

Молекулярная биология, 2020, T. 54, № 5, стр. 802-812
Идентификация и характеристика кольцевых РНК из камбия развивающегося стебля проростков тополя
W. Q. Zheng a, b, Y. Zhang a, b, B. Chen a, b, M. Wei c, X. W. Wang a, b, L. Du a, b, *
a Beijing Advanced Innovation Center of Tree Breeding by Molecular Design, Beijing Forestry University
100083 Beijing, China
b College of Biological Sciences and Technology, Beijing Forestry University
100083 Beijing, China
c COFCO Nutrition & Health Research Institute
102209 Beijing, China
* E-mail: duliang@bjfu.edu.cn
Поступила в редакцию 02.01.2020
После доработки 27.02.2020
Принята к публикации 05.03.2020
Аннотация
Некодирующие РНК выполняют множество функций в жизни растений. Недавно показали, что ковалентно замкнутые кольцевые молекулы РНК (цирРНК) образуют группу изоформ с тканеспецифичной экспрессией, которые часто взаимодействуют с линейными мРНК, регулируя функции генов растений. Однако цирРНК из камбия тополя и их потенциальная роль в развитии камбия и ксилемы ранее не были изучены. Нами секвенирован транскриптом камбия на двух стадиях развития проростков тополя, идентифицированы и охарактеризованы 4912 цирРНК. Транскрипты 87 генов, находящихся на разных хромосомах, подвергаются альтернативному бэксплайсингу с циркуляризацией, ведущей к образованию цирРНК. Всего описано 928 генов, продуцирующих 1138 цирРНК, которые с помощью GO-анализа отнесены к трем основным функциональным категориям. Выявлены 39 цирРНК, дифференциально экспрессирующихся в камбии на двух стадиях развития проростков. Предполагается, что 24 дифференциально экспрессируемых цирРНК взаимодействуют с 98 микроРНК-мишенями, часть которых связана с ростом и развитием камбия. Полученные результаты указывают на важную роль цирРНК камбия в регуляции роста и развития проростков тополя.
ВВЕДЕНИЕ
Сосудистая система, которая является продуктом адаптации растений к наземной жизни, включает все сосудистые ткани растения. Эта система пронизывает все растение, она выполняет как механическую функцию, так и участвует в передвижении растворенных питательных веществ [1, 2]. Сосудистую систему по последовательности ее развития и составу можно разделить на первичную и вторичную. Первичная система представлена преимущественно в первичных зрелых тканях, а вторичная – во вторичных. Сосудистая система состоит в основном из камбия, ксилемы и флоэмы. Сосудистый камбий расположен между ксилемой и флоэмой. В результате деления клеток камбия образуются новые ксилема и флоэма (вторичная ксилема и вторичная флоэма), что приводит к росту стеблей и корней в ширину. Слияние пучкового и межпучкового камбия с образованием непрерывного камбиального кольца (называемого сосудистым камбием) вызывает вторичный рост [3], в результате которого происходит прирост ствола в толщину, что в случае древесных культур имеет важное экономическое значение [4, 5].
Тополь (Populus trichocarpa) из-за его быстрого роста, быстрого формирования древесной массы и известной полной геномной последовательности часто используется для систематического изучения вторичного роста [6]. Развитие технологии высокопроизводительного секвенирования и анализ больших массивов данных позволили идентифицировать много некодирующих РНК, вовлеченных во вторичный рост, особенно микроРНК. Например, AtmiR857 участвует в регуляции вторичного роста сосудистых тканей у Arabidopsis thaliana, влияя на экспрессию гена-мишени LACCASE7 [7]. Кроме того, показано, что miR397a тополя участвует в синтезе лигнина, негативно регулируя гены лакказы [8]. Установлено также, что HB4 и HB7 – гены-мишени miR166, вовлечены в развитие камбия тополя [9, 10]. Однако роль кольцевых РНК в регуляции вторичного роста тополя не изучена.
Ковалентно замкнутые кольцевые РНК (цирРНК) – стабильные эндогенные некодирующие РНК, широко распространенные у эукариот, образуются путем ковалентного замыкания 3'- и 5'-концов некодирующих РНК [11]. цирРНК могут действовать как губки для микроРНК, конкурентно подавляя ингибирование их генов-мишеней. [12]. Таким образом, взаимодействия между цирРНК, микроРНК и мРНК можно рассматривать как один из основных механизмов транскрипционной и посттранскрипционной регуляции экспрессии генов [12]. цирРНК изучены у различных видов растений, включая рис [13], A. thaliana [14], Triticum aestivum [15], томаты [16], сою [17] и кукурузу [18]. Ранее показали, что цирРНК играют важную роль в ответе растений на биотический и абиотический стресс, а также в процессах роста и развития [13, 19–24]. Например, выявлена дифференциальная экспрессия 27 цирРНК экзонного типа в растениях риса в условиях фосфорного стресса [22]. В плодах сладкого перца при действии холодового стресса дифференциально экспрессируются 36 цирРНК [25]. Участие длинных некодирующих РНК во вторичном росте P. tomentosa обнаружено с помощью аллельной дискриминации [26]. Реконструирован также ряд регуляторных сетей (длинные некодирующие РНК/цирРНК−микроРНК−мРНК), участвующих в регуляции развития корней и меристем проростков Populus [27]. Тем не менее, участие цирРНК в камбий-опосредованном росте и развитии ствола практически не изучено.
Сначала, чтобы установить роль цирРНК в камбии и их участие в развитии и вторичном росте ствола тополя, с помощью технологии секвенирования РНК идентифицировали цирРНК в апикальном и базальном камбии. Всего идентифицировано 4912 цирРНК, 1138 из которых кодируются 928 генами. Эти гены в ходе GO-анализа были классифицированы по подкатегориям биологического процесса, клеточного компонента и молекулярных функций. Показана дифференциальная экспрессия 39 цирРНК в двух образцах стволов. Предсказано взаимодействие 24 дифференциально экспрессирующихся цирРНК (ДЕцирРНК) и 98 микроРНК в качестве мишеней. Получены профили экспрессии цирРНК в стволовом камбии молодых проростков тополя, установлена важная роль цирРНК во вторичном росте и развитии тополя.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Подготовка растительных образцов. У молодых побегов тополя сорта 84K (Populus alba × P. glandulosa) вырезали участки между первым и третьим узлом и культивировали на полунасыщенной агаризованной среде Мурасиге и Скуга (1/2 MS) в течение примерно 6 недель. После того, как растения-регенеранты достигали примерно 10 см в высоту, их переносили в смесь вегетативной почвы и вермикулита (1 : 1 по объему) и растили в теплице при 25°C с циклом освещения день/ночь 16 ч/8 ч. Растения, выращенные в теплице в течение примерно 1 мес. и имеющие одинаковую высоту, отбирали для дальнейшего анализа. Камбий из апикальной (T) и базальной (B) частей стебля отдельных деревьев отбирали с помощью нити (CM1850 Cryostat, “Leica”, Германия), быстро замороженной в жидком азоте, и хранили при −80°C до выделения РНК.
Выделение РНК, приготовление библиотек и секвенирование. Суммарную РНК выделяли с помощью набора RNAprep Pure Plant Kit (DP419, “TIANGEN”, КНР) в соответствии с инструкциями производителя. Очищенные образцы РНК после определения их чистоты, концентрации и целостности использовали для получения РНК-библиотек с помощью набора NEB Next® Ultra™ Directional RNA Library Prep Kit (E7775, КНР). Концентрацию и размер фрагментов в полученных библиотеках определяли с помощью флуориметра Qubit® 2.0 и биоанализатора Agilent 2100 соответственно. Качество библиотек и их точную концентрацию подтверждали с помощью количественной ПЦР в реальном времени. Библиотеки, прошедшие проверку, использовали в высокопроизводительном секвенировании с помощью платформы HiSeq X Ten. Длина парных прочтений последовательностей составила 150 п.н.
Характеристика и идентификация цирРНК. Из исходных прочтений удаляли последовательности адапторов, прочтения низкого качества и прочтения, содержащие более 5% N (неопределенная информация об основании). Очищенные данные выравнивали с референсной последовательностью генома P. trichocarpa (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Ptri-chocarpa_er) с помощью Tophat2 (v2.1.0) [28]. цирРНК идентифицировали с помощью программного обеспечения find_circ [20]. Кандидатные цирРНК удовлетворяли следующим критериям: (1) GU/AG присутствует с обеих сторон участка сплайсинга [29]; (2) имеют четкий участок ветвления; (3) имеют только два основания, не соответствующие референсной последовательности; (4) участки ветвления не могут находиться вне двух якорных нуклеотидов; (5) как минимум два прочтения содержат участок стыка; (6) выравнивание с правильным участком в геноме оценивается как минимум на 35 пунктов выше, чем выравнивание с другими участками.
Анализ дифференциальной экспрессии цирРНК. цирРНК, уровень экспрессии которых изменился в 2 раза и более при значении P менее 0.05, рассматривали как ДЕцирРНК. Изменение экспрессии рассчитывали как соотношение уровней экспрессии в двух образцах. Учитывая, что анализ дифференциальной экспрессии цирРНК это независимый статистический тест проверки гипотез, можно ожидать появление ложноположительных результатов при определении уровней экспрессии многих цирРНК. Следовательно, при поиске ДЕцирРНК с помощью программы DEseq2 использовали значения P, скорректированные для множественных сравнений. В экспериментах по выявлению ДЕцирРНК использовали три биологических повторности.
GO и COG-анализ хозяйских цирРНК. Анализ обогащения GO-онтологий генами цирРНК выполнен с помощью пакета программ GOseq, написанного на языке R [30]. Базу данных COG использовали для выявления функционально значимых обогащений генов цирРНК [31].
Предсказание микроРНК в качестве мишеней цирРНК и функциональная аннотация. Взаимодействия цирРНК–микроРНК и микроРНК–мРНК предсказывали с помощью программы psRNATarget [32]. Сеть взаимодействий строили с использованием Cytoscape 3.4.0 [33]. Обнаруженные мРНК разделяли на три функциональные группы, а именно, биологический процесс, молекулярная функция и клеточный компонент, на основе аннотаций GO с помощью программного обеспечения BLAST2GO (https://www.blast2go.com/) [34]. Функции мРНК анализировали с помощью программного обеспечения KOBAS на основе аннотаций базы данных KEGG [35].
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Идентификация цирРНК в камбии развивающегося стебля
Из двух частей развивающегося стебля (T и B, взятых из апикальной и базальной частей стебля соответственно) с целью идентификации цирРНК в камбии получили шесть библиотек РНК, свободных от рРНК. Проведено полное секвенирование транскриптомов шести образцов, и в итоге получили 113.47 Гб отфильтрованных данных. Чистые данные каждого образца составляли 15.50 Гб, содержание прочтений с качеством оснований Q30 – 90.81% и выше. Наблюдали высокую степень сходства между биологическими повторами. Чистые прочтения, полученные после секвенирования библиотек, использовали в картировании на референсный геном тополя. Доля выровненных прочтений на референсный геном варьировала от 98.12 до 98.34%. Всего с использованием программного обеспечения find_circ обнаружено 4912 цирРНК. Из идентифицированных цирРНК 3024 и 3138 найдены в T и B участках стебля соответственно (табл. 1).
Свойства цирРНК в камбии стебля
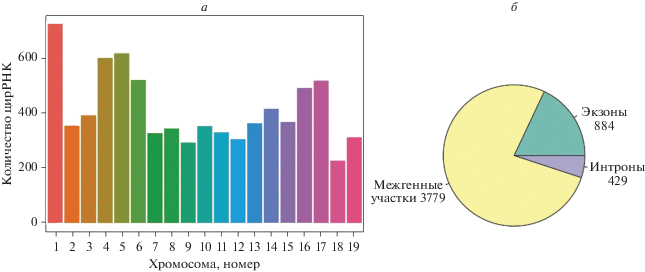
Согласно результатам картирования, цирРНК неравномерно распределены по хромосомам тополя (рис. 1а). Например, количество цирРНК на хромосомах 1, 4, 5, 6, 16 и 17 значительно больше, чем на других хромосомах с количеством прочтений, картированных на хромосомы, более 500. Меньше всего цирРНК обнаружено на хромосоме 18, на которую картировано менее 200 прочтений. Выделяют три типа цирРНК, а именно, экзонные, интронные и межгенные [19, 36, 37]. Большинство цирРНК (76.93%) происходит из межгенных участков (рис. 1б). Оставшаяся часть цирРНК соответствует экзонам (18.00%) и интронам (5.07%) (рис. 1б). В целом, 1133 цирРНК образуется из 928 кодирующих генов.
Рис. 1.
Типы цирРНК и их распределение по хромосомам. а – Количество цирРНК, обнаруженных на хромосомах тополя; б – классификация типов цирРНК по участкам генов.
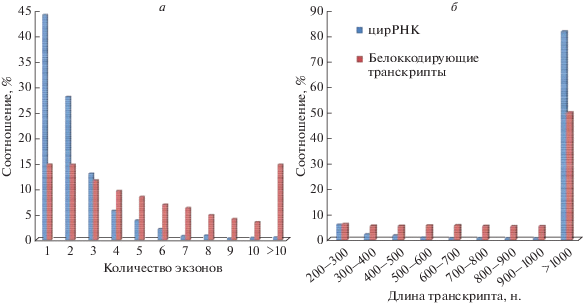
С целью анализа геномных локусов цирРНК сопоставили число экзонов и длину транскриптов кандидатных цирРНК с соответствующими характеристиками белоккодирующих генов в референсном геноме P. trichocarpa. Большинство цирРНК из камбия стебля (72.17%) содержат 1–2 экзона. В то же время, в линейных генах число экзонов варьирует от 1 до 10 (рис. 2а). Длина цирРНК из камбия стебля варьирует от 150 до 99650 н., и у большинства из них (85.27%) длина превышает 1000 н. Напротив, длина приблизительно 52.19% линейных генов превышает 1000 п.н. Среди транскриптов длиной менее 1000 н. преобладают транскрипты длиной 200–300 н. (6.34%) (рис. 2б).
Рис. 2.
Сопоставление числа экзонов, длины транскриптов цирРНК и белоккодирующих генов из стеблевого камбия. а – Большинство цирРНК содержат 1–2 экзона, тогда как количество экзонов в генах варьирует от 1 до 10. б – Длина цирРНК варьирует от 150 до 99650 н. Длина большинства цирРНК превышает 1000 н., тогда как только почти половина транскриптов белоккодирующих генов длиннее 1000 н.
Альтернативная циркуляризация цирРНК стеблевого камбия
В процессе биогенеза цирРНК растения могут продуцировать различные альтернативные кольцевые формы из одного и того же локуса. Эти формы подразделяют на два основных типа – формы с циркуляризацией в результате альтернативного бэксплайсинга и формы с циркуляризацией после альтернативного сплайсинга [13, 38]. Поэтому анализировали формы альтернативной циркуляризации цирРНК, происходящих из хозяйских генов. В транскриптах 87 из 928 генов идентифицирована циркуляризации путем альтернативного бэксплайсинга. Из них 48 локусов продуцировали две циклические изоформы, 20 – три, семь – четыре изоформы, семь – пять изоформ, два – шесть изоформ, а оставшиеся три локуса – восемь и более изоформ (табл. 2). Более подробная информация о событиях циркуляризации продуктов альтернативного бэксплайсинга представлена в табл. S1 (см. Приложение).
Идентификация дифференциально экспрессируемых цирРНК
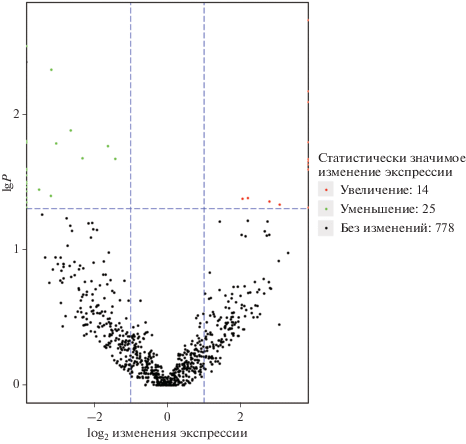
Чтобы построить профили экспрессии цирРНК в камбии из разных частей стебля проростков тополя, в T и B образцах выявлили ДЕцирРНК. (табл. S2 , см. Приложение). Из 4912 цирРНК, обнаруженных в стеблевом камбии, 3024 и 3138 были специфичными для T и B образцов соответственно (рис. 3). В целом, 1250 цирРНК были представлены в обоих образцах, а уровень экспрессии 818 цирРНК варьировал. В соответствии с критериями отбора (уровень изменения > 2 раз, P < 0.05) 39 цирРНК определены как ДЕцирРНК. Экспрессия 14 из них была повышена, а 25 снижена в образце T по сравнению с образцом B (рис. 4).
Рис. 3.
Диаграмма Венна, показывающая количество цирРНК в камбии апикального (T) и базального (B) сегментов молодых побегов тополя.
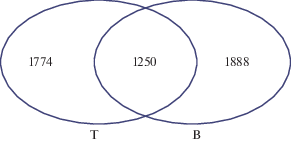
Рис. 4.
Вулканная диаграмма ДЕцирРНК. Точки на диаграмме соответствуют индивидуальным цирРНК. По оси абсцисс отложен логарифм изменения уровня экспрессии цирРНК между двумя образцами. По оси ординат – отрицательные значения логарифма частоты обнаружения ошибок. Зелеными точками (верхняя левая часть рисунка) обозначены цирРНК с пониженной экспрессией; красными точками (верхняя правая часть рисунка) – цирРНК с повышенной экспрессией, черными точками (нижняя часть рисунка) – цирРНК, не проявляющие значимых отличий в уровне экспрессии.
Биологические функции генов, кодирующих цирРНК
Функциональные характеристики ДЕцирРНК недостаточно изучены, и анализ генов этих РНК может дать ценную информацию об их функциях в раннем развитии ствола тополя. Поскольку обнаружена положительная корреляция между экспрессией цирРНК и их генов, то цирРНК, по-видимому, могут влиять на транскрипцию этих генов. Например, Ac_circ04842 активно регулирует экспрессию Achn372061 у киви [39]. Более того, сверхэкспрессия Os08circ16564 в растениях риса существенно подавляет экспрессию ее гена, указывая на то, что цирРНК могут негативно влиять на экспрессию собственных генов [13]. Как показано, всего 1133 цирРНК образуется из 928 линейных генов (табл. S3 , см. Приложение). Далее провели GO-анализ, чтобы исследовать биологические функции всех цирРНК и ДЕцирРНК, в частности. Гены цирРНК, классифицировали по трем GO-категориям: биологический процесс, клеточный компонент и молекулярная функция. Гены ДЕцирРНК, в категории биологический процесс сосредоточены преимущественно в подкатегории метаболический процесс; в категории клеточный компонент – в подкатегориях клетка, часть клетки и органелла; а в категории молекулярная функция – связывание, каталитическая активность и активность транспортера (рис. 5). GO-аннотации могут отражать то, что митотическая активность камбия требует метаболической активности клеток, чтобы обеспечить их энергией. Кроме того, выполнили COG-классификацию функций ДЕцирРНК, чтобы оценить влияние цирРНК на развитие камбия [31] (рис. 6). Гены, продуцирующие ДЕцирРНК, сосредоточены в функциональном классе O (посттрансляционная модификация, обмен белка и шапероны), что позволяет предположить вовлеченность ДЕцирРНК в регуляцию роста и развития камбия.
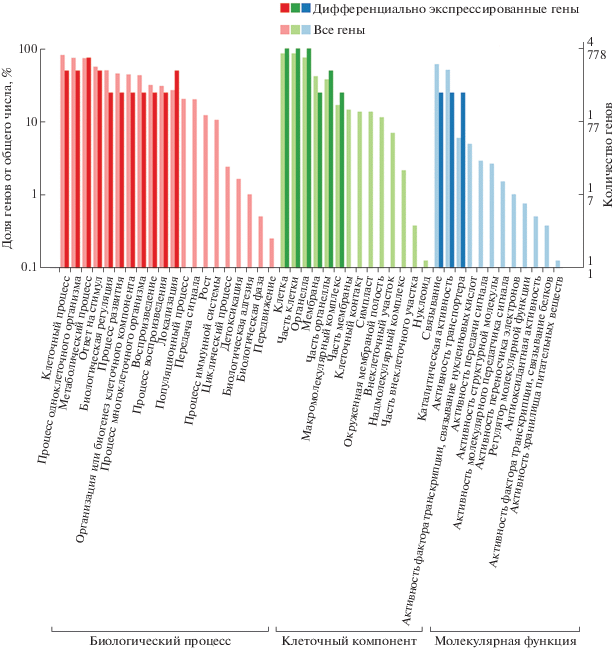

Предсказание микроРНК-мишеней цирРНК
Действуя, как губки для микроРНК, цирРНК играют регуляторную роль в процессе роста и развития растений. С помощью программного обеспечения Target Finder проведен поиск вероятных микроРНК-мишеней среди цирРНК, чтобы определить, способны ли экспрессируемые цирРНК связываться с микроРНК. В целом, выявлены вероятные взаимодействия между 2608 цирРНК и 391 микроРНК (табл. S4 , см. Приложение). В ходе анализа взаимодействий ДЕцирРНК–микроРНК установлено, что 24 ДЕцирРНК взаимодействуют с 98 микроРНК в качестве мишеней (рис. 7 и табл. S5 Приложения). Согласно предсказаниям, некоторые из этих микроРНК могут участвовать в процессе вторичного роста или регулировать время перехода между ювенильной и зрелой фазами роста (табл. 3). МикроРНК-мишени включают miR397, которая участвует в регуляции развития ксилемы у древесных растений [8], и miR475b, влияющую на свойства древесины тополя [40]. Кроме того, подавление miR156 с участием miR159 регулирует переход между ювенильной и зрелой фазами роста у Arabidopsis [41, 42]. Следовательно, цирРНК могут функционировать в качестве губок микроРНК, участвуя в регуляции роста и развития стеблевого камбия.
Рис. 7.
Сеть предсказанных взаимодействий ДЕцирРНК–микроРНК. Красными и зелеными кругами обозначены ДЕцирРНК и микроРНК соответственно.
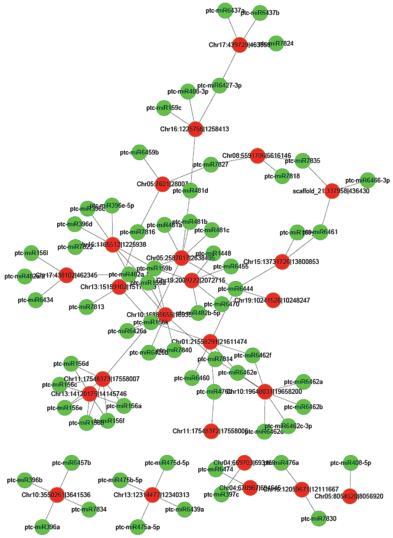
Таблица 3.
цирРНК и соответствующие микроРНК, вовлеченные в рост и развитие камбия апикальной и базальной частей стебля
| Номер | Дифференциально экспрессируемые цирРНК | микроРНК | Функция |
|---|---|---|---|
| 1 | Chr05:2587017|2638495 Chr11:17548373|17558007 Chr13:14120175|14145746 Chr13:15159102|15170879 |
miR156 | Регулирует сроки перехода между ювенильной и зрелой фазами роста |
| 2 | Chr10:16885655|16935749 Chr15:1165512|1225938 Chr16:1225756|1258413 Chr19:2009222|2072715 |
miR159 | Регулирует сроки ветегативного роста у A. thaliana |
| 3 | Chr04:669703|693169 Chr04:670987|694645 |
miR397 | Регулирует синтез лакказы |
| 4 | Chr05:8054520|8056920 Chr16:1225756|1258413 |
miR408 | Регулирует синтез лакказы |
| 5 | Chr13:12314477|12340313 | miR475 | Влияет на рост и свойства древесины |
Далее изучили функциональные аннотации GO и KEGG генов-мишеней микроРНК, чтобы получить более детальное представление о функциях ДЕцирРНК в регуляции вторичного роста (рис. S1 и S2 , см. Приложение). Функциональные аннотации GO показывают, что эти гены-мишени относятся в основном к подкатегориям метаболический процесс и клеточный процесс категории биологический процесс. Функциональные аннотации KEGG указывают на преимущественную кластеризацию мРНК в метаболизме углеводов. Согласно полученным результатам, цирРНК могут участвовать в регуляции роста и развития камбия в качестве губок, оттитровывающих микроРНК некоторых генов-мишеней.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
С использованием технологий высокопроизводительного секвенирования в сочетании с методами биоинформатики у модельных организмов и человека идентифицированы тысячи цирРНК [21, 43]. Вслед за развитием технологий высокопроизводительного секвенирования созданы базы данных цирРНК растений, в которых депонированы цирРНК риса, A. thaliana, томатов, кукурузы и ячменя [44]. Тем не менее, информация о цирРНК у видов Populus отсутствовала. В нашей работе выявлены в общей сложности 4912 цирРНК в камбии апикальной и базальной частей стеблей проростков тополя. Количество идентифицированных цирРНК сопоставимо с их количеством у других видов растений, например, 12037 цирРНК обнаружено в корнях риса, 6012 в листьях A. thaliana [22]. Размер генома A. thaliana равен 120 млн.п.н., риса – 389 млн.п.н. [14, 45]. Геном P. trichocarpa значительно больше (550 млн.п.н.), чем у A. thaliana и риса, но количество цирРНК, идентифицированных у тополя, меньше, чем у A. thaliana и риса. Это различие может быть связано с тем, что мы исследовали только ткань ствола тополя и не анализировали другие ткани. Другой причиной меньшего количества цирРНК может быть то, что мы не использовали обогащение цирРНК в библиотеке путем обработки фракции РНК РНКазой R.
Для изучения функции ДЕцирРНК, связанных с развитием камбия, провели GO- и COG-анализ продуцирующих их генов. Гены цирРНК классифицированы по трем основным функциональным категориям, а именно, биологический процесс, клеточный компонент и молекулярная функция. В ходе COG-анализа установлено, что ДЕцирРНК сосредоточены в классе O-функций. Мы предполагаем, что непрерывное деление клеток камбия, от которого дифференцируются ксилема и флоэма, может потребовать существенной модификации или обмена белков. Подобные результаты впервые представлены для Populus. Предполагается, что развитие стволового камбия тополя может регулироваться цирРНК.
Ранее установили, что цирРНК, выступая в роли губок для микроРНК, выполняют регуляторные функции в процессе роста и развития животных, людей и растений [13, 22, 46]. Однако отмечено, что лишь несколько цирРНК участвуют в адсорбции микроРНК [47]. Показано, что сверхэкспрессия цирРНК Os08circ16564 в растениях риса не привела к изменению уровня экспрессии OsmiR172 – мишени этой цирРНК [13]. Детальный анализ регуляторных функций цирРНК еще отсутствует, поэтому необходимо их дальнейшее экспериментальное изучение. У пшеницы обнаружено шесть цирРНК, потенциальных мишеней 26 микроРНК [15], а в растениях томата выявлены 102 цирРНК, которые могут служить мишенями для 24 микроРНК [48]. Однако в настоящее время цирРНК с потенциальной адсорбционной способностью составляют лишь небольшую часть от обнаруженных у разных видов растений. Например, согласно предсказанию, только небольшая доля цирРНК A. thaliana и риса (5.0 и 6.6% соответственно) рассматривается в качестве потенциальных мишеней микроРНК [22]. Еще меньшая доля (0.67%) предсказана у A. thaliana [14]. Однако в настоящем исследовании выявлено взаимодействие между 2608 цирРНК и 391 микроРНК, что составляет 53% от общего числа цирРНК у P. trichocarpa. Мы предполагаем, что большинство идентифицированных у Populus цирРНК являются новыми, отличными от цирРНК, обнаруженных ранее у A. thaliana и риса. Это может быть связано с местом отбора проб (камбий) и тем самым указывает на то, что камбий может содержать отдельную регуляторную сеть цирРНК. Однако подтверждение этой гипотезы требует проведения дополнительных экспериментов.
Примечание: Исходные файлы прочтений в формате fastq загружены на SRA NCBI (https:// submit.ncbi.nlm.nih.gov/), идентификационный номер PRJNA608835.
Авторы выражают благодарность Huimin Xu из Университета Пекина за помощь в подготовке образцов, а также Robert McKenzie из Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac) за редактирование черновой версии текста манускрипта на английском языке.
Работа выполнена при финансовой поддержке Fundamental Research Funds for the Central Universities [Grant No. BLX201418].
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы сообщают об отсутствии конфликта интересов.
Дополнительные материалы к статье размещены на сайте https://doi.org/10.31857/S0026898420050134.
Список литературы
Hertzberg M., Aspeborg H., Schrader J., Andersson A., Erlandsson R., Blomqvist K., Bhalerao R., Uhlén M., Teeri T.T., Lundeberg J., Nilsson P., Sandberg G. (2001) A transcriptional roadmap to wood formation. Proc. Natl. Acad. Sci. USA. 98, 14732–14737.
Schrader J., Nilsson J., Mellerowicz E., Berglund A., Nilsson P., Hertzberg M., Sandberg G. (2004) A high-resolution transcript profile across the wood-forming meristem of poplar identifies potential regulators of cambial stem cell identity. Plant Cell. 16, 2278–2292.
Baucher M., El Jaziri M., Vandeputte O. (2007) From primary to secondary growth: origin and development of the vascular system. J. Exp. Bot. 58, 3485–3501.
Savidge R. (2001) Intrinsic regulation of cambial growth. J. Plant Growth Regul. 20, 52–77.
Mellerowicz E.J., Sundberg B. (2008) Wood cell walls: biosynthesis, developmental dynamics and their implications for wood properties. Curr. Opin. Plant Biol. 11, 293–300.
Tuskan G.A., Difazio S., Jansson S., Bohlmann J., Grigoriev I., Hellsten U., Putnam N., Ralph S., Rombauts S., Salamov A. (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 313, 1596–1604.
Zhao Y., Lin S., Qiu Z., Cao D., Wen J., Deng X., Wang X., Lin J., Li X. (2015) MicroRNA857 is involved in the regulation of secondary growth of vascular tissues in Arabidopsis. Plant Physiol. 169, 2539–2552.
Lu S., Li Q., Wei H., Chang M.-J., Tunlaya-Anukit S., Kim H., Liu J., Song J., Sun Y.-H., Yuan L., Yeh T.-F., Peszlen I., Ralph J., Sederoff R.R., Chiang V.L. (2013) Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. USA. 110, 10848–10853.
Zhu Y., Song D., Sun J., Wang X., Li L. (2013) PtrHB7, a class III HD-zip gene, plays a critical role in regulation of vascular cambium differentiation in Populus. Mol. Plant. 6, 1331–1343.
Zhu Y., Song D., Xu P., Sun J., Li L. (2018) A HD-ZIP III gene, PtrHB4, is required for interfascicular cambium development in Populus. Plant Biotechnol. J. 16, 808–817.
Suzuki H., Tsukahara T. (2014) A view of pre-mRNA splicing from RNase R resistant RNAs. Int. J. Mol. Sci. 15, 9331–9342.
Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. (2013) Natural RNA circles function as efficient microRNA sponges. Nature. 495, 384–388.
Lu T., Cui L., Zhou Y., Zhu C., Fan D., Gong H., Zhao Q., Zhou C., Zhao Y., Lu D., Luo J., Wang Y., Tian Q., Feng Q., Huang T., Han B. (2015) Transcriptome-wide investigation of circular RNAs in rice. RNA. 21, 2076–2087.
Chen G., Cui J., Wang L., Zhu Y., Lu Z., Jin B. (2017) Genome-Wide identification of circular RNAs in Arabidopsis thaliana. Front. Plant Sci. 8, 1678.
Wang Y., Yang M., Wei S., Qin F., Zhao H., Suo B. (2016) Identification of circular RNAs and their targets in leaves of Triticum aestivum L. under dehydration stress. Front. Plant Sci. 7, 2024.
Tan J., Zhou Z., Niu Y., Sun X., Deng Z. (2017) Identification and functional characterization of tomato circRNAs derived from genes involved in fruit pigment accumulation. Sci. Rep. 7, 8594.
Zhao W., Cheng Y., Zhang C., You Q., Shen X., Guo W., Jiao Y. (2017) Genome-wide identification and characterization of circular RNAs by high throughput sequencing in soybean. Sci. Rep. 7, 5636.
Chen L., Zhang P., Fan Y., Lu Q., Li Q., Yan J., Muehlbauer G.J., Schnable P.S., Dai M., Li L. (2018) Circular RNAs mediated by transposons are associated with transcriptomic and phenotypic variation in maize. New Phytol. 217, 1292–1306.
Stoffelen R., Jimenez M.I., Dierckxsens C. Salzman J., Gawad C., Wang P.L., Lacayo N., Brown P.O. (2012) Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 7, 30733.
Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M., Loewer A., Ziebold U., Landthaler M., Kocks C., le Noble F., Rajewsky N. (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 495, 333–338.
Wang P.L., Yun B., Muh-Ching Y., Barrett S.P., Hogan G.J., Olsen M.N., Dinneny J.R., Brown P.O., Julia S. (2014) Circular RNA is expressed across the eukaryotic tree of life. PloS One. 9, e90859.
Ye C.-Y., Chen L., Liu C., Zhu Q.-H., Fan L.J. (2015) Widespread noncoding circular RNAs in plants. New Phytol. 208, 88–95.
Yang W., Zefeng W. (2015) Efficient backsplicing produces translatable circular mRNAs. RNA. 21, 172–179.
Lee S.M., Kong H.G., Ryu C.M. (2017) Are circular RNAs new kids on the block? Trends Plant Sci. 22, 357–360.
Zuo J., Wang Y., Zhu B., Luo Y., Wang Q., Gao L. (2018) Analysis of the coding and non-coding RNA transcriptomes in response to bell pepper chilling. Int. J. Mol. Sci. 19(7), pii: E2001. https://doi.org/10.3390/ijms19072001
Zhou D., Du Q., Chen J., Wang Q., Zhang D. (2017) Identification and allelic dissection uncover roles of lncRNAs in secondary growth of Populus tomentosa. DNA Res. 24, 473–486.
Liu S., Wu L., Qi H., Xu M. (2019) LncRNA/circRNA–miRNA–mRNA networks regulate the development of root and shoot meristems of Populus. Ind. Crop. Prod. 133, 333–347.
Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome. Biol. 14, 36.
Sun X., Wang L., Ding J., Wang Y., Wang J., Zhang X., Che Y., Liu Z., Zhang X., Ye J. (2016) Integrative analysis of Arabidopsis thaliana transcriptomics reveals intuitive splicing mechanism for circular RNA. FEBS Lett. 590, 3510–3516.
Young M.D., Wakefield M.J., Smyth G.K., Oshlack A. (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, 14.
Tay Y., Rinn J., Pandolfi P.P. (2014) The multilayered complexity of ceRNA crosstalk and competition. Nature. 505, 344–352.
Dai X., Zhao P.X. (2011) psRNATarget: a plant small RNA target analysis server. Nucl. Acids Res. 39, 155–159.
Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504.
Conesa A., Gotz S. (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics. 2008, 619832.
Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C.Y., Wei L. (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucl. Acids Res. 39, 316–322.
Salzman J., Chen R.E., Olsen M.N., Wang P.L., Brown P.O. (2013) Cell-type specific features of circular RNA expression. PloS Genet. 9, 1003777.
Zhang Y., Zhang X.O., Chen T., Xiang J.F., Yin Q.F., Xing Y.H., Zhu S., Yang L., Chen L.L. (2013) Circular intronic long noncoding RNAs. Mol. Cell. 51, 792–806.
Ye C.Y., Zhang X., Chu Q., Liu C., Yu Y., Jiang W., Zhu Q.H., Fan L., Guo L. (2016) Full-length sequence assembly reveals circular RNAs with diverse non-GT/AG splicing signals in rice. RNA Biol. 14, 1055–1063.
Wang Z.P., Liu Y.F., Li D.W., Li L., Zhang Q., Wang S.B., Huang H.W. (2017) Identification of circular RNAs in kiwi fruit and their species-specific response to bacterial canker pathogen invasion. Front. Plant Sci. 8, 413.
Xiao L., Quan M., Du Q., Chen J., Xie J., Zhang D. (2017) Allelic interactions among Pto-MIR475b and its four target genes potentially affect growth and wood properties in Populus. Front. Plant Sci. 8, 1055.
Wu G., Park M.Y., Conway S.R., Wang J.-W., Weigel D., Poethig R.S. (2009) The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell. 138, 750–759.
Guo C., Xu Y., Shi M., Lai Y., Wu X., Wang H., Zhu Z., Poethig R.S., Wu G. (2017) Repression of miR156 by miR159 regulates the timing of the juvenile-to-adult transition in Arabidopsis. Plant Cell. 29, 1293–1304.
Abe N., Matsumoto K., Nishihara M, Nakano Y., Shibata A., Maruyama H., Shuto S., Matsuda A., Yoshida M., Ito Y., Abe H. (2015) Rolling circle translation of circular RNA in living human cells. Sci. Rep. 5, 16435.
Chu Q., Zhang X., Zhu X., Liu C., Mao L., Ye C., Zhu Q.H., Fan L. (2017) PlantcircBase: a database for plant circular RNAs. Mol. Plant. 10, 1126–1128.
Goff S.A., Darrell R., Lan T.H., Presting G., Wang R., Dunn M., Glazebrook J., Sessions A., Oeller P., Varma H., Hadley D., Hutchison D., Martin C., Katagiri F., Lange B.M., Moughamer T., Xia Y., Budworth P., Zhong J., Miguel T., Paszkowski U., Zhang S., Colbert M., Sun W.L., Chen L., Cooper B., Park S., Wood T.C., Mao L., Quail P., Wing R., Dean R., Yu Y., Zharkikh A., Shen R., Sahasrabudhe S., Thomas A., Cannings R., Gutin A., Pruss D., Reid J., Tavtigian S., Mitchell J., Eldredge G., Scholl T., Miller R.M., Bhatnagar S., Adey N., Rubano T., Tusneem N., Robinson R., Feldhaus J., Macalma T., Oliphant A., Briggs S. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science. 296, 92–100.
Huntzinger E., Izaurralde E. (2011) Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 12, 99–110.
Guo J.U., Agarwal V., Guo H., Bartel D.P. (2014) Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 15, 409.
Zuo J., Wang Q., Zhu B., Luo Y., Gao L. (2016) Deciphering the roles of circRNAs on chilling injury in tomato. Biochem. Biophys. Res. Commun. 479, 132–138.
Дополнительные материалы
- скачать Zheng_ESM_1.tif
- Figure S1. The GO function annotations of the predicted miRNA target genes.
- скачать Zheng_ESM_1.tif
- Figure S2. The KEGG function annotations of the predicted miRNA target genes.
- скачать Zheng_ESM_2.xls
- Dataset S1. The details of the alternative back-splicing circularization events.
- скачать Zheng_ESM_3.xls
- Dataset S2. DEcircRNAs
- скачать Zheng_ESM_4.xls
- Dataset S3. A total of 1133 circRNAs originated from 928 host linear genes.
- скачать Zheng_ESM_5.xls
- Dataset S4. In total, 2608 circRNAs and 391 miRNAs interactions were detected.
- скачать Zheng_ESM_6.xls
- Dataset S5. Table S5 24 DEcircRNAs interacted with 98 miRNAs as targets.
Инструменты
Молекулярная биология