 в крови [15]. Кроме того, сами нейровоспалительные процессы могут усиливать резистентность к медикаментозной терапии. Показана корреляция между увеличением экспрессии генов провоспалительных медиаторов и снижением ответа на терапию антидепрессантами [100].
Потенциально перспективными для лечения депрессии могут стать препараты, действие которых направлено на иммунную систему. Например, блокада рецепторов к PAMP и DAMP снижает выраженность депрессивной симптоматики [92], а у мышей с нокаутом гена инфламмасомы отсутствуют проявления депрессивно-подобного поведения после ХНМС [99]. Таким образом, препараты, обладающие противовоспалительным действием, могут быть перспективными при депрессивных расстройствах, в первую очередь, при терапевтически резистентной депрессии.
Свой вклад в патогенез депрессивных состояний вносит и окислительный стресс [101, 102]. Активные формы кислорода и азота играют важную роль в функционировании клеток. Однако их чрезмерное образование может быть причиной структурных и функциональных изменений, повреждающих клетку. Результаты клинических и доклинических исследований показали, что униполярная и биполярная депрессия ассоциированы с окислительным стрессом [103]. Так, при депрессивных состояниях увеличивается продукция малонового диальдегида (MDA), маркера перекисного окисления липидов, и 8-изо-простагландина F2, маркера окисления арахидоновой кислоты [103, 104]. У лиц с депрессивными расстройствами зафиксировано нарушение работы антиоксидантной системы. В частности, наблюдается снижение концентрации витаминов Е и С в плазме крови, уровней цинка, глутатиона, коэнзима Q10, понижение активности супероксиддисмутазы и глутатионпероксидазы [105]. Роль окислительного стресса в депрессии подтверждается генетическими исследованиями, показавшими ассоциацию SNP генов, вовлеченных в контроль окислительной системы, с проявлениями депрессии [103].
Предполагается, что вклад окислительного стресса в патогенез депрессии тесно связан с нейровоспалением [103]. В различных типах клеток такие цитокины, как IL-1, TNFα и IFN-α индуцируют образование активных форм кислорода, задействуя комплекс NADPH-оксидазы (NOX) [105]. С другой стороны, сам окислительный стресс также может индуцировать процессы нейровоспаления через активацию путей NF-κB и MAPK [103]. Окислительный стресс может также приводить к развитию депрессивной симптоматики через активацию пути катаболизма триптофана (TRYCAT), что индуцирует повышение образования NO и повреждение клеток [106]. Окислительный стресс также может вызывать изменения в системе глутамата, снижая уровень внутриклеточного глутатиона и индуцируя окислительную глутаматную токсичность [107]. Кроме того, стресс провоцирует нарушения в работе митохондрий, что стимулирует образование активных форм кислорода [103]. Таким образом, окислительный стресс также играет роль в возникновении депрессивных состояний, тесно пересекаясь с процессами нейровоспаления.
В целом, патогенез депрессии регулируется множеством процессов, которые затрагивают различные уровни организации и разнообразные молекулярные каскады. Тем не менее, невозможно выделить единственную причину развития патологических процессов, определяющую течение и развитие заболевания. Гетерогенная природа развития депрессивных патологий существенно осложняет поиск подходов к терапии депрессии как таковой, так и к лечению депрессивных состояний, ассоциированных с другими заболеваниями. Детальное исследование комплекса процессов, вызывающих депрессию, а также их взаимодействие между собой может способствовать поиску новых и усовершенствованию известных методов лечения депрессивных состояний.
ДЕПРЕССИВНЫЕ СОСТОЯНИЯ, АССОЦИИРОВАННЫЕ С ДРУГИМИ ПСИХОНЕВРОЛОГИЧЕСКИМИ ЗАБОЛЕВАНИЯМИ
Аффективные нарушения, особенно депрессивные симптомы, часто сопровождают психические и неврологические заболевания, такие как шизофрения, БАР, нейродегенеративные заболевания головного мозга и многие другие. Получены данные, указывающие на сходные молекулярные механизмы развития депрессивных состояний при БА, шизофрении, БАР и даже БП [4]. Например, нарушение моноаминергической передачи и ГГА-оси, усиление окислительных и нейровоспалительных явлений и нарушение трофических функций, как полагают, способствуют атрофии и гибели нейронов при всех этих заболеваниях. Кроме того, результаты нейровизуализационных исследований отражают особенности структурных и функциональных изменений, связанных с депрессией, коморбидной другим формам психоневрологической патологии. Общность патогенетических механизмов подтверждают установленные генетические корреляции между депрессией и другими психическими заболеваниями. В частности, подтверждены данные о генетических корреляциях между депрессивной симптоматикой в рамках клинической депрессии и шизофренией, БАР, болезнью коронарных артерий, повышенным уровнем триглицеридов, увеличением массы жировой ткани, а также нейродегенеративными заболеваниями [108]. В табл. 1 отражены некоторые особенности нейробиологических механизмов депрессии как самостоятельного психического расстройства, так и клинического синдрома при других психоневрологических заболеваниях.
Таблица 1.
Особенности патогенеза психических и неврологических заболеваний
| Характерные особенности | Депрессия | Шизофрения | БАР | БА | БП |
|---|---|---|---|---|---|
| Структурные нейроанатомические изменения | Атрофия в области гиппокампа, миндалины, области передней фронтальной коры и поясной извилины [111] | Атрофия серого вещества в лобных долях в префронтальной коре, в затылочно-височной и височных бороздах, в гиппокампе, миндалине и таламусе [112] | Нарушения фронто-кортикального контура (фронтальная кора, стриатум, бледный шар, черная субстанция, миндалина, гиппокамп и таламус) [113], увеличение латеральных желудочков мозга [114] | Атрофия коры в области передней поясной извилины и гиппокампа [115] | Снижение префронтально-лимбических связей и атрофия лобно-височных долей, гиппокампа, передней поясной извилины, миндалины, мозжечка [116] |
| ГГА-система | ↑Кортизол ↑Кортикотропин-рилизинг-фактор [19] |
↑Кортизол [117]. Часть исследователей предполагает, что повышение кортизола предшествует острой фазе эпизода [118] | ↑Кортизол в маниакальной фазе заболевания [117] | ↑Кортизол [119]. Коморбидность с депрессией может быть обусловлена накоплением кортизола, обуславливающего возрастные изменения тканей гиппокампа [120] | Стресс – фактор риска развития заболевания. ↑Кортизол [121] |
| Нейровоспаление | ↑IL-1β, ↑IL-6, ↑TNF-α, ↑IL1R1, ↑TNFR1, ↑TNFR2 [122], ↑NLRP3 [92] | ↑ IL-6, ↑IL-8, ↑IL-1β
в дорсолатеральной префронтальной коре [123], ↑IL-6, ↑IL-12, ↑TNF-α, ↑IL-1β, ↑IFN-γ в фазе обострения [124] |
↑IL-1β, ↑IL-6, ↑TNF-α, ↑IL-1β, отсутствие коррекции после терапии [125, 126] | ↑IL-1β, ↑IL-6 ↑TNF-α, [122]. Процесс нейровоспаления принимает участие в отложении β-амилоида и образовании клубков, повреждении нейронов [127] | ↑IL-1β, ↑IL-6, ↑TNF-α, аутоиммунный ответ приводит к повреждению нейронов [128] |
| Нейротрофины и нейропластичность | ↓BDNF, ↓GDNF, ↓NGF, ↓VEGF, ↓FGF-2 [75] |
Снижение пролиферации клеток гиппокампа ↓BDNF [129] |
↓BDNF, ↓GDNF, ↓NGF [130–132] |
↓BDNF, ↓VEGF, ↓NGF [133]. Снижение нейрогенеза [134] |
↓BDNF, ↓VEGF, ↓NGF ↓CDNF [133]. Угнетение формирования новых нейронов в гиппокампе коррелирует с депрессивной симптоматикой [135] |
| Генетические полиморфизмы | 5HTTLRP, 5HT1A, МАОА, TPH2, BDNF, DRD2, DRD4, APOE, CREB1, NEGR1, FKBP5 [54–56] | DRD2, GRM3, GRIN2A, SRR, GRIA1, CACNA1C, CACNA1I, KCNB1, SLC39A8, MIR137, MIR548AJ2, MAD1L1, FAM5B, CD46, СR1L [136] | BDNF, DRD4, DAOA, TPH1, TRANK1 [137] | ADAM10, BCKDK/KAT8, ACE, VKORC1, TOMM40 [139] APOE [55] | ITPKB, IL1R2, CDC71, ALAS1, TLR9, DNAH1, BAP1, PHF7, SORBS3, GALC, COQ7, PSMC3IP, TUBG2 [139] |
К особенностям аффективных нарушений при шизофрении относится возникновение дефицита механизмов контроля дофаминовой нейромедиации в области гиппокампа и дорсолатеральной префронтальной коре. При депрессии же дефицит контроля высвобождения дофамина, по-видимому, возникает в вентромедиальных лобных областях коры и в последующем вовлекает миндалину [109]. Кроме того, важную роль в развитии депрессивной симптоматики при шизофрении играет дисфункция интернейронов – общий патофизиологический паттерн, характерный как для шизофрении, так и для депрессии [110].
Депрессии и шизофрении свойственны общие генетические мутации [140]. Недавно показано, что гены, мутации в которых традиционно связывали с риском развития шизофрении, также ассоциированы с риском депрессии. Эти данные подтверждены с помощью мета-анализа результатов полногеномного поиска ассоциаций (Genome wide-association studies, GWAS) [140]. Так, 10 из 44 генов, связанных с риском развития депрессии [141], повышают также риск шизофрении [140]. В качестве примера можно привести SNP в гене дофаминового D2-рецептора (DRD2), связь которого как с шизофренией, так и с депрессией наиболее хорошо изучена [108, 141]. Кроме того, ассоциации между шизофренией и мутациями в гене RSRC1, кодирующем белок с богатым аргинином и серином мотивом “coiled-coil” 1, и в гене MEF2C (Myocyte-specific enhancer factor 2C) находят и при депрессии [142].
Что касается нейробиологии БАР, то у этого расстройства также есть много общего с депрессией. Основными биомаркерами этих заболеваний служат изменения в содержании BDNF, GDNF, кортизола и цитокинов. Снижение концентрации BDNF в сыворотке крови наблюдается у пациентов с БАР как в фазе депрессии, так и в фазе мании в отличие от тех же пациентов в эутимной фазе или от контрольной группы здоровых испытуемых [131]. Уровень BDNF повышается при назначении пациентам с БАР нормотимиков из группы вальпроатов или препаратов лития в результате активации вторичных мессенджеров, связанных с усилением внутриклеточной сигнализации и экспрессии G-белка [143]. Механизм действия как антидепрессантов, так и нормотимиков состоит в способности увеличивать экспрессию рецепторов TrkB и за счет этого повышать синтез BDNF в гиппокампе. Тем не менее, назначение нормотимиков эффективно только при БАР. GDNF, подобно BDNF, отрицательно коррелирует с симптомами БАР, и его концентрация снижается во время депрессии, а в фазу мании или в эутимном состоянии остается в пределах нормы [130].
Нарушение баланса цитокинов также играет роль в патофизиологии БАР [126]. Изменения в профиле TNF-α и C-реактивного белка, а также IL-1 и IL-6 связывают с аффективными и другими психическими расстройствами [126]. Согласно Goldstein и соавт. [144], хронический воспалительный процесс прямо связан со снижением экспрессии генов нейротрофических факторов (BDNF и NGF) и увеличением содержания циркулирующих в крови провоспалительных цитокинов. Предполагается, что именно этот механизм связан с астроглиозом и снижением нейропластичности в гиппокампе, что характерно для депрессии. А хроническое нейровоспаление, наблюдаемое как при БАР, так и при депрессии, со временем приводит к демиелинизации, апоптозу нейронов и глиальных клеток, к тяжелым нейротрофическим нарушениям, которые коррелируют с тяжестью симптомов [132].
Нарушение именно трофической функции мозга характерно для начала нейродегенерации при таких заболеваниях, как БА и БП. Более того, в последнее время депрессия рассматривается многими как часть продромального периода нейродегенеративных состояний [145].
Отличительная особенность депрессивных явлений при БА – их резистентность к антидепрессантам, что объясняется механизмом сочетанных нарушений моноаминергической нейротрансмиссии, особенно обусловленной гибелью дофаминергических нейронов в мезолимбической системе [146]. Хронический стресс считается основным фактором риска депрессии, он также усугубляет накопление β-амилоида, гиперфосфорилирование тау-белка и снижение когнитивных функций, что показано в экспериментах на моделях БА [147]. Нейровоспаление, как еще одно общее звено нейробиологии БА и депрессии, приводит к реактивному астроглиозу и повышению концентрации провоспалительных цитокинов IL-1β, IL-6 и TNF-α [122], что негативно влияет на рост, дифференцировку и выживание нейронов, а также на синаптическую пластичность. Показано, что медиаторы воспаления, выделяемые микроглиальными клетками, способствуют отложению β-амилоида и дальнейшему развитию бляшек. Гибель нейронов еще более активирует TLR, индуцирует рецепторы RAGE и MAPK, нарушая таким образом синаптическую связь в коре [148]. Считается, что молекулярные каскады, активируемые этими событиями, вызывают нейродегенерацию за счет увеличения глутаматергической эксайтотоксичности, дисфункции митохондрий и окислительного стресса, а также путем снижения моноаминергической нейротрансмиссии и нейропластичности [149]. Общий итог, к которому приводит воспаление, дисфункция ГГА-оси и снижение активности трофических факторов, таких как BDNF и VEGF – нарушение корковых и подкорковых функций. Однако, несмотря на доказанную наследуемость как депрессии (31–42%), так и БА (60–80%) [141], вопрос об общих генетических паттернах этих заболеваний остается спорным. Исследования на нокаутных животных в качестве моделей позволяют получить некоторое представление о возможном молекулярно-генетическом механизме, объясняющем коморбидность депрессии и БА. Изучение специфической изоформы APOEε4 выявило связь с нарушением как моноаминергической системы (повышение экспрессии 5-HT2A), так и нейротрофической функции (уменьшение BDNF), что согласуется и с моноаминергической, и с нейротрофической гипотезой патогенеза депрессии [150].
В настоящее время существуют две гипотезы, объясняющие коморбидность депрессии и БП. Одна из них предполагает, что депрессия – это раннее проявление и часть естественного течения БП [151]. В соответствии с другой, депрессия и БП рассматриваются не как стадии одного патологического процесса, а как самостоятельные расстройства, причем депрессия считается фактором БП [151].
Серотонинергическая система непосредственно вовлечена в патофизиологию депрессии при БП. Обнаружено снижение уровня метаболитов серотонина в спинномозговой жидкости при БП, коморбидной с депрессией [152]. Кроме того, в ходе прогрессии БП на ядрах дорсального шва (место локации тел серотонинергических нейронов) происходит отложение телец Леви [153], что также свидетельствует о вовлеченности серотонинергической системы в патофизиологию БП [153]. Хорошо известна роль и дофаминергической, и норадренергической иннервации в лимбической системе и их нарушение как при БП, так и при депрессии [154]. С использованием ПЭТ выявлено значительное снижение плотности переносчиков дофамина и норадреналина в locus coeruleus и нескольких структурах, относящихся к лимбической системе, у пациентов с БП и симптомами депрессии [154]. Кроме того, низкий уровень гомованилиновой кислоты (маркера дофаминергической активности) в спинномозговой жидкости коррелирует с тяжестью депрессии при БП и БА [4].
Наряду с изменениями нейротрансмиссии, общими для этих заболеваний являются изменения в уровне нейротрофинов. Так, у пациентов с БП, коморбидной с депрессией, количество BDNF в сыворотке крови ниже, чем у пациентов без нее, причем выявлена отрицательная корреляция с выраженностью симптомов [155].
Роль генетики в развитии депрессии при БП широко обсуждается. Однако несмотря на то, что эти состояния тесно связаны, в настоящее время нет никаких доказательств их генетической общности. При этом идентифицирован ряд генов-кандидатов (например, PARK1–8), тесно связанных с повышенным риском БП, но эта связь, по-видимому, исключает случаи раннего проявления заболевания (10–20%) [156]. В качестве возможного фактора риска развития предложено рассматривать функциональный полиморфизм в промоторной области гена транспортера серотонина, связанный со снижением серотонинергической активности [157].
Поскольку исходом каждого из рассмотренных состояний является нейродегенерация, к которой приводят преимущественно нарушения трофической функции головного мозга, то ключевым фактором патогенеза этих состояний следует считать BDNF. Как сказано выше, этот нейротрофин в той или иной мере ассоциирован со всеми описанными заболеваниями. Так, при клинической депрессии и при БАР выявлено снижение концентрации BDNF в сыворотке крови, корректируемое соответствующими препаратами. Нарушение синаптической пластичности, вызванное дефицитом нейротрофина, инициирует появление признаков, соответствующих шизофрении [158], а морфологические изменения гиппокампа, сопутствующие снижению функции BDNF, наблюдаются и при депрессивных, и при нейродегенеративных расстройствах. Кроме того, предполагается, что снижение концентрации BDNF в раннем возрасте может влиять на развитие каких-либо психических расстройств в более позднее время [159]. Все эти данные позволяют рассматривать BDNF в качестве маркера депрессивных состояний.
Множество исследований подтверждают связь между снижением синтеза BDNF и нарушением когнитивных функций как у пациентов с психическими расстройствами, так и у лиц, не имеющих подтвержденного диагноза, но жалующихся на снижение памяти и внимания [160]. BDNF рассматривают как возможный маркер тяжести состояния и при депрессии, и при шизофрении [159], и при БА [133], и даже при БП [133]. Тем не менее, поскольку снижение концентрации данного белка находят при всех этих состояниях, то при очевидной существующей связи BDNF с нейропсихическими нарушениями, этот фактор нельзя использовать в качестве маркера какого-либо расстройства из-за его неспецифичности [129]. Однако не вызывает сомнения, что снижение синтеза BDNF в головном мозге, выявляемое при нейровизуализационных исследованиях, а также уменьшение его концентрации в сыворотке крови является маркером нарушения когнитивных функций [161].
Тщательное исследование депрессивных состояний, сопровождающих многие психические и неврологические заболевания, и поиск мишеней, отражающих общие и отличающиеся молекулярные и биохимичекие механизмы, может способствовать повышению качества диагностики и разработке персонализированной терапии, направленной на патогенетически обоснованные мишени.
СОВРЕМЕННЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ПАТОГЕНЕЗА ДЕПРЕССИВНЫХ СОСТОЯНИЙ
Современные экспериментальные методы открывают большие перспективы в изучении патогенеза депрессивных состояний [162, 163]. Чаще всего для исследования биохимических и молекулярно-генетических механизмов различных заболеваний используют лабораторных грызунов – мышей и крыс [164, 165]. К настоящему моменту существует большое разнообразие экспериментальных моделей депрессии, основанных на разных типах стрессора, таких как физический стресс [162], эмоциональный стресс [166], хирургическое вмешательство [167] и т.д. Роль генетических факторов в развитии депрессивных состояний изучают с использованием трансгенных животных и искусственно выведенных линий, предрасположенных к стрессу [163, 168].
Методы создания нокаутных и трансгенных животных
Благодаря достижениям генетики и молекулярной биологии становится возможным изучение роли отдельных генов, молекул и нейронов в развитии пcихоневрологических нарушений. Современные экспериментальные инструменты позволяют выборочно “включать” или “выключать” искомые гены в живых организмах и исследовать их роль в различных процессах. Методы получения трансгенных животных и животных с “выключенным” геном (нокаутом) базируются на феномене гомологичной рекомбинации – способности обмениваться участками ДНК. С помощью этого процесса можно встраивать в геном поврежденный участок ДНК (чтобы выключить ген) или нужный новый участок (чтобы получить трансгенных животных) [169]. В основе нокаута гена лежат несколько процессов, таких как получение и клонирование необходимого гена, применение генетических конструкций для селекции, использование линии эмбриональных стволовых клеток ES [170], внедрение измененных клеток в гаметы и выведение генно-модифицированного потомства [171]. В качестве конструкций, внедряемых в ДНК, чаще всего применяют ген резистентности к неомицину (ген neo) и ген тимидинкиназы вируса простого герпеса (HSV-tk), помещаемый вне гомологичной области и чувствительный к воздействию противовирусного препарата ганцикловира [171]. Полученную генетическую конструкцию вводят в клетки ES, которые помещают на питательную среду, содержащую аналог неомицина G418 и ганцикловир, что позволяет отбирать клетки с необходимыми свойствами. Клетки, в которые конструкция не встроилась, погибают от G418; клетки, в геном которых конструкция встроилась случайным образом и несет оба гена (neo и HSV-tk), погибают от ганцикловира. Только клетки, содержащие подходящий гомологичный ген, в котором и произошла гомологичная рекомбинация, выживают на питательной среде. Подобные модифицированные клетки вводят в эмбрионы мыши на стадии бластулы или морулы и имплантируют в матку самке, стимулированной гормональными препаратами. От такой самки рождаются мышата, часть половых клеток которых содержит полноценный испорченный ген. После инбридинга получают мышей, гомозиготных по данному гену, от которых можно получить нокаутную линию [169].
Для создания трансгенных животных используют векторы, содержащие последовательность ДНК искомого гена, а также промоторные и энхансерные участки [172]. Вектор в большом количестве копий вводят в оплодотворенную яйцеклетку самки мыши. Он быстро встраивается в гомологичные участки генома или в случайные места, если гомология отсутствует. Подобную измененную яйцеклетку вводят в половые пути другой самки. Часть полученных мышат несет искомый трансген, но в гетерозиготном состоянии [172]. Затем мышат скрещивают между собой для получения гомозиготных трансгенов. Использование индуцибельных и тканеспецифичных промоторов позволяет изменять гены только в определенных типах клеток или только под влиянием определенных факторов-индукторов, что позволяет исследовать тканеспецифическую роль генов в различных процессах [169].
Среди различных специфичных промоторов можно выделить два основных, наиболее часто используемых в современной нейробиологии. Это промоторы генов CaMKIIα и EMX1 [169]. Промотор CaMKIIα позволяет мутации проявляться только в возбуждающих нейронах неокортекса и гиппокампа на ранних этапах развития организма [173]. EXM1 экспрессируется также на ранних стадиях развития и затрагивает такие зоны мозга, как кора, обонятельные луковицы и гиппокамп. Использование данного промотора может быть полезным для изучения механизма гиппокампзависимого формирования памяти [169]. Существование подобных механизмов позволяет значительно усовершенствовать подходы к изучению функций различных молекул в ЦНС.
Разработка систем сайт-специфической прижизненной рекомбинации ДНК открыла возможности более детального изучения работы отдельных генов в мозгу на протяжении жизни. Первой подобной системой стала система Cre/lox-рекомбинации [174], наиболее распространенная и по сей день. В данной системе по краям интересующего гена встраивают сайты loxP, которые узнаются Cre-рекомбиназой и заменяют ген, что делает возможным получение “условных нокаутов” [175]. Технология позволяет заменять ген при помощи Cre только в тех тканях, где Cre экспрессируется, что обеспечивает пространственную специфичность. Помимо создания условных нокаутов эту систему можно использовать и для активации или подавления экспрессии генов в определенных структурах. Так, например, за счет рекомбинации участка, содержащего терминаторную последовательность, можно добиться селективного повышения экспрессии гена в областях, экспрессирующих Cre [175].
Таким образом, к настоящему моменту разработано большое количество инструментов, способных регулировать работу генов в различных структурах мозга или в организме в целом. Так, создаются линии грызунов с нокаутом генов, ассоциированных с развитием депрессии: 5-HT1A, 5-HT1B, SERT, МАО, ГАМК, нейропептида Y (NPY) и др. [176]. Одной из часто используемых генетических моделей депрессии являются мыши, трансгенные по GR. Так, мутантные гетерозиготы (GR+/–) характеризуются более высоким уровнем беспомощности и меньшим процентом избеганий тока в челночной камере [177], что свидетельствует о более низкой устойчивости к стрессу. Кроме того, мышы с селективным нокаутом гена GR в области переднего мозга проявляют признаки депрессивно-подобного поведения и нарушения работы обратной связи в системе ГГА [177]. Таким образом, подобные исследования позволяют связать функционирование отдельных генов с поведенческими реакциями и изучать патогенез патологических состояний, в том числе, оценивать взаимосвязь между развитием депрессии и нарушением определенных структур мозга.
Метод CRISPR/Cas редактирования генома
Одна из самых перспективных технологий редактирования генома – система CRISPR/Cas. Похожие методы существовали и до разработки CRISPR/Cas (нуклеазы, содержащие цинковые пальцы (ZFN) и эндонуклеазы TAL (Transcription activator-like effector nucleases, TALEN)), однако они оказались менее эффективными и более дорогими [178]. Среди разнообразных типов CRISPR/Cas наиболее удобной считается система CRISPR/Cas второго типа, включаяющая эндонуклеазу Cas9 (CRISPR-ассоциированный белок 9) [179]. В наиболее частом варианте CRISPR/Cas9 состоит из двух основных компонентов: Cas9, которая разрезает ДНК, и единой направляющей РНК (sgРНК) – химерного комплекса из crРНК и trackРНК [180]. Этот комплекс помогает получать нокаутных животных следующим образом: sgРНК обеспечивает поиск гена-мишени, а ассоциированная с ней Cas9 производит двухцепочечный разрыв ДНК [180]. Для корректной работы эндонуклеазы выбранная мишень должна соседствовать с последовательностью протоспейсерного смежного мотива PAM (5‵NGG, где N – любой нуклеотид) [169]. В дальнейшем репаративная система клетки удаляет разрыв ДНК с помощью системы негомологичного соединения концов (Non-homologous end joining, NHEJ) или путем гомологичной рекомбинации [173]. В результате NHEJ возникают вставки или делеции, которые нарушают рамку считывания и приводят к нарушению работы гена-мишени [180]. Вариант NHEJ может быть удобен для получения нокаута, в то время как гомологичную рекомбинацию, заменяющую участок ДНК, можно использовать для замены поврежденного гена на здоровый или другой искомый ген.
Показано, что наряду с удалением участка ДНК, воздействие системы CRISPR/Cas9 может приводить к одноцепочечному разрыву молекулы. Это достигается с помощью никазы Cas9 (nCas9), которая вносит одноцепочечный разрыв [178]. Кроме того, использование поврежденной Cas9 (dCas9 – dead Cas9) способствует регуляции транскрипционной активности определенных белков. Подобная эндонуклеаза не способна разрезать ДНК, но может служить своего рода матрицей для транскрипционных активаторов, репрессоров и модификаций хроматина для изменения экспрессии гена-мишени [181]. Интересно, что dCas служит также для создания конструкций с флуоресцентыми белками, что позволяет метить нужные мишени [181].
Таким образом, технология CRISPR/Cas открывает большие перспективы в дальнейших научных и медицинских разработках, в том числе, способствует выявлению механизмов патогенеза депрессии. Несмотря на ряд преимуществ, связанных с удобством и быстротой метода, а также его относительно невысокой стоимостью, технология имеет некоторые недостатки, такие как неполная избирательность эндонуклеазы Cas9 при формировании разрывов ДНК. Так, система может не учитывать некоторые расхождения в искомой последовательности и приводить к повреждению генома. Еще один ограничивающий фактор – необходимость расположения последовательности PAM рядом с геном-мишенью, что в некоторой степени сужает выбор потенциального гена-кандидата для нокаута. Несмотря на приведенные рамки, этот метод предоставляет большие возможности для изучения заболеваний человека и разработки эффективных методов терапии.
Оптогенетические и хемогенетические подходы
Один из наиболее перcпективных современных методов исследования как патогенеза психических расстройств, так и работы ЦНС – оптогенетика. Этот метод основан на внедрении методами генетической инженерии в нейроны специальных ионных каналов – опсинов, активирующихся в ответ на возбуждение светом определенной длины волны [182, 183]. Активация канала может привести к деполяризации или гиперполяризации клетки в зависимости от типа используемого опсина. Таким образом, воздействуя на определенные нейроны, можно индуцировать появление специфического типа поведения и исследовать вклад определенных групп нейронов в различные реакции, включая депрессивно-подобное поведение [184]. Разработка метода оптогенетики стала возможной благодаря обнаружению в клетках водоросли Chlamidomonas reinhardtii светоуправляемых анионных каналов – каналродопсинов, способных деполяризовать культуры клеток млекопитающих в ответ на свет и контролировать таким образом уровень их активности [185]. К настоящему моменту в оптогенетике наиболее часто используют два опсина – каналродопсин 2 (СhR2) и галородопсин из архебактерии Natromonas pharaonic [184]. Воздействие голубого света (примерно 470 нм) на ChR2 приводит к открытию Na+-каналов и деполяризации нейронов, что увеличивает уровень их активности. Галородопсин активируют желтым светом (примерно 580 нм), который вызывает открытие Cl-канала и соответствующую гиперполяризацию клеток [184]. Ген опсина можно встраивать в нейроны определенного типа, используя клеточноспецифичный промотор и лентивирусный или аденовирусный вектор [186]. Для изучения определенной структуры мозга конструкция вводится стереотаксически [186]. Повысить специфичность встраивания каналов в нейроны определенного типа можно с помощью технологии Cre-рекомбинации. При использовании трансгенных линий грызунов, экспрессирующих Cre-рекомбиназу только в определенных популяциях клеток, можно добиться встраивания опсинов только в данные клетки [185]. После встраивания каналов в структуры мозга крыс прижизненно облучают светом определенной длины волны при помощи оптоволокна или лазера. Для проникновения облучения на нужную глубину оптоволокно имплантируют внутрь тканей мозга. При проведении поведенческих тестов, направленных на оценку депрессивно-подобного поведения, подобные манипуляции могут показать вклад определенных структур мозга в поведенческие реакции.
Наряду с оптогенетическими технологиями в последние годы активно развивается метод прижизненного контроля нейронов, основанный на воздействии химических веществ – хемогенетика. В этом случае специальные белки генетически встраиваются в определенные клетки и селективно активируются небольшими триггерными молекулами [184]. Наиболее часто используются оригинальные рецепторы, эксклюзивно активируемые оригинальными препаратами (Desighner Receptors Exclusevely Activated by Desighner Drugs, DREADD) [187]. Они представляют собой модифицированные мускариновые рецепторы, связанные с G-белком, которые также встраивают в нужные нейроны при помощи вирусных векторов [186]. Первые DREADD представляли собой три Gq-связанных мускариновых рецептора: hM1Dq, hM3Dq, hM5Dq. Gq DREADD – это деполяризующие рецепторы [186], которые активируются клозапин-N-оксидом (CNO) и используют внутриклеточный кальций в качестве вторичного посредника [184]. Gi-связанные рецепторы (hM2Di, hM4Di, KORD – k-opioid-derived DREADD), наоборот, подавляют активность нейронов. Рецепторы hM2Di и hM4Di также активируются CNO, а рецепторы KORD – сальвинорином B [184]. В целом, реакция нейронов зависит от типа G-белка, ассоциированного с рецептором, и каскада реакций, вызываемого им. Технология встраивания рецепторов в специфичные клетки мозга аналогична технологии, используемой в оптогенетике, в ней также применяют вирусные векторы, Cre-рекомбинацию и стереотаксическое введение конструкций. В отличие от оптогенетических подходов она не требует имплантирования оптоволоконной канюли в ткани мозга, ее действие опосредуется введением веществ-лигандов.
Технологии оптогенетики и хемогенетики пролили свет на многие функции ЦНС и поведенческие реакции, ассоциированные с различными расстройствами, в том числе с депрессией. Например, выявлен вклад активации вентромедиальной префронтальной коры и путей, ассоциированных с ней, в депрессивное поведение [184]. Оптогенетическая стимуляция глутаматергических нейронов вентромедиальной префронтальной коры индуцировала противодепрессивные эффекты в тесте подвешивания за хвост [188], а хемогенетическая стимуляция коры предотвратила развитие депрессивно-подобного состояния, вызванного ранним отлучением мышей от матери [188]. В оптогенетических и хемогенетических исследованиях показана роль и других структур в формировании депрессивно-подобного поведения, таких как прилежащее ядро, вентральная область покрышки, гиппокамп, миндалина, ядро шва и др. [188, 189].
В целом, эти подходы открывают большие перспективы в исследовании нейрональных процессов и имеют ряд преимуществ по сравнению со многими другими методами [182, 187, 190]. Так, появляется возможность прижизненного контроля работы нейронов и изучения вклада тех или иных популяций клеток в поведенческие процессы. Кроме того, эти методы высокоспецифичны. Тем не менее, и они имеют свои недостатки. Например, в оптогенетике значительным ограничением является низкая степень проникновения света в ткани мозга, что затрудняет эксперименты с более крупными живыми организмами, такими как нечеловекообразные приматы [186]. Также открытым остается вопрос о длительности и частоте стимуляции, так как подчас разный тип воздействия на одни и те же структуры может приводить к различным поведенческим реакциям [184]. Хемогенетические методы отличаются более низкой скоростью реакций из-за способа активации рецепторов, кроме того, возможно влияние самого лиганда на поведение животных, что ограничивает применение данного подхода. Тем не менее, подобные методы однозначно вносят существенный вклад в понимание работы ЦНС и нейробиологических процессов, ассоциированных с тревогой, депрессией и другими эмоциональными проявлениями.
Клинический анализ данных с использованием GWAS
Механизмы депрессивных состояний изучают также с использованием методов анализа клинических данных. Они основаны, в первую очередь, на поиске генетических основ депрессии. Существует несколько подходов к поиску таких мишеней. В частности, анализ групп сцепления, изучение генов-кандидатов и расшифровка последовательности генома [191]. Тем не менее, в связи с тем, что депрессия представляет собой полиморфное и полигенное заболевание [192], эти подходы оказываются малоэффективными для понимания ее патогенеза. Наиболее перспективным считается GWAS [193], в основе которого лежит сравнение геномов контрольной популяции и больных, в частности, выявление SNP, играющих большую роль в возникновении разнообразия генома человека [136]. Исследования охватывают огромные массивы данных в связи с большим (сотни тысяч) разнообразием возможных SNP.
Для исследований используют образцы ДНК, полученные из слюны, крови и других биологических материалов. Сам анализ проводят методом логистической регрессии с зависимым статусом переменной случай–контроль (0=контроль, 1=случай) и SNP в качестве независимой переменной, что позволяет выявлять ассоциации различных полиморфизмов с патологией. Так как GWAS выполняют для каждого SNP с целью исключения ошибки первого рода (ложноположительные результаты), то требуется соответствие кумулятивного значения вероятности (p) значению не более 5 × 10–8, точная репликация участков в разных выборках и тесная ассоциация показателей [136].
Предполагается, что генетические полиморфизмы могут приводить к развитию психических расстройств в результате совместного действия сотен и тысяч различных распространенных SNP, каждый из которых по отдельности оказывает незначительный эффект, или воздействия множества редких SNP, которые нарушают структуру гена и поэтому обладают сильным эффектом [136]. В связи со значительной полигенностью депрессии ее возникновение вызвано, скорее всего, воздействием множества распространенных полиморфизмов [192]. В целом, к настоящему моменту при помощи GWAS выявлены 102 генетические вариации, ассоциированные с клинической депрессией [194]. Предположительно, расширение анализируемых выборок будет способствовать выявлению новых генетических ассоциаций.
Таким образом, данные GWAS могут способствовать пониманию механизмов патогенеза депрессии. Кроме того, изучение локальных причин формирования депрессивных состояний может способствовать определению причин различных форм депрессии и подбору направленной терапии с высокой степенью эффективности.
ЗАКЛЮЧЕНИЕ
К настоящему моменту известно множество биологических механизмов, так или иначе участвующих в развитии депрессивной симптоматики. К ним можно отнести дисбаланс в моноаминовой системе, нарушение нейропластичности, нейровоспаление и др. Тем не менее, невозможно выделить единый механизм развития депрессивных состояний. Для возникновения патологии важно комбинированное влияние различных факторов из изученных звеньев патогенеза. В этом проявляется гетерогенность депрессивных расстройств.
Многообразие нейробиологических нарушений, которые обнаруживаются при депрессии, в сочетании с возникновением депрессивной симптоматики в структуре многих психических расстройств, совершенно различных по своим причинам и клиническим проявлениям, позволяет предполагать, что развитие аффективной патологии в каждом случае может идти разными путями. Это несомненно снижает прогнозируемость течения депрессии и эффективность ее лечения, так как один и тот же антидепрессант, действующий на определенный нейрохимический механизм, может быть эффективным в одном случае, но не вызывать выраженного ответа в другом. Вследствие этого необходим персонализированный подход к выбору средств терапии, основанный на выявлении биомаркеров заболевания в биологических образцах пациента.
Постановка диагноза по клинической картине должна быть усовершенствована набором молекулярно-биологических инструментов, позволяющих детально оценивать специфичные биомаркеры, указывающие на развитие патологии еще до проявления первых клинических симптомов. Несмотря на то, что GWAS-анализ, проведенный на больших выборках, не привел к ясному пониманию молекулярных механизмов патогенеза депрессии, изучение уникальных редких SNP могло бы способствовать более точному установлению генетических ассоциаций и определению частных причин развития депрессивных состояний. Вполне вероятно, что генетические исследования будут сосредоточены на изучении небольших выборок с помощью методов секвенирования нового поколения (NGS). Одной из главных задач может стать также изучение эпигенетической регуляции депрессии.
Для поиска персонализированных терапевтических мишеней и изучения особенностей формирования депрессивных расстройств важным становятся моделирование на животных специфической и контролируемой экспрессии генов, а также интеграция гуманизированных генных вариантов, например, с помощью CRISPR/Cas9. Выявлению путей и направлений поиска биомаркеров депрессии способствует и изучение роли отдельных групп нейронов в патогенезе аффективных расстройств. Определение специфичных биомаркеров, основанное на различиях в профиле экспрессии генов, белков, паттернов активности метаболизма, электроэнцефалографических и томографических данных, позволит создать качественные тест-системы, способствующие подбору персонализированных препаратов в нужные сроки и в необходимых дозах. Такие системы помогут вывести современную психиатрию на новый уровень диагностики и лечения психических заболеваний.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований в рамках научного проекта № 19-115-50458.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Wang Q., Timberlake M.A., Prall K., Dwivedi Y. (2017) The recent progress in animal models of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry. 77, 99–109.
Felmet K., Zisook S., Kasckow J. (2010) Elderly patients with schizophrenia and depression: diagnosis and treatment. Clin. Schizophr. Relat. Psychoses. 4, 239–250.
Upthegrove R., Marwaha S., Birchwood M. (2017) Depression and schizophrenia: cause, consequence, or trans-diagnostic issue? Schizophr. Bull. 43, 240—244.
Galts C., Bettio L., Jewett D.C., Yang C.C., Brocardo P.S., Rodrigues A., Thacker J.S., Gil-Mohapel J. (2019) Depression in neurodegenerative diseases: common mechanisms and current treatment options. Neurosci. Biobehav. Rev. 102, 56–84.
Treadway M.T., Zald D.H. (2011) Reconsidering anhedonia in depression: lessons from translational neuroscience. Neurosci. Biobehav. Rev. 35, 537–555.
Otte C., Gold S.M., Penninx B.W., Pariante C.M., Etkin A., Fava M., Mohr D.C., Schatzberg A.F. (2016) Major depressive disorder. Nat. Rev. Dis. Primers. 2, 160–165.
Jesulola E., Micalos P., Baguley I.J. (2018) Understanding the pathophysiology of depression: from monoamines to the neurogenesis hypothesis model – are we there yet? Behav. Brain Res. 341, 79–90.
Saavedra K., Molina-Márquez A.M., Saavedra N., Zambrano T., Salazar L.A. (2016) Epigenetic modifications of major depressive disorder. Int. J. Mol. Sci. 17, 1279. https://doi.org/10.3390/ijms17081279
Lee S., Jeong J., Kwak Y., Park S.K. (2010) Depression research: where are we now? Mol. Brain. 3, 8. https://doi.org/10.1186/1756-6606-3-8
Смулевич А.Б. (1997) Депрессии и коморбидные расстройства. М.: РАМН – НЦПЗ.
Koolschijn P.C., van Haren N.E., Lensvelt-Mul-ders G.J., Hulshoff Pol H.E., Kahn R.S. (2009) Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum. Brain Map. 30, 3719–3735.
Bora E., Fornito A., Pantelis C., Yucel M. (2012) Gray matter abnormalities in major depressive disorder: a meta-analysis of voxel based morphometry studies. J. Affect. Disord. 138, 9–18.
Du X., Pang T.Y. (2015) Is dysregulation of the HPA-axis a core pathophysiology mediating comorbid depression in neurodegenerative diseases? Front. Psychiatry. 6, 32. https://doi.org/10.3389/fpsyt.2015.00032
Nikkheslat N., Pariante C.M., Zunszain P.A. (2018) Neuroendocrine abnormalities in major depression: an insight into glucocorticoids, cytokines, and the kynurenine pathway. Inflammation and Immunity in Depression: Basic Science and Clinical Application. M: Acad. Press. 45–60.
Villas Boas G.R., Boerngen de Lacerda R., Paes M.M., Gubert P., Almeida W., Rescia V.C., de Carvalho P., de Carvalho A., Oesterreich S.A. (2019) Molecular aspects of depression: a review from neurobiology to treatment. Eur. J. Pharmacol. 851, 99–121.
de Kloet E.R., Meijer O.C., de Nicola A.F., de Rijk R.H., Joëls M. (2018) Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuroinflammation. Front. Neuroendocrinol. 49, 124–145.
Binder E.B. (2009) The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 34, 186–195.
Xu J., Wang R., Liu Y., Wang W., Liu D., Jiang H., Pan, F. (2019) Short- and long-term alterations of FKBP5-GR and specific microRNAs in the prefrontal cortex and hippocampus of male rats induced by adolescent stress contribute to depression susceptibility. Psychoneuroendocrinology. 101, 204–215.
Bouwknecht J.A. (2015) Behavioral studies on anxiety and depression in a drug discovery environment: keys to a successful future. Eur. J. Pharmacol. 753, 158–176.
Landgraf R. (2006) The involvement of the vasopressin system in stress-related disorders. CNS Neurol. Disord. Drug Targets. 5, 167–179.
Li J., Chen J., Ma N., Yan D., Wang Y., Zhao X., Zhang Y., Zhang C. (2019) Effects of corticosterone on the expression of mature brain-derived neurotrophic factor (mBDNF) and proBDNF in the hippocampal dentate gyrus. Behav. Brain Res. 365, 150–156.
Simon N.G., Guillon C., Fabio K., Heindel N.D., Lu S.F., Miller M., Ferris C.F., Brownstein M.J., Garripa C., Koppel G.A. (2008). Vasopressin antagonists as anxiolytics and antidepressants: recent developments. Recent Pat. CNS Drug Discov. 3, 77–93.
Lang U.E., Borgwardt S. (2013) Molecular mechanisms of depression: perspectives on new treatment strategies. Cell. Physiol. Biochem. 31, 761–777.
Базян А.С., Григорян Г.А. (2006) Молекулярно-химические основы эмоциональных состояний и подкрепления. Усп. физиол. наук. 37, 68–83.
Goddard A.W., Ball S.G., Martinez J., Robinson M.J., Yang C.R., Russell J.M., Shekhar A. (2010) Current perspectives of the roles of the central norepinephrine system in anxiety and depression. Depress. Anxiety. 27, 339–350.
Shen L.H., Liao M.H., Tseng Y.C. (2012) Recent advances in imaging of dopaminergic neurons for evaluation of neuropsychiatric disorders. J. Biomed. Biotechnol. 2012, 259349. https://doi.org/10.1155/2012/259349
Hannon J., Hoyer D. (2008) Molecular biology of 5-HT receptors. Behav. Brain Res. 195, 198–213.
Nautiyal K.M., Hen R. (2017) Serotonin receptors in depression: from A to B. F1000Res. 6, 123. https://doi.org/10.12688/f1000research.9736.1
Yohn C.N., Gergues M.M., Samuels B.A. (2017) The role of 5-HT receptors in depression. Mol. Brain. 10, 28. https://doi.org/10.1186/s13041-017-0306-y
Anttila S., Huuhka K., Huuhka M., Rontu R., Hurme M., Leinonen E. (2007) Interaction between 5-HT1A and BDNF genotypes increases the risk of treatment-resistant depression. J. Neural. Transm. (Vienna). 114, 1065–1068.
Ohtsuki T., Ishiguro H., Detera-Wadleigh S.D., Toyota T., Shimizu H., Yamada K., Yoshitsugu K., Hattori E., Yoshikawa T., Arinami T. (2002) Association between serotonin 4 receptor gene polymorphisms and bipolar disorder in Japanese case-control samples and the NIMH Genetics Initiative Bipolar Pedigrees. Mol. Psychiatry. 7, 954–961.
Lucas G., Rymar V.V., Du J., Mnie-Filali O., Bisgaard C., Manta S., Lambas-Senas L., Wiborg O., Haddjeri N., Piñeyro G., Sadikot A.F., Debonnel G. (2007) Serotonin(4) (5-HT(4)) receptor agonists are putative antidepressants with a rapid onset of action. Neuron. 55, 712–725.
Попова Н.К., Ильбаичева Т.В., Науменко В.С. (2017) Нейротрофические факторы (BDNF, GDNF) и серотонинергическая система мозга. Биохимия. 82, 449–459.
Caspi A., Sugden K., Moffitt T.E., Taylor A., Craig I. W., Harrington H., McClay J., Mill J., Martin J., Braithwaite A., Poulton R. (2003) Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 301, 386–389.
Vinkers C.H., Joels M., Milaneschi Y., Kahn R.S., Penninx B.W., Boks M.P. (2014) Stress exposure across the life span cumulatively increases depression risk and is moderated by neuroticism. Depress. Anxiety. 31, 737–745.
Landau A.M., Phan J.A., Iversen P., Lillethorup T.P., Simonsen M., Wegener G., Jakobsen S., Doudet D.J. (2015) Decreased in vivo α2 adrenoceptor binding in the Flinders Sensitive Line rat model of depression. Neuropharmacology. 91, 97–102.
Seki K., Yoshida S., Jaiswal M.K. (2018) Molecular mechanism of noradrenaline during the stress-induced major depressive disorder. Neural Regen. Res. 13, 1159–1169.
Pitsillou E., Bresnehan S.M., Kagarakis E.A., Wijoyo S.J., Liang J., Hung A., Karagiannis T.C. (2020) The cellular and molecular basis of major depressive disorder: towards a unified model for understanding clinical depression. Mol. Biol. Rep. 47, 753–770.
Belujon P., Grace A.A. (2017) Dopamine system dysregulation in major depressive disorders. Int. J. Neuropsychopharmacol. 20, 1036–1046.
Sarchiapone M., Carli V., Camardese G., Cuomo C., Di Giuda D., Calcagni M.L., Focacci C., De Risio S. (2006) Dopamine transporter binding in depressed patients with anhedonia. Psychiatry Res. 147, 243–248.
Beaulieu J.M. (2012) A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J. Psychiatry Neurosci. 37, 7–16.
Weng Y.T., Chien T., Kuan I.I., Chern Y. (2018) The TRAX, DISC1, and GSK3 complex in mental disorders and therapeutic interventions. J. Biomed. Sci. 25, 71. https://doi.org/10.1186/s12929-018-0473-x
Costemale-Lacoste J.F., Guilloux J.P., Gaillard R. (2016) The role of GSK-3 in treatment-resistant depression and links with the pharmacological effects of lithium and ketamine: a review of the literature. Encephale. 42, 156–164.
Kram M.L., Kramer G.L., Ronan P.J., Steciuk M., Petty F. (2002) Dopamine receptors and learned helplessness in the rat: an autoradiographic study. Prog. Neuropsychopharmacol. Biol. Psychiatry. 26, 639–645.
Furuyashiki T. (2012) Roles of dopamine and inflammation-related molecules in behavioral alterations caused by repeated stress. J. Pharmacol. Sci. 120, 63–69.
Drago A., Crisafulli C., Sidoti A., Serretti A. (2011) The molecular interaction between the glutamatergic, noradrenergic, dopaminergic and serotoninergic systems informs a detailed genetic perspective on depressive phenotypes. Prog. Neurobiol. 94, 418–460.
Duman R.S., Sanacora G., Krystal J.H. (2019) Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. 102, 75–90.
Kang H.J., Voleti B., Hajszan T., Rajkowska G., Stockmeier C.A., Licznerski P., Lepack A., Majik M.S., Jeong L.S., Banasr M. (2012) Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 18, 1413–1417.
Li N., Liu R.J., Dwyer J.M., Banasr M., Lee B., Son H., Li X.Y., Aghajanian G., Duman R.S. (2011) Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol. Psychiatry. 69, 754–761.
Banasr M., Lepack A., Fee C., Duric V., Maldonado-Aviles J., DiLeone R., Sibille E., Duman R.S., Sanacora G. (2017) Characterization of GABAergic marker expression in the chronic unpredictable stress model of depression. Chronic Stress (Thousand Oaks). 1, 10. https://doi.org/10.1177/2470547017720459
Luscher B., Fuchs T. (2015) GABAergic control of depression-related brain states. Adv. Pharmacol. 73, 97–144.
Glue P., Medlicott N.J., Harland S., Neehoff S., Anderson-Fahey B., Le Nedelec M., Gra A., McNaughton N. (2017) Ketamine’s dose-related effects on anxiety symptoms in patients with treatment refractory anxiety disorders. J. Psychopharmacol. (Oxford.). 31, 1302–1305.
Lesch K.P., Bengel D., Heils A., Sabol S.Z., Greenberg B.D., Petri S., Benjamin J., Muller C.R., Hamer D.H., Murphy D.L. (1996) Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 274, 1527–1531.
Xia L., Yao S. (2015) The involvement of genes in adolescent depression: a systematic review. Front. Behav. Neurosci. 9, 329. https://doi.org/10.3389/fnbeh.2015.00329
Tsang R.S., Mather K.A., Sachdev P.S., Reppermund S. (2017) Systematic review and meta-analysis of genetic studies of late-life depression. Neurosci. Biobehav. Rev. 75, 129–139
Northoff G. (2013) Gene, brains, and environment-genetic neuroimaging of depression. Curr. Opin. Neurobiol. 23, 133–142.
Liu Y., Yu J.-T., Wang H.-F., Han P.-R., Tan C.-C., Wang C., Meng X.-F., Risacher S.L., Saykin A.J., Tan L. (2015) APOE genotype and neuroimaging markers of Alzheimer’s disease: Systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry. 86, 127–134.
Liu R-J., Lee F.S., Li X-Y., Bambico F., Duman R.S., Aghajanian G.K. (2012) Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol. Psychiatry. 71, 996–1005.
Piechaczek C.E., Greimel E., Feldmann L., Pehl V., Allgaier A.K., Frey M., Freisleder F.J., Halldorsdottir T., Binder E.B., Ising M., Schulte-Körne G. (2019) Interactions between FKBP5 variation and environmental stressors in adolescent major depression. Psychoneuroendocrinology. 106, 28–37.
Dalton V.S., Kolshus E., McLoughlin D.M. (2014) Epigenetics and depression: return of the repressed. J. Affec. Disord. 155, 1–12.
Chistiakov D.A., Bobryshev Y.V., Chekhonin V.P. (2017) Epigenetic alterations in DNA and histone modifications caused by depression and antidepressant drugs: Lessons from the rodent models. Curr. Pharm. Des. 23, 6828–6840.
Park C., Rosenblat J.D., Brietzke E., Pan Z., Lee Y., Cao B., Zuckerman H., Kalantarova A., McIntyre R.S. (2019) Stress, epigenetics and depression: A systematic review. Neurosci. Biobehav. Rev. 102, 139–152.
Chen D., Meng L., Pei F., Zheng Y., Leng J. (2017) A review of DNA methylation in depression. J. Clin. Neurosci. 43, 39–46.
Kim G.H., Ryan J.J., Marsboom G., Archer S.L. (2011) Epigenetic mechanisms of pulmonary hypertension. Pulm. Circ. 1, 347–356.
Gruzdev S.K., Yakovlev A.A., Druzhkova T.A., Guekht A.B., Gulyaeva N.V (2019) The missing link: how exosomes and miRNAs can help in bridging psychiatry and molecular biology in the context of depression, bipolar disorder and schizophrenia. Cell Mol. Neurobiol. 39, 729–750.
Pruunsild P., Kazantseva A., Aid T., Palm K., Timmusk T. (2007) Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics. 90, 397–406.
Kang H.J., Kim J.M., Stewart R., Kim S.Y., Bae K.Y., Kim S.W., Yoon J.S. (2013). Association of SLC6A4 methylation with early adversity, characteristics and outcomes in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry. 44, 23–28.
Murgatroyd C., Quinn J.P., Sharp H.M., Pickles A., Hill J. (2015) Effects of prenatal and postnatal depression, and maternal stroking, at the glucocorticoid receptor gene. Transl. Psychiatry. 5, e560. https://doi.org/10.1038/tp.2014.140
Covington H.E. 3rd, Maze I., LaPlant Q.C., Vialou V.F., Ohnishi Y.N., Berton O., Fass D.M., Renthal W., Rush A.J. 3rd, Wu E.Y., Ghose S., Krishnan V., Russo S.J., Tamminga C., Haggarty S.J., Nestler E.J. (2009). Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 29, 11451–11460.
Hobara T., Uchida S., Otsuki K., Matsubara T., Funato H., Matsuo K., Suetsugi M., Watanabe Y. (2010) Altered gene expression of histone deacetylases in mood disorder patients. J. Psychiatr. Res. 44, 263–270.
Tavakolizadeh J., Roshanaei K., Salmaninejad A., Yari R., Nahand J.S., Sarkarizi H.K., Mousavi S.M., Salarinia R., Rahmati M., Mousavi S.F., Mokhtari R., Mirzaei H. (2018) MicroRNAs and exosomes in depression: potential diagnostic biomarkers. J. Cell. Biochem. 119, 3783–3797.
Gururajan A., Naughton M., Scott K., O’Connor R., Moloney G., Clarke G., Dowling J., Walsh A., Ismail F., Shorten G., Scott L., McLoughlin D., Cryan J., Dinan T. (2016) MicroRNAs as biomarkers for major depression: a role for Let-7b and Let-7c. Transl. Psychiatry. 6, e862. https://doi.org/10.1038/tp.2016.131
O'Connor R.M., Grenham S., Dinan T.G., Cryan J.F. (2013) MicroRNAs as novel antidepressant targets: converging effects of ketamine and electroconvulsive shock therapy in the rat hippocampus. Int. J. Neuropsychopharmacol. 16, 1885–1892.
Liu W., Ge T., Leng Y., Pan Z., Fan J., Yang W., Cui R. (2017) The role of neural plasticity in depression: from hippocampus to prefrontal cortex. Neural Plast. 2017, https://doi.org/10.1155/2017/6871089
Levy M.J.F., Boulle F., Steinbusch H.W., van den Hove D.L.A., Kenis G., Lanfumey L. (2018) Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology (Berl.). 235, 2195–2220.
Базовкина Д.В., Кондаурова Е.М., Цыбко А.С., Ковецкая А.И., Ильчибаева Т.В., Науменко В.С. (2017) Влияние хронической алкоголизации на экспрессию гена нейротрофического фактора мозга (BDNF) и его рецепторов в мозге мышей с генетической предрасположенностью к “депрессивно-подобному” поведению. Молекуляр. биология. 51, 647–655.
Notaras M., van den Buuse M. (2020) Neurobiology of BDNF in fear memory, sensitivity to stress, and stress-related disorders. Mol. Psychiatry. https://doi.org/10.1038/s41380-019-0639-2
Leal G., Bramham C.R., Duarte C.B. (2017) BDNF and hippocampal synaptic plasticity. Vitam. Horm. 104, 153–195.
Huang E.J., Reichardt L.F. (2003) Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642.
Garraway S.M., Huie J.R. (2016) Spinal plasticity and behavior: BDNF-induced neuromodulation in uninjured and injured spinal cord. Neural. Plast. 2016, https://doi.org/10.1155/2016/9857201
Jin W. (2020) Regulation of BDNF-TrkB signaling and potential therapeutic strategies for Parkinson’s disease. J. Clin. Med. 9, https://doi.org/10.3390/jcm9010257
Wang J.Q., Mao L. (2019) The ERK pathway: molecular mechanisms and treatment of depression. Mol. Neurobiol. 56, 6197–6205.
Lynch M.A. (2004) Long-term potentiation and memory. Physiol. Rev. 84, 87–136.
Eisch A.J., Petrik D. (2012) Depression and hippocampal neurogenesis: a road to remission? Science. 338, 72–75.
Losenkov I.S., Mulder N., Levchuk L.A., Vyalova N.M., Loonen A., Bosker, F.J., Simutkin G.G., Boiko A.S., Bokhan N.A., Wilffert B., Hak E., Schmidt A.F., Ivanova S.A. (2020). Association between BDNF gene variant Rs6265 and the severity of depression in antidepressant treatment-free depressed patients. Front. Psychiatry. 11, 38. https://doi.org/10.3389/fpsyt.2020.00038
Иванова С.А., Лосенков И.Н., Левчук Л.А., Бойко А.Н., Вялова Н.М., Симуткин Г.Г., Бохан Н.А. (2018) Депрессивные расстройства: гипотезы патогенеза и потенциальные биологические маркеры. М.: Изд-во Сибирского отделения РАН.
Chourbaji S., Brandwein C., Gass P. (2011) Altering BDNF expression by genetics and/or environment: impact for emotional and depression-like behaviour in laboratory mice. Neurosci. Biobehav. Rev. 35, 599–611.
Evans S.J., Choudary P.V., Neal C.R., Li J.Z., Vawter M.P., Tomita H. (2004) Dysregulation of the fibroblast growth factor system in major depression, Proc. Natl. Acad. Sci. USA. 101, 15506–15511.
Elsayed M., Banasr M., Duric V., Fournier N.M., Licznerski P., Duman R.S. (2012) Antidepressant effects of fibroblast growth factor-2 in behavioral and cellular models of depression. Biol. Psychiatry. 72, 258–265.
Angelucci F., Aloe L., Jimenez-Vasquez P., Mathe A.A. (2003). Electroconvulsive stimuli alter nerve growth factor but not brainderived neurotrophic factor concentrations in brains of a rat model of depression. Neuropeptides. 37, 51–56.
Jeon S.W., Kim Y.K. (2017) Inflammation-induced depression: Its pathophysiology and therapeutic implications. J. Neuroimmunol. 313, 92–98.
Wohleb E.S., Franklin T., Iwata M., Duman R.S. (2016) Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 17, 497–511.
Zhan Y., Paolicelli R.C., Sforazzini F., Weinhard L., Bolasco G., Pagani F., Vyssotski A.L., Bifone A., Gozzi A., Ragozzino D., Gross C.T. (2014) Deficient neuron–microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406.
Musselman D.L., Lawson D.H., Gumnick J.F., Manatunga A.K., Penna S., Goodkin R.S., Greiner K., Nemeroff C.B., Miller A.H. (2001) Paroxetine for the prevention of depression induced by high-dose interferon alfa. N. Engl. J. Med. 344, 961–966.
Franklin T.C., Xu C., Duman R.S. (2018) Depression and sterile inflammation: essential role of danger associated molecular patterns. Brain. Behav. Immun. 72, 2–13.
Trottier M.D., Newsted M.M., King L.E., Fraker, P.J. (2008) Natural glucocorticoids induce expansion of all developmental stages of murine bone marrow granulocytes without inhibiting function. Proc. Natl Acad. Sci. USA. 105, 2028–2033.
Milenkovic V.M., Stanton E.H., Nothdurfter C., Rupprecht R., Wetzel C.H. (2013) The role of chemokines in the pathophysiology of major depressive disorder. Int. J. Mol. Sci. 20, 2283. https://doi.org/10.3390/ijms20092283
Kierdorf K., Prinz M. (2013) Factors regulating microglia activation. Front. Cell. Neurosci. 7, 44. https://doi.org/10.3389/fncel.2013.00044
Guo H., Callaway J.B., Ting J.P. (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687.
Kulmatycki K.M., Jamali F. (2005) Drug disease interactions: role of inflammatory mediators in disease and variability in drug response. J. Pharm. Pharm. Sci. 8, 602–625.
Black C.N., Bot M., Scheffer P.G., Cuijpers P., Penninx B.W. (2015) Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology. 51, 164–175.
Gorlova A.V., Zorkina Ya.A., Zubkov E.A., Morozova A.Yu., Inozemtsev A.N., Chekhonin V.P. (2019) Alteration of oxidative stress markers and behavior of rats in a novel model of depression. Acta Neurobiol. Exp. (Wars.). 79, 232–237
Moylan S., Berk M., Dean O.M., Samuni Y., Williams L.J., O’Neil A., Hayley A.C., Pasco J. A., Anderson G., Jacka F.N., Maes M. (2014) Oxidative and nitrosative stress in depression: why so much stress? Neurosci. Biobehav. Rev. 45, 46–62.
Yager S., Forlenza M.J., Miller G.E. (2010) Depression and oxidative damage to lipids. Psychoneuroendocrinology. 35, 1356–1362.
Maes M., Galecki P., Chang Y.S., Berk M. (2011) A review on the oxidative andnitrosative stress (O&NS) pathways in major depression and their possible contribution to the neurodegenerative processes in that illness. Prog. Neuropsychopharmacol. Biol. Psychiatry. 35, 676–692.
Wigner P., Czarny P., Galecki P., Su K.P., Sliwinski T. (2018) The molecular aspects of oxidative and nitrosative stress and the tryptophan catabolites pathway (TRYCATs) as potential causes of depression. Psychiatry Res. 262, 566–574.
Schubert D., Piasecki D. (2001) Oxidative glutamate toxicity can be a component ofthe excitotoxicity cascade. J. Neurosci. 21, 7455–7462.
Howard D.M., Adams M.J., Clarke T.K., Hafferty J.D., Gibson J., Shirali M., Coleman J.R.I., Hagenaars S.P., Ward J., Wigmore E.M., Alloza C., Shen X., Barbu M.C., Xu E.Y., Whalley H.C., Marioni R.E., Porteous D.J., Davies G., Deary I.J., Hemani G., Berger K., Teismann H., Rawal R., Arolt V., Baune B.T., Dannlowski U., Domschke K., Tian C., Hinds D.A., 23andMe Research Team; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium, Trzaskowski M., Byrne E.M., Ripke S., Smith D.J., Sullivan P.F., Wray N.R., Breen G., Lewis C.M., McIntosh A.M. (2019) Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352.
Grace A.A. (2016) Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 17, 524–532.
Buzsaki G., Draguhn A. (2004) Neuronal oscillations in cortical networks. Science. 304, 1926–1929.
Bora E., Fornito A., Pantelis C., Yucel M. (2012) Gray matter abnormalities in major depressive disorder: a meta-analysis of voxel based morphometry studies. J. Affect. Disord. 138, 9–18.
Herman J.P., Mueller N.K. (2016) Role of the ventral subiculum in stress integration. Behav. Brain Res. 174, 215–224.
Marchand W.R., Lee J.N., Thatcher G.W., Jensen C., Stewart D., Dilda V., Thatcher J., Creem-Regehr S.H. (2007) A functional MRI study of a paced motor activation task to evaluate frontalsubcortical circuit function in bipolar depression. Psychiatry Res. 1553, 221–230.
Hibar D.P., Westlye L.T., van Erp T.G., Rasmussen J, Leonardo C.D., Faskowitz J., Haukvik U.K., Hartberg C.B., Doan N.T., Agartz I., Dale A.M., Gruber O., Krämer B., Trost S., Liberg B., Abé C., Ekman C.J., Ingvar M., Landén M., Fears S.C., Freimer N.B., Bearden C.E.; Costa Rica/Colombia Consortium for Genetic Investigation of Bipolar Endophenotypes, Sprooten E., Glahn D.C., Pearlson G.D., Emsell L., Kenney J., Scanlon C., McDonald C., Cannon D.M., Almeida J., Versace A., Caseras X., Lawrence N.S., Phillips M.L., Dima D., Delvecchio G., Frangou S., Satterthwaite T.D., Wolf D., Houenou J., Henry C., Malt U.F., Bøen E., Elvsåshagen T., Young A.H., Lloyd A.J., Goodwin G.M., Mackay C.E., Bourne C., Bilderbeck A., Abramovic L., Boks M.P., van Ha-ren N.E., Ophoff R.A., Kahn R.S., Bauer M., Pfennig A., Alda M., Hajek T., Mwangi B., Soares J.C., Nickson T., Dimitrova R., Sussmann J.E., Hagenaars S., Whalley H.C., McIntosh A.M., Thompson P.M., Andreassen O.A. (2016) Subcortical volumetric abnormalities in bipolar disorder. Mol. Psychiatry. 21, 1710–1716.
Sacuiu S., Eckerström M., Johansson L., Kern S., Sigström R., Xinxin G., Östling S., Skoog I. (2018) Increased risk of dementia in subjective cognitive decline if CT crain changes are present. J. Alzheimers Dis. 66, 483–495.
Wen C., Chan L.L., Tan L.C., Tan E.K. (2016) Depression, anxiety, and apathy in Parkinson’s disease: insights from neuroimaging studies. Eur. J. Neurol. 23, 1001–1019.
Girshkin L., Matheson S.L., Shepherd A., Green M.J. (2017) Morning cortisol levels in schizophrenia and bipolar disorder: a meta-analysis. Psychoneuroendocrinology. 49, 187–206.
Karanikas E., Garyfallos G. (2015) Role of cortisol in patients at risk for psychosis mental state and psychopathological correlates: A systematic review. Psychiatry Clin. Neurosci. 69, 268–282.
Ouanes S., Popp J. (2019) High cortisol and the risk of dementia and Alzheimer’s disease: A review of the literature. Front. Aging Neurosci. 11, 43. https://doi.org/10.3389/fnagi.2019.00043
Bao A.M., Meynen G., Swaab D.F. (2008) The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res. Rev. 57, 531–553.
Soares N., Pereira G.M., Altmann V., de Almeida R.M., Rieder C.R. (2019) Cortisol levels, motor, cognitive and behavioral symptoms in Parkinson’s disease: a systematic review. J. Neural. Transm. (Vienna). 126, 219–232.
Santos L.E., Beckman D., Ferreira S.T. (2016) Microglial dysfunction connects depression and Alzheimer’s disease. Brain. Behav. Immun. 55, 151–165.
Fillman S., Cloonan N., Catts V., Miller L.C., Wong J., McCrossin T., Cairns M., Weickert C.S. (2013) Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry. 18, 206–214.
Miller B.J., Buckley P., Seabolt W., Mellor A., Kirkpatrick B. (2011) Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol. Psychiatry. 70, 663–671.
Benedetti F., Aggio V., Pratesi M.L., Greco G., Furlan R. (2020) Neuroinflammation in bipolar depression. Front. Psychiatry. 11, 71. https://doi.org/10.3389/fpsyt.2020.00071
Kunz M., Cereser K.M., Goi P.D. (2011) Serum levels of IL-6, IL-10 and TNF-alpha in patients with bipolar disorder and schizophrenia: differences in pro- and anti-inflammatory balance. Rev. Bras. Psiquiatr. 333, 268–274.
Calsolaro V., Edison P. (2016) Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732.
De Virgilio A., Greco A., Fabbrini G., Inghilleri M., Rizzo M.I., Gallo A., Conte M., Rosato C., Ciniglio Appiani M., de Vincentiis M. (2016) Parkinson’s disease: autoimmunity and neuroinflammation. Autoimmun. Rev. 15, 1005–1011.
Peng S., Li W., Lv L., Zhang Z., Zhan X. (2018) BDNF as a biomarker in diagnosis and evaluation of treatment for schizophrenia and depression. Discov. Med. 26, 127–136.
Barbosa I.G., Huguet R.B., Sousa L.P. (2011) Circulating levels of GDNF in bipolar disorder. Neurosci Lett. 5022, 103–106.
Barbosa I.G., Rocha N.P., Miranda A.S., Huguet R.B., Bauer M.E., Reis H.J., Teixeira, A.L. (2013) Increased BDNF levels in long-term bipolar disorder patients. Rev. Bras. Psiquiatr. 351, 67–69.
Kraft A.D., McPherson C.A., Harry G.J. (2009) Heterogeneity of microglia and TNF signaling as determinants for neuronal death or survival. Neurotoxicology. 305, 785–793.
Sampaio T.B., Savall A.S., Gutierrez M.E.Z., Pinton S. (2017) Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: implications for pathogenesis and therapy. Neural. Regen. Res. 12, 549–557.
Valero J., Bernardino L., Cardoso F.L., Silva A.P., Fontes-Ribeiro C., Ambrósio A.F., Malva J.O. (2017) Impact of neuroinflammation on hippocampal neurogenesis: relevance to aging and Alzheimer’s Disease. J. Alzheimers Dis. 60, 161–168.
Lim J., Bang Y., Choi H.J. (2018) Abnormal hippocampal neurogenesis in Parkinson’s disease: relevance to a new therapeutic target for depression with Parkinson’s disease. Arch. Pharm. Res. 41, 943–954.
Зоркина Я.А., Морозова А.Ю., Андреюк Д.С. (2019) Перспективы использования молекулярно-генетических методов для исследования патофизиологии шизофрении. Психическое здоровье. 12, 26–33.
Ikeda M., Saito T., Kondo K., Iwata N. (2018) Genome-wide association studies of bipolar disorder: A systematic review of recent findings and their clinical implications. Psychiatry Clin. Neurosci. 72, 52–63.
Marioni R.E., Harris S.E., Zhang Q, McRae A.F., Hagenaars S.P., Hill W.D., Davies G., Ritchie C.W., Gale C.R., Starr J.M., Goate A.M., Porteous D.J., Yang J., Evans K.L., Deary I.J., Wray N.R., Visscher P.M. (2019) GWAS on family history of Alzheimer’s disease. Transl. Psychiatry. 9, 161. https://doi.org/10.1038/s41398-018-0150-6
Chang D., Nalls M.A., Hallgrímsdóttir I.B., Hunkapiller J., van der Brug M., Cai F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team, Kerchner G.A., Ayalon G., Bingol B., Sheng M., Hinds D., Behrens T.W., Singleton A.B., Bhangale T.R., Graham R.R. (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516.
Amare A.T., Vaez A., Hsu Y.H., Direk N., Kamali Z., Howard D.M., McIntosh A.M., Tiemeier H., Bültmann U., Snieder H., Hartman C.A. (2019) Bivariate genome-wide association analyses of the broad depression phenotype combined with major depressive disorder, bipolar disorder or schizophrenia reveal eight novel genetic loci for depression. Mol. Psychiatry. 1, 18–33.
Wray N.R., Ripke S., Mattheisen M., Trzaskowski M., Byrne E.M., Abdellaoui A. Adams M.J., Agerbo E., Air T.M., Andlauer T.M.F., Bacanu S.A., Bækvad-Hansen M., Beekman A.F.T., Bigdeli T.B., Binder E.B., Blackwood D.R.H., Bryois J., Buttenschøn H.N., Bybjerg-Grauholm J., Cai N., Castelao E., Christensen J.H., Clarke T.K., Coleman J.I.R., Colodro-Conde L., Couvy-Duchesne B., Craddock N., Crawford G.E., Crowley C.A., Dashti H.S., Davies G., Deary I.J., Degenhardt F., Derks E.M., Direk N., Dolan C.V., Dunn E.C., Eley T.C., Eriksson N., Escott-Price V., Kiadeh F.H.F., Finucane H.K., Forstner A.J., Frank J., Gaspar H.A., Gill M., Giusti-Rodríguez P., Goes F.S., Gordon S.D., Grove J., Hall L.S., Hannon E., Hansen C.S., Hansen T.F., Herms S., Hickie I.B., Hoffmann P., Homuth G., Horn C., Hottenga J.J., Hougaard D.M., Hu M., Hyde C.L., Ising M., Jansen R., Jin F., Jorgenson E., Knowles J.A., Kohane I.S., Kraft J., Kretzschmar W.W., Krogh J., Kutalik Z., Lane J.M., Li Y., Li Y., Lind P.A., Liu X., Lu L., MacIntyre D.J., MacKinnon D.F., Maier R.M., Maier W., Marchini J., Mbarek H., McGrath P., McGuffin P., Medland SE, Mehta D, Middeldorp C.M, Mihailov E, Milaneschi Y., Milani L., Mill J., Mondimore F.M., Montgomery G.W., Mostafavi S., Mul-lins N., Nauck M., Ng B., Nivard M.G., Nyholt D.R., O’Reilly P.F., Oskarsson H., Owen M.J., Painter J.N., Pedersen C.B., Pedersen M.G., Peterson R.E., Pettersson E., Peyrot W.J., Pistis G., Posthuma D., Purcell S.M., Quiroz J.A., Qvist P., Rice J.P., Riley B.P., Rivera M., Saeed Mirza S., Saxena R., Schoevers R., Schulte E.C., Shen L., Shi J., Shyn S.I., Sigurdsson E., Sinnamon G.B.C., Smit J.H., Smith D.J., Stefansson H., Steinberg S., Stockmeier C.A., Streit F., Strohmaier J., Tansey K.E., Teismann H., Teumer A., Thompson W., Thomson P.A., Thorgeirsson T.E., Tian C., Traylor M., Treutlein J., Trubetskoy V., Uitterlinden A.G., Umbricht D., Van der Auwera S., van Hemert A.M., Viktorin A., Visscher P.M., Wang Y., Webb B.T., Weinsheimer S.M., Wellmann J., Willemsen G., Witt S.H., Wu Y., Xi H.S., Yang J., Zhang F.; eQTLGen; 23andMe, Arolt V., Baune B.T., Berger K., Boomsma D.I., Cichon S., Dannlowski U., de Ge-us E.C.J., DePaulo J.R., Domenici E., Domschke K., Esko T., Grabe H.J., Hamilton S.P., Hayward C., Heath A.C., Hinds D.A., Kendler K.S., Kloiber S., Lewis G., Li Q.S., Lucae S., Madden P.F.A., Magnusson P.K., Martin N.G., McIntosh A.M., Metspalu A., Mors O., Mortensen P.B., Müller-Myhsok B., Nordentoft M., Nöthen M.M., O’Donovan M.C., Paciga S.A., Pedersen N.L., Penninx B.W.J.H., Perlis R.H., Porteous D.J., Potash J.B., Preisig M., Rietschel M., Schaefer C., Schulze T.G., Smoller J.W., Stefansson K., Tiemeier H., Uher R., Völzke H., Weissman M.M., Werge T., Winslow A.R., Lewis C.M., Levinson D.F., Breen G., Børglum A.D., Sullivan P.F.; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium (2018) Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681.
Hyde C.L., Nagle M.W., Tian C., Chen X., Paciga S.A., Wendland J.R., Tung J.Y., Hinds D.A., Perlis R.H., Winslow A.R. (2016) Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 48, 1031–1036.
Frey B.N., Fonseca M., Machado-Vieira R., Soares J.C., Kapczinski F. (2004) Neuropathological and neurochemical abnormalities in bipolar disorder. Rev. Bras. Psiquiatr. 26, 180–188.
Goldstein B.I., Kemp D.E., Soczynska J.K., McIntyre R.S. (2009) Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J. Clin. Psychiatry. 708, 1078–1090.
Petersen J.D., Waldorff F.B., Siersma V.D., Phung T.K.T., Bebe A.C.K.M., Waldemar, G. (2017) Major depressive symptoms increase 3-year mortality rate in patients with mild dementia. Int. J. Alzheimers. Dis. 21, 45–58.
Trillo L., Das D., Hsieh W., Medina B., Moghadam S., Lin B., Dang V., Sanchez M.M., De Miguel Z., Ashford J.W., Salehi A. (2013) Ascending monoaminergic systems alterations in Alzheimer’s disease. Translating basic science into clinical care. Neurosci. Biobehav. Rev. 37, 1369–1379.
Dong H., Murphy K.M., Meng L., Montalvo-Ortiz J., Zeng Z., Kolber B.J., Zhang S., Muglia L.J., Csernansky J.G. (2012) Corticotrophin releasing factor accelerates neuropathology and cognitive decline in a mouse model of Alzheimer’s disease. J. Alzheimers. Dis. 28, 579–592.
Glass C.K., Saijo K., Winner B., Marchetto M.C., Gage F.H. (2010) Mechanisms underlying inflammation in neurodegeneration. Cell. 140, 918–934.
Wuwongse S., Chang R.C.C., Law A.C.K. (2010) The putative neurodegenerative links between depression and Alzheimer’s disease. Prog. Neurobiol. 12, 32–57.
Chhibber A., Zhao L. (2017) ER β and ApoE isoforms interact to regulate BDNF – 5-HT 2A signaling and synaptic function in the female brain. Alzheimers. Res. Ther. 21, 9–49.
Leentjens A.F. (2015) Parkinson disease: depression—risk factor or early symptom in Parkinson disease? Nat. Rev. Neurol. 18, 432–433.
Olivola E., Pierantozzi M., Imbriani P., Liguori C., Stampanoni Bassi M., Conti M., D’Angelo V., Mercuri N.B., Stefani A. (2014) Serotonin impairment in CSF of PD patients, without an apparent clinical counterpart. PLoS One. 9 (7), e101763.https://doi.org/10.1371/journal.pone.0101763
Borgonovo J., Allende-Castro C., Laliena A., Guerrero N., Silva, H., Concha M.L., Guerrero E., An Silva H., Concha M.L. (2017) Changes in neural circuitry associated with depression at pre-clinical, pre-motor and early motor phases of Parkinson’s disease. Parkinsonism Relat. Disord. 35, 17–24.
Remy P., Doder M., Lees A., Turjanski N., Brooks D. (2005). Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain. 128, 1314–1322.
Wang Y., Liu H., Du X.D., Zhang Y., Yin G., Zhang B.S., Soares J.C., Zhang X.Y. (2017). Association of low serum BDNF with depression in patients with Parkinson’s disease. Parkinsonism Relat. Disord. 41, 73–78.
Foltynie T., Sawcer S., Brayne C., Barker R.A. (2002). The genetic basis of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry. 73, 363–370.
Burn D.J., Tiangyou W., Allcock L.M., Davison J., Chinnery P.F. (2006). Allelic variation of a functional polymorphism in the serotonin transporter gene and depression in Parkinson’s disease. Parkinsonism Relat. Disord. 12, 139–141.
Gören J.L. (2016) Brain-derived neurotrophic factor and schizophrenia. Ment. Health Clin. 6, 285–288.
Toll A., Mane A. (2015) Brain-derived neurotrophic factor levels in first episode psychosis: a systematic review. World J. Psychiatry. 5, 154–159.
Zhang X.Y., Chen D.C., Xiu M.H., Haile C.N., Luo X., Xu K. (2012) Cognitive and serum BDNF correlates of BDNF Val66Met gene polymorphism in patients with schizophrenia and normal controls. Hum. Genet. 131, 1187–1195.
Miranda M., Morici J.F., Zanoni M.B., Bekinschtein P. (2019) Brain-derived neurotrophic factor: a key molecule for memory in the healthy and the pathological brain. Front. Cell Neurosci. 13, 363. https://doi.org/10.3389/fncel.2019.00363
Slattery D.A., Cryan J.F. (2014) The ups and downs of modelling mood disorders in rodents. ILAR. J. 55, 297–309.
Jacobsen J.P., Medvedev I.O., Caron M.G. (2012) The 5-HT deficiency theory of depression: perspectives from a naturalistic 5-HT deficiency model, the tryptophan hydroxylase 2Arg439His knockin mouse. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 367, 2444–2459.
Morozova A., Zubkov E., Strekalova T., Kekelidze Z., Storozeva Z., Schroeter C., Bazhenova N., Lesch K., Cline B., Chekhonin V. (2016) Ultrasound of alternating frequencies and variable emotional impact evokes depressive syndrome in mice and rats. Prog. Neuropsychopharmacol. Biol. Psychiatry. 68, 52–63.
Zorkina Y.A., Zubkov E.A., Morozova A.Y., Ushakova V.M., Chekhonin V.P. (2019) The comparison of a new ultrasound-induced depression model to the chronic mild stress paradigm. Front. Behav. Neurosci. 13, 146. https://doi.org/10.3389/fnbeh.2019.00146
Ушакова В.М., Горлова А.В., Зубков Е.А., Морозова А.Ю., Зоркина Я.А., Павлов Д.А., Иноземцев А.Н., Чехонин В.П. (2019) Экспериментальные модели депрессивного состояния. Журн. ВНД им. И.П. Павлова. 69, 230–247.
Jarosik J., Legutko B., Unsicker K., von Bohlen Und Halbach O. (2007) Antidepressant-mediated reversal of abnormal behavior and neurodegeneration in mice following olfactory bulbectomy. Exp. Neurol. 204, 20–28.
Саркисова К.Ю., Куликов М.А., Кудрин В.С., Наркевич В.Б., Мидзяновская И.С., Бирюкова Л.М., Фоломкина А.А., Базян А.С. (2013) Нейрохимические механизмы депрессивно-подобного поведения у крыс линии WAG/RIJ. Журн. ВНД им. И.П. Павлова. 63, 303–315.
Navabpour S., Kwapis J.L., Jarome T.J. (2020) A neuroscientist’s guide to transgenic mice and other genetic tools. Neurosci. Biobehav. Rev. 108, 732–748.
Evans M.J., Kaufman M.H. (1981) Establishment in culture of pluripotential cells from mouse embryos. Nature. 292, 154–156.
Mortensen R. (2011) Production of a heterozygous mutant cell line by homologous recombination (single knockout). Curr. Protoc. Neurosci. Ch. 4. https://doi.org/10.1002/0471142301.ns0430s55
Babinet C., Morello D., Renard J.P. (1989) Transgenic mice. Genome. 31, 938–949.
Achterberg K.G., Buitendijk G.H., Kool M.J., Goorden S.M., Post L., Slump D.E., Silva A.J., van Woerden G.M., Kushner S.A., Elgersma Y. (2014) Temporal and region-specific requirements of alpha CaMKII in spatial and contextual learning. J. Neurosci. 34, 11180–11187.
Orban P.C., Chui D., Marth J.D. (1992) Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. USA. 89, 6861–6865.
Kim H., Kim M., Im S.K., Fang S. (2018) Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 34, 147–159.
Canavello P.R., Egan R.J., Bergner C.L., Hart P.C., Cachat J.M., Kalueff A.V. (2009) Genetic animal models of depression. Neurometh. 44, 191–200.
Chourbaji S., Gass P. (2008) Glucocorticoid receptor transgenic mice as models for depression. Brain Res. Rev. 57, 554–560.
Knott G.J., Doudna J.A. (2018) CRISPR-Cas guides the future of genetic engineering. Science. 361, 866–869.
Hussain W., Mahmood T., Hussain J., Ali N., Shah T., Qayyum S., Khan I. (2019) CRISPR/Cas system: A game changing genome editing technology, to treat human genetic diseases. Gene. 685, 70–75.
Zhang F., Wen Y., Guo X. (2014) CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum. Mol. Genet. 23, 40–46.
Adli M. (2018) The CRISPR tool kit for genome editing and beyond. Nat. Commun. 9, 1911. https://doi.org/10.1038/s41467-018-04252-2
Rost B.R., Schneider-Warme F., Schmitz D., Hegemann P. (2017) Optogenetic tools for subcellular applications in neuroscience. Neuron. 96, 572–603.
Leopold A.V., Chernov K.G., Verkhusha V.V. (2018) Optogenetically controlled protein kinases for regulation of cellular signaling. Chem. Soc. Rev. 47, 2454–2484.
Biselli T., Lange S.S., Sablottny L., Steffen J., Walther A. (2019) Optogenetic and chemogenetic insights into the neurocircuitry of depression-like behaviour: A systematic review. Eur. J. Neurosci. https://doi.org/10.1111/ejn.14603
Boyden E.S., Zhang F., Bamberg E., Nagel G., Deisseroth K. (2005) Millisecond-timescale, genetically 719 targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268.
Muir J., Lopez J., Bagot R.C. (2019) Wiring the depressed brain: optogenetic and chemogenetic circuit interrogation in animal models of depression. Neuropsychopharmacology. 44, 1013–1026.
Roth B.L. (2016) DREADDs for neuroscientists. Neuron. 89, 683–694.
Teissier A., Magueresse C.L., Olusakin J., Costa B.L.S.A. da, Stasi A.M.D., Bacci A., Kawasawa Y.I., Vaidya V.A., Gaspar P. (2019) Early-life stress impairs postnatal oligodendrogenesis and adult emotional behaviour through activity-dependent mechanisms. Mol. Psychiatry. https://doi.org/10.1038/s41380-019-0493-2
Cheng J., Umschweif G., Leung J., Sagi Y., Greengard P. (2019) HCN2 channels in cholinergic interneurons of nucleus accumbens shell regulate depressive behaviors. Neuron. 101, 662–672.
Leopold A.V., Chernov K.G., Shemetov A.A., Verkhusha V.V. (2019) Neurotrophin receptor tyrosine kinases regulated with near-infrared light. Nat. Commun. 10, 1129. https://doi.org/10.1038/s41467-019-08988-3
McIntosh A.M., Sullivan P.F., Lewis C.M. (2019) Uncovering the genetic architecture of major depression. Neuron. 102, 91–103.
Mullins N., Lewis C.M. (2017) Genetics of depression: progress at last. Curr. Psychiatry Rep. 19, 43. https://doi.org/10.1007/s11920-017-0803-9
Maul S., Giegling I., Fabbri C., Corponi F., Serretti A., Rujescu D. (2020) Genetics of resilience: implications from genome-wide association studies and candidate genes of the stress response system in posttraumatic stress disorder and depression. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 183, 77–94.
Ripke S., Wray N.R., Lewis C.M., Hamilton S.P., Weissman M.M., Breen G., Byrne E.M., Blackwood D.H., Boomsma D.I., Cichon S., Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, Heath A.C., Holsboer F., Lucae S., Madden P.A., Martin N.G., McGuffin P., Muglia P., Noethen M.M., Penninx B.P., Pergadia M.L., Potash J.B., Rietschel M., Lin D., Müller-Myhsok B., Shi J., Steinberg S., Grabe H.J., Lichtenstein P., Magnusson P., Perlis R.H., Preisig M., Smoller J.W., Stefansson K., Uher R., Kutalik Z., Tansey K.E., Teumer A., Viktorin A., Barnes M.R., Bettecken T., Binder E.B., Breuer R., Castro V.M., Churchill S.E., Coryell W.H., Craddock N., Craig I.W., Czamara D., De Geus E.J., Degenhardt F., Farmer A.E., Fava M., Frank J., Gainer V.S., Gallagher P.J., Gordon S.D., Goryachev S., Gross M., Guipponi M., Henders A.K., Herms S., Hickie I.B., Hoefels S., Hoogendijk W., Hottenga J.J., Iosifescu D.V., Ising M., Jones I., Jones L., Jung-Ying T., Knowles J.A., Kohane I.S., Kohli M.A., Korszun A., Landen M., Lawson W.B., Lewis G., Macintyre D., Maier W., Mattheisen M., McGrath P.J., McIntosh A., McLean A., Middeldorp C.M., Middleton L., Montgomery G.M., Murphy S.N., Nauck M., Nolen W.A., Nyholt D.R., O’Donovan M., Oskarsson H., Pedersen N., Scheftner W.A., Schulz A., Schulze T.G., Shyn S.I., Sigurdsson E., Slager S.L., Smit J.H., Stefansson H., Steffens M., Thorgeirsson T., Tozzi F., Treutlein J., Uhr M., van den Oord E.J., Van Grootheest G., Völzke H., Weilburg J.B., Willemsen G., Zitman F.G., Neale B., Daly M., Levinson D.F., Sullivan P.F. (2013). A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry. 18, 497–511.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология